

Abstract
Appropriate control of infection depends on the generation of lymphocytes armed with a particular array of cytokine and chemokine effector molecules. The differentiation of naïve T cells into functionally distinct effector subsets is regulated by signals from the T cell receptor (TCR) and cytokine receptors. Using gene knock-out approaches, the initiation of discrete effector programs appears differentially sensitive to the loss of individual TCR signaling components; likely due to differences in the transcription factors needed to activate individual cytokine genes. Less well understood however are the signal requirements for the execution of effector function. With a focus on Th2 cells and the kinase Itk, we review recent observations that point to differences between the signals needed for the initiation and implementation of cytokine programs in CD4+ T cells. Indeed, Th2 effector cells signal differently from both their naïve counterparts and from Th1 effectors suggesting they may transduce activation signals differently or may be selectively receptive to different activation signals. Potential regulation points for effector function lie at the level of transcription and translation of cytokine genes. We also discuss how provision of these execution signals may be spatially segregated in vivo occurring at tissue sites of inflammation and subject to modulation by the pathogen itself.
Introduction
The control of microbial infection by CD4+ T cells depends on the acquisition and delivery of appropriate effector function to the infection site. Gain of function, the ability to produce a restricted set of effector molecules such as cytokines and chemokines, is attained upon initial T cell activation and differentiation in the lymph node draining the site of infection. Once armed, the effector cells home to the site of infection guided by chemokine and adhesion cues and require re-activation at the infection site to exert their anti-microbial functions. Some of those effector cells will receive additional signals (as yet ill-defined) that support long term survival as functional memory cells. Since the initial description of functionally distinct CD4+ T cell subsets (Th1 and Th2) by Mosmann and Coffman in the 1980’s, we have gained enormous molecular insight into the signaling and transcriptional regulation that controls the initial differentiation of naive CD4+ T cells into distinct subsets: Th1, Th2, Th17 and induced regulatory T cells (iTreg) [1]. However the signals that the effector T cells require for the synthesis and secretion of effector molecules at the infected tissue site are poorly understood. Nonetheless, a number of studies highlight that the signaling requirements for expression of a given cytokine gene differ in naïve and effector/memory T cells [2–4]. A better understanding of the regulation of effector T cell function should help in the design of therapeutic strategies to promote or suppress immune function at peripheral sites of inflammation.
Rapid cytokine production by effectors
The hallmark of effector and memory cells is their ability to rapidly express and secrete high levels of effector cytokines in response to antigen stimulation. High-level cytokine production appears critical for effective immune function. Indeed, in a detailed study by the Seder group designed to define the immune criteria for effective vaccine strategies, the amount of cytokine produced by individual effector cells positively correlated with vaccine efficacy [5]. The mechanisms that facilitate this heightened response are poorly defined. Production of Th2 cytokines at high levels has been linked to chromatin remodeling at three conserved noncoding sequences in the IL-4 locus: the CNS-1 region, the inducible DNase I hypersensitivity (DHS) site VA and adjacent CNS-2 region and the conserved intron 1 sequence of IL-4 (CIRE) [6–8]. Deletion of the CNS-1 regulatory region in mice led to a modest (2–4 fold) reduction in IL-4, IL-5 and IL-13 production by Th2 cells (but not in mast cells) that compromised the ability of mice to mount Th2 responses to a variety of infections, implicating a general role for the region in transcription of cytokines in the IL-4 locus in CD4+ T cells [6]. The inducible 3′ enhancer (DHS VA) is essential for high-level IL-4 production in both ‘mature’ Th2 cells and mast cells [7]. This region binds both GATA3 and NFAT1 suggesting TCR signals that fail to support the activation of these transcription factors will strongly impact the amount of IL-4 produced by Th2 effectors. Indeed, continued expression of GATA3 in Th2 effectors is critical for high-level Th2 cytokine production revealed by a number of conditional deletion approaches [9–11]. GATA3 does not appear to be essential for the enforcement of the remodeled chromatin structure in Th2 cells but plays an important role in the transcription of Th2 cytokine genes, possibly through binding to the inducible 3′ enhancer and the intronic regulatory element CIRE for IL-4 and the promoter region of IL-5 and IL-13 [12]. In contrast, the Mixed-Lineage Leukemia (MLL) gene, a mammalian homologue of the Drosophila trithorax a epigenetic transcriptional regulator, has been implicated in function of Th2 memory cells at the chromatin level [13]. MLL was not required for the induction of the Th2 lineage or IL-4 production in effector cells but was required for maintaining histone modifications at the GATA3 and Th2 locus necessary for optimal IL-4 production in memory Th2 cells. In addition, a recent study shows that memory T cells maintain higher levels of NFAT protein expression than do naïve cells [2], suggesting that resting memory T cells may be poised for transcription by virtue of an enhanced pool of available transcription factors. Thus a combination of greater accessibility for transcription of specific cytokine genes and the increased expression of transcription factors such as NFAT may assist in the rapid production of cytokines by effector T cells.
Distinct biochemical responses to TCR engagement in Th2 effectors
During the differentiation process from naïve to effector, Th1 and Th2 cells begin to utilize different TCR-driven signaling components (Figure 1). Unlike Th1 cells, Th2 cells loose the ability to induce a high and sustained calcium flux and have reduced TCR-triggered tyrosine phosphorylation [14–17]. Both a difference in Ca2+ clearance from the cytosol and smaller Ca2+-activated K+ currents contribute to the lower Ca2+ response in Th2 cells [18]. Poor proximal signaling in Th2 cells can partly be explained by a decrease in the expression of CD4 on the cell surface. Th2 cells have been found to express 2-fold less CD4 on their cell surface than Th1 cells [17] and to poorly recruit CD4 to lipid rafts on TCR ligation [19]. The functional significance of this reduction in CD4 was revealed by restoration of CD4 expression levels in Th2 cells using retroviral transfer [17]. Th2 cells expressing CD4 levels comparable to that of Th1 cells showed more robust protein tyrosine phosphorylation and elevated Ca2+ signaling. The rationale for decreased CD4 expression in Th2 cells remains to be determined. Interestingly, targeted deletion of CD4 renders CD4 cells unable to differentiate into Th2 cells but leaves Th1 responses intact [20]. Therefore while Th2 cells express less CD4 on their surface they appear more dependent on that CD4 pool for effector function than Th1 cells.
Figure 1.

Signaling differences in Th1 and Th2 effectors.
Expression of distinct TEC-family kinase members in T effectors provides an additional level of differential control of TCR signaling. The TEC-family kinases are important amplifiers of the calcium flux through activation of PLCγ, amongst other functions [21, 22]. Naive CD4+ T cells express predominantly ITK and to a lesser extent RLK and TEC. On T cell activation RLK expression is downregulated and is only re-expressed in Th1 effectors. During Th2 differentiation, loss of RLK is accompanied by an increase in ITK expression [23]. The resulting effector cells express ITK if Th2 and RLK and ITK if Th1. Indeed, Th2 cells are heavily dependent on ITK for the calcium flux: ITK-deficient Th2 cells have severely compromised calcium fluxes that correlate with the abrogation of Th2, but not Th1, effector function [23–26]. In addition, RLK appears to directly regulate IFNγ cytokine gene expression by translocation to the nucleus and DNA-binding to a region upstream of the IFNγ transcriptional start-site [27, 28]. Although a specific role for RLK in Th1 function remains unclear; given RLK-deficient mice show little attenuation of Th1-dependent immune response [29] and ectopic expression of RLK in Itk-deficient cells can rescue some Th2 effector function [30]. Nevertheless, during differentiation the expression levels of signal components are re-set and accompany specific effector functions on subsequent re-stimulation.
Differences in the signal transduction patterns between Th1 and Th2 cells may partly be the result of altered immunological synapse formation. A number of studies have observed a difference between Th1 and Th2 cells in the organization of molecules at the interface between the effector T cell and the antigen presenting cell. The T cell receptor and CD4 were efficiently recruited to lipid rafts in Th1 cells but not Th2 cells [19] and correlated with a decrease ability of Th2 cells to respond to low-affinity peptide stimulation. The synapse structure in Th2 cells differs from naïve and Th1 cells in the distribution of a number of signaling molecules including TCR, PKCθ, CD45 and talin. In general, the synapse in Th2 cells is less organized with a failure to form the classic ‘bulls-eye’ patterning of central TCR and peripheral ICAM/LFA-1/Talin seen in non-polarized CD4 T cells and Th1 effectors [31]. Interestingly, these differences may stem from expression of higher levels of CTLA-4 in Th2 cells [32]. Manipulation of CTLA-4 expression levels correlated with altered synapse organization: CTLA-4-deficient Th2 cells were able to form ordered synapses with central TCR clustering while reintroduction of CTLA-4 disrupted TCR clustering [32]. The functional outcome of these experimental changes in Th2 synapse structure with respect to effector cytokine production were not explored; thus the linkage between synapse structure, downstream signaling and cytokine production remains obscure.
An interesting alternative explanation for differences in synapse structure in Th1 and Th2 cells is the role of the synapse in delivery of effector molecules [33]. Seminal studies in the 1980s provided evidence to support the idea that cytokines and cytolytic molecules were synthesized and secreted in a directional fashion, providing a mechanism to limit bystander damage [34, 35]. Recent studies suggest that effector molecules utilize directionally distinct pathways for secretion [36, 37]. Cytokines including IFNγ appear to be secreted at the synapse while TNF and Mip1α are secreted multidirectionally [37]. The regulation of directional secretion is not fully understood but does correlate with the co-localization of particular cytokines with distinct trafficking proteins of the Rab and SNARE families [37] and is aided by activation of the actin cytoskeletal regulator the Wiskott Aldrich Syndrome protein [36]. Interestingly, IL-4 could be secreted both synaptically and multidirectionally [37]. Extrapolating to the biological functions of IFNγ and IL-4 (IFNγ exerting anti-microbial activity on infected target cells and IL-4 (and IL-5) providing help for B cells and recruiting innate Type2 effectors), the synapse structure in Th1 and Th2 cells may differ to facilitate delivery of functionally distinct effector molecules.
How these differential signaling patterns become established and their role in effector function is not clear. Differences in TCR affinity or amount of antigen may modulate these pathways in natural responses to pathogens in vivo. Indeed, the extent of the differences in synapse structure between Th1 and Th2 cells was very much context dependent in in vitro studies. The synapse structure of Th2 effectors could be modified by the number of MHC/peptide complexes, extent of CD80/86 expression and the type of APC [31, 32]. Whether the changes in signaling profiles play an active role in the establishment of the distinct effector fate or are a consequence of adopting a particular effector fate has not been resolved. What the observations do suggest however, is that Th1 and Th2 effectors (and likely Th17 and Tregs) will have different signaling needs for activation to exert their effector function. The infected tissue milieu will likely differentially regulate the efficiency of cytokine production, and perhaps directionality of cytokine secretion, by each effector subset through provision of signals by different types of antigen presenting cells, antigen dose, inflammatory cytokines and chemokines.
Signal requirements for Th2 cells to exert effector function
Distinct signaling molecules have been identified that play critical roles in Th2 differentiation including Itk, PKCθ, SAP, SLAM, LAT and VAV1 (reviewed in [38]). Targeted disruption of these genes leads to defects in Th2 cytokine production in vitro and in vivo following infection with Type 2 pathogens. Commonality in these signaling components comes from their ability to support the activation of key transcription factors, NFATc1, NFkB p50 and/or GATA3, essential for IL-4 production. The subsequent role these signaling molecules play in Th2 effector function is largely unknown.
We have recently shown that the interleukin-2-inducible T cell kinase (ITK) plays a critical role in the execution, but not gain, of Th2 effector function [3] (Figure 2). ITK is a member of the Tec family of non-receptor tyrosine kinases and plays a key role in the activation of phospholipase C (PLC)- γ1. Therefore ITK-deficient T cells show defects in the calcium flux, MAP kinase activation and activation of NFAT. In addition, ITK links to a number of other signal pathways including TCR-induced actin polymerization via activation of Cdc42 and WASp and chemokine receptor and adhesion molecule signaling for migration [21]. Mice deficient in ITK show impaired Th2 function following infection by a number of Th2-inducing pathogens such as Schistosoma mansoni, the nematode Nippostronglyus brasilensis and Leishmania major on the susceptible BALB/c background [24, 25] and are resistant to experimental induced allergy/asthma [26, 39]. While these in vivo models highlighted the important requirement for ITK in Th2 responses they did not scrutinize the point at which ITK regulated Th2 responses in the complex in vivo setting of disease. Using IL-4 reporter mice, ITK was found to be dispensable for early IL-4 expression during Th2 differentiation but was critical for IL-4 production in Th2 effectors both in vitro and in vivo [3]. In the absence of ITK, the initial upregulation of gene expression for IL-4 and GATA3 during Th2 priming was intact. Nonetheless, ITK-deficient Th2 effectors failed to upregulate IL-4 gene transcription in response to re-stimulation through the TCR. The inability to upregulate IL-4 mRNA correlated with a decrease in GATA3 gene expression consistent with a requirement for GATA3 in Th2 effector function. However, neither GATA3 nor NFAT retroviral add-back could singularly restore IL-4 production in Th2 effectors suggesting multi-factoral control (Figure 2 summarizes possible Itk-dependent regulation points). As previously discussed, changes in transcription in Th2 primed cells may reflect a role for the signaling molecule in accelerated cytokine gene transcription or alternatively for the maintenance of cytokine gene accessibility to transcription. Indeed, in addition to potential transcriptional regulation of IL-4 gene expression, preliminary analysis of histone modifications in ITK-deficient Th2-primed cells supports a role for ITK in acetylated histone 3 at the IL-4 promoter [40].
Figure 2. Possible ITK-dependent control points in Th2 effector function.
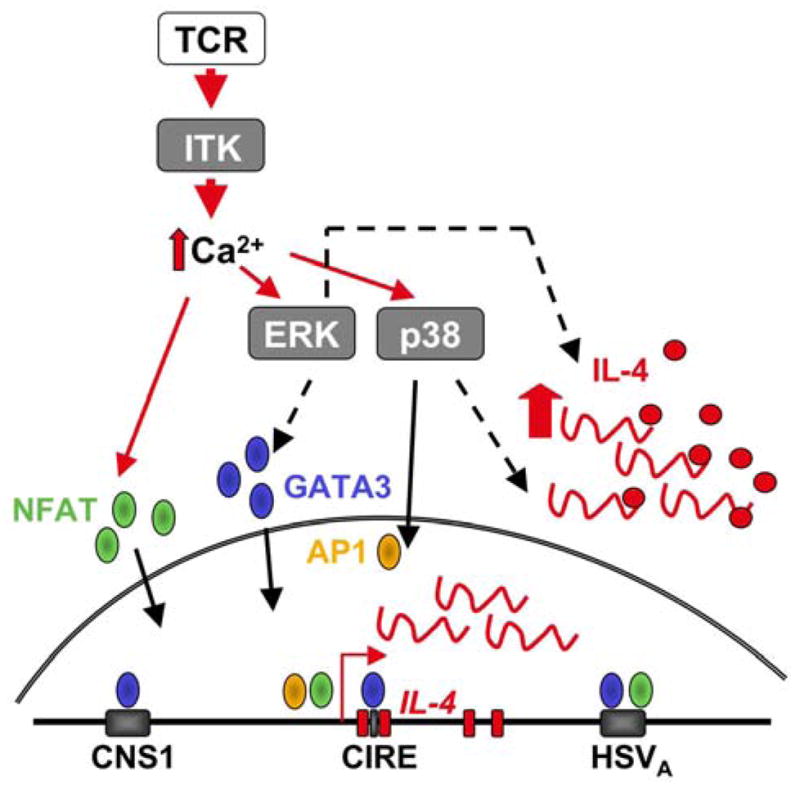
ITK is an amplifier of the TCR-dependent calcium flux and could modulate Th2 effector function at a number of levels including: 1) Activation of NFAT for increased cytokine transcription through binding to the IL-4 promoter and HSVA 3′ enhancer element; 2) Maintenance of GATA3 expression at the level of transcription or protein stability via ERK activation; 3) Translational enhancement via ERK; 4) Expression of AP1 and/or IL-4 mRNA stability via p38. CNS, conserved non-coding sequence; CIRE, conserved element in the first intron of the il4 gene; HS, DNase hypersensitivity site.
Our results of a stage-specific requirement for ITK in IL-4 gene expression parallel the data from mice transgenic for a dominant negative NFAT [2]. Surprisingly, NFAT was not required for IL-2 production in naïve cells but was essential for IL-2 production in memory T cells. Stage-specific regulation of both IL-4 and IL-2 cytokine genes was controlled in part at the level of transcription predicting that transcriptional requirements for a given cytokine gene can change as the cell acquires its effector program. In support of this notion, the transcription factor interferon regulatory factor (IRF)-4 has recently been shown to inhibit Th2 cytokine production in naïve CD4+ T cells but to promote cytokine production in Th2 effector cells [4]. IRF-4 can function as either a transcriptional activator or repressor depending on the context of the DNA-binding sequences and/or the protein-interacting partners. In the context of IL-4 gene regulation in Th2 effectors, IRF-4 in association with NFAT is predicted to act as a transcriptional enhancer.
In addition to transcriptional control of accelerated cytokine production in effector cells, TCR stimulation increases translational efficiency [41, 42]. A detailed study by the Luban group revealed the coordinated upregulation of translational machinery along with increased cytokine gene expression in Th2 effector cells [41]. Increased translational efficiency was characterized by increased transcription of rRNA and a family of genes involved in rRNA processing and post-transcriptional regulation of the ribosomal protein subunit (RBS) through promoting polysome loading. Both transcriptional and translational regulation of the translational machinery was dependent on the ERK-MAPK pathway [41]. Interestingly, ERK also post-transcriptionally controls the level of GATA3 protein in Th2 cells through inhibition of the ubiquitin-proteasome degradation pathway [43]. Thus ERK is an important regulator of Th2 effector function at multiple levels. Another MAP kinase, p38, has also been shown to promote cytokine production in Th2 effector cells in part post-transcriptionally through regulating IL-4 mRNA stability [44].
Additional post-transcriptional control of effector function was suggested by a provocative study by the Locksley group proposing that during differentiation T cells may evoke the integrated stress response (ISR) to optimize cytokine expression [42]. The ISR is conserved from yeast to mammals and is initiated in times of stress to modulate protein biosynthesis, down regulating translation of many mRNAs and conserving translation for key mRNAs critical for adapting to the new environment. Translation is attenuated in the ISR through phosphorylation of the translation initiation factor eIF2α. During Th2 differentiation, T cells were found to activate the ISR (phosphorylation of eIF2α and increased expression of stress-response genes) leading to translational attenuation of cytokine mRNAs [42]. Biologically, this resulted in effector cells that contained IL-4/eGFP transcripts but produced no IL-4 protein. On re-stimulation of the cells, presumably at sites of infection in vivo, TCR signals reverse this translational block by de-phosphoryating eIF2α and allowing rapid cytokine synthesis. In future studies, it will be important to define the TCR-signaling pathways critical to the alleviation of this translation block and the status of antigen presenting cell that provides such signals.
Regulation at sites of inflammation
While competency for IL-4 production is gained in the draining lymph node, high level IL-4 protein production appears to be spatially segregated. The use of a dual reporter system for readouts of IL-4 transcription and protein production (eGFP and huCD2 respectively) enabled analysis of the fidelity of transcription and protein production following infection with the Th2-inducing pathogen Heligmosomoides polygyrus [45]. eGFP+ Th2 cells expressing constitutive IL-4 transcripts homed from the draining lymph node to many non-lymphoid peripheral tissue sites but did not produce IL-4 protein unless re-exposed to antigen locally in infected tissues. We have found a similar correlation in Leishmania major infected mice, transcriptional upregulation and high-level protein production being restricted to the infected dermis and not found in the draining lymph node (Katzman and Fowell, unpublished). Thus the acquisition of effector function and the execution of effector function are kinetically, and likely spatially, uncoupled. The infected tissue micro-environment must provide multiple signals for Th2 effector function including signals for the release of a possible translation block and for increasing transcription and translation efficiencies. The inflammatory milieu therefore has the potential to further edit the lymph node-generated cytokine repertoire through differential activation of effector functions [46].
A number of extrinsic factors have recently been found to influence the magnitude of the Th2 response in peripheral tissues. The interleukin-1-like cytokine IL-33 is produced at mucosal surfaces including the stomach, lung and skin at higher levels than the lymphoid tissue. IL-33 can potently upregulate IL-5 and IL-13 production by Th2 cells and potentiate mast cell function [47] by virtue of binding to the IL-1 receptor ST2 that is preferentially expressed by these cell types [48, 49]. Two independent studies of allergic inflammation have also revealed additional local control of Th2 function in non-lymphoid tissues. In one study, the thymic stromal lymphopoietin (TSLP), a cytokine produced by epithelial cells, was found to be a potent enhancer of Th2 cytokine secretion via the TSLP receptor expressed on T cell effectors [50]. CD4 T cells from TSLP-receptor deficient mice were primed efficiently for Th2 cytokine production in the LN and homed to the skin normally but failed to exert effector function in the skin. Consistent with a key role for TSLP in allergic skin inflammation, local blockade of TSLP in the skin disrupted Th2 cytokine secretion [50]. In a second study of lung inflammation, the tumor necrosis receptor superfamily (TNFRSF) pair TNFRSF25/TNFSF15 (TL1A) was identified as an important trigger of both NKT and Th2 effector function [51]. TNFR25 and its ligand TL1A can be expressed by activated T cells and NKT cells. Blocking the pathway with an antagonistic anti-TL1A diminished Th2 cytokine production while TNFR25 co-stimulation amplified Th2 cytokine production [51]. The potent effect of TNFR25 blockade on lung inflammation was compounded by defects in NKT and Th2 cytokine production and the recruitment of eosinophils. It will be interesting to determine whether the downstream signaling pathways in CD4+ T cells activated by these cytokines feed into the regulation points for Th2 effector function described in this review.
Most pathogens have co-evolve with their mammalian hosts therefore it is likely that the pathogen itself will ultimately determine the efficiency of effector T cell activation in the infected tissue. Toll-like receptor engagement on Th1 cells has been found to directly activate IFNγ production and could be further enhanced by IL-12 [52]. There are also examples of pathogen attenuation of Th1 effector function [46, 53]. For instance, in studies of cytokine production in the schistosome granuloma, IFNγ-producers were present the granuloma milieu but were functionally silent until provided with activation signals ex vivo [53]. IL-4 has been implicated in the negative regulation of these Th1 effectors [54] possibly by modulating the magnitude of IFNγ production by Th1 effectors [55]. Less well understood is pathogen regulation of Th2 effector function; though there are examples of both positive and negative regulation by TLR ligands. Intrinsic to the effector cell, response to direct TLR-ligation appears to differ between Th1 and Th2 cells. Athough TLR2 is expressed by both Th1 and Th2 cells, TLR2 ligands directly induced effector cytokines only in Th1 cells [52]. Therefore the response to TLR-ligation is probably context dependent. Mechanistic studies on the therapeutic application of CpGs for the treatment of Th2-mediated allergic asthma demonstrated that CpG treatment indirectly inhibited Th2 effector function through modifying lung antigen presenting cells (APC) [56]. CD11c+ antigen presenting cells from the lungs of CpG-treated mice supported the production of IFNγ but not IL-13 or IL-5 from effector/memory CD4+ T cells. The failure to support Th2 effector function was linked to an inhibition of MHC Class II expression and reduced costimulatory molecule expression by APC including PD-1, CD40 and CD80/86. The specific attenuation of APC signals that support Th2 effector function by CpG, raises the possibility that Th1 and Th2 effector function maybe reciprocally regulated by TLRs similarly to their cross-regulation by cytokines. Interestingly, TLR3 triggering by dsRNA and rhinovirus infection upregulates TSLP production by epithelial cells and can amplify Th2 inflammation [57]. Therefore, respiratory viruses may exacerbate allergic inflammation despite also inducing Th1-type immune response. Clearly the fine-tuning of efffector responses at tissue sites of infection will be subject to many counterbalances in immune function [58].
Lately, an additional level of control has been proposed based on reprogramming of the cytokine repertoire in Th2-primed cells. Once differentiated, the Th1 and Th2 effector subsets have always been considered functionally inflexible, at least with respect to reciprocal cytokine production [59]. However, a recent paper by the Stockinger group suggests that Th2 effector cells can be further functionally modified by transforming growth factor-β (TGFβ) [60]. In vitro generated Th2 effector cells that retained expression of IL-4 and GATA3 when restimulated with IL-12 (Th1-conditions), lost expression of IL-4 and GATA3 and gained expression of IL-9 when restimulated in the presence of TGFβ. The ability of TGFβ to reprogrammed Th2 effector function, by suppressing IL-4 and IL-13 production and promoting IL-9 production, suggests Th2 cells retain some functional flexibility. The next important step will be to begin to define the in vivo conditions that promote this re-tuning of Th2 responses and to define the new functional activities that accompany the production of IL-9 in the absence of IL-4. It is tempting to speculate that the Th1 cells found at chronic infection sites that also make IL-10 represent a similar ‘reprograming’ event for type 1 immune responses [61–63]. Moreover, current studies suggest that Th1 effector cells can autoregulate their effector activity in chromic inflammatory settings through expression of the transcriptional negative regulator twist1 [64]. Identification of similar mechanisms of internal control of Th2 effector function are likely to be forth coming.
Conclusion
We know very little about the signals that regulate CD4+ T cell effector function. However data are accumulating to support the idea that effector cells signal differently from their naïve counterparts and that different effector cytokines require distinct signals for high-level production. Stage-specific signaling events for high-level cytokine production may control effector function through the maintenance of gene accessibility for transcription, transcription enhancement and/or translational efficiency. In addition, while gain of function results from priming in the lymph node, implementation of effector function occurs at the infected tissue site. In the context of the inflammatory milieu, effector function is likely to be modulated by the type and activation status of antigen presenting cells, cytokines, TLR ligands and the presence of regulatory T cells. Understanding the cellular and molecular requirements for full Th2 effector function (and Th1, Th17, Treg cells) in inflammatory settings will be essential for fine-tuning immune responses. The idea that effector cell function is under a different set of controls to naïve and differentiating cells makes site-directed targeting of immune function an attractive and highly selective therapeutic strategy for existing pathologies.
Acknowledgments
The author thanks members of the lab and the CVBI for thoughtful discussions. This work was supported by grants to DJF from the NIH; AI50201 and AI072690.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dienz O, Eaton SM, Krahl TJ, Diehl S, Charland C, Dodge J, Swain SL, Budd RC, Haynes L, Rincon M. Accumulation of NFAT mediates IL-2 expression in memory, but not naive, CD4+ T cells. Proc Natl Acad Sci U S A. 2007;104:7175–7180. doi: 10.1073/pnas.0610442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Au-Yeung BB, Katzman SD, Fowell DJ. Cutting edge: Itk-dependent signals required for CD4+ T cells to exert, but not gain, Th2 effector function. J Immunol. 2006;176:3895–3899. doi: 10.4049/jimmunol.176.7.3895. [DOI] [PubMed] [Google Scholar]
- 4.Honma K, Kimura D, Tominaga N, Miyakoda M, Matsuyama T, Yui K. Interferon regulatory factor 4 differentially regulates the production of Th2 cytokines in naive vs. effector/memory CD4+ T cells. Proc Natl Acad Sci U S A. 2008;105:15890–15895. doi: 10.1073/pnas.0803171105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darrah PA, Patel DT, De Luca PM, Lindsay RW, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL, Roederer M, Seder RA. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13:843–850. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- 6.Mohrs M, Blankespoor CM, Wang ZE, Loots GG, Afzal V, Hadeiba H, Shinkai K, Rubin EM, Locksley RM. Deletion of a coordinate regulator of type 2 cytokine expression in mice. Nat Immunol. 2001;2:842–847. doi: 10.1038/ni0901-842. [DOI] [PubMed] [Google Scholar]
- 7.Solymar DC, Agarwal S, Bassing CH, Alt FW, Rao A. A 3′ enhancer in the IL-4 gene regulates cytokine production by Th2 cells and mast cells. Immunity. 2002;17:41–50. doi: 10.1016/s1074-7613(02)00334-5. [DOI] [PubMed] [Google Scholar]
- 8.Tykocinski LO, Hajkova P, Chang HD, Stamm T, Sozeri O, Lohning M, Hu-Li J, Niesner U, Kreher S, Friedrich B, Pannetier C, Grutz G, Walter J, Paul WE, Radbruch A. A critical control element for interleukin-4 memory expression in T helper lymphocytes. J Biol Chem. 2005;280:28177–28185. doi: 10.1074/jbc.M502038200. [DOI] [PubMed] [Google Scholar]
- 9.Pai SY, Truitt ML, Ho IC. GATA-3 deficiency abrogates the development and maintenance of T helper type 2 cells. Proc Natl Acad Sci U S A. 2004;101:1993–1998. doi: 10.1073/pnas.0308697100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamashita M, Ukai-Tadenuma M, Miyamoto T, Sugaya K, Hosokawa H, Hasegawa A, Kimura M, Taniguchi M, DeGregori J, Nakayama T. Essential role of GATA3 for the maintenance of type 2 helper T (Th2) cytokine production and chromatin remodeling at the Th2 cytokine gene loci. J Biol Chem. 2004;279:26983–26990. doi: 10.1074/jbc.M403688200. [DOI] [PubMed] [Google Scholar]
- 11.Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, Killeen N, Urban JF, Jr, Guo L, Paul WE. Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol. 2004;5:1157–1165. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- 12.Nakayama T, Yamashita M. Initiation and maintenance of Th2 cell identity. Curr Opin Immunol. 2008;20:265–271. doi: 10.1016/j.coi.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 13.Yamashita M, Hirahara K, Shinnakasu R, Hosokawa H, Norikane S, Kimura MY, Hasegawa A, Nakayama T. Crucial role of MLL for the maintenance of memory T helper type 2 cell responses. Immunity. 2006;24:611–622. doi: 10.1016/j.immuni.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 14.Sloan-Lancaster J, Steinberg TH, Allen PM. Selective loss of the calcium ion signaling pathway in T cells maturing toward a T helper 2 phenotype. J Immunol. 1997;159:1160–1168. [PubMed] [Google Scholar]
- 15.Tamura T, Nakano H, Nagase H, Morokata T, Igarashi O, Oshimi Y, Miyazaki S, Nariuchi H. Early activation signal transduction pathways of Th1 and Th2 cell clones stimulated with anti-CD3. Roles of protein tyrosine kinases in the signal for IL-2 and IL-4 production. J Immunol. 1995;155:4692–4701. [PubMed] [Google Scholar]
- 16.Gajewski TF, Schell SR, Fitch FW. Evidence implicating utilization of different T cell receptor-associated signaling pathways by TH1 and TH2 clones. J Immunol. 1990;144:4110–4120. [PubMed] [Google Scholar]
- 17.Itoh Y, Wang Z, Ishida H, Eichelberg K, Fujimoto N, Makino J, Ogasawara K, Germain RN. Decreased CD4 expression by polarized T helper 2 cells contributes to suboptimal TCR-induced phosphorylation and reduced Ca2+ signaling. Eur J Immunol. 2005;35:3187–3195. doi: 10.1002/eji.200526064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fanger CM, Neben AL, Cahalan MD. Differential Ca2+ influx, KCa channel activity, and Ca2+ clearance distinguish Th1 and Th2 lymphocytes. J Immunol. 2000;164:1153–1160. doi: 10.4049/jimmunol.164.3.1153. [DOI] [PubMed] [Google Scholar]
- 19.Balamuth F, Leitenberg D, Unternaehrer J, Mellman I, Bottomly K. Distinct patterns of membrane microdomain partitioning in Th1 and th2 cells. Immunity. 2001;15:729–738. doi: 10.1016/s1074-7613(01)00223-0. [DOI] [PubMed] [Google Scholar]
- 20.Fowell DJ, Magram J, Turck CW, Killeen N, Locksley RM. Impaired Th2 subset development in the absence of CD4. Immunity. 1997;6:559–569. doi: 10.1016/s1074-7613(00)80344-1. [DOI] [PubMed] [Google Scholar]
- 21.Schwartzberg PL, Finkelstein LD, Readinger JA. TEC-family kinases: regulators of T-helper-cell differentiation. Nat Rev Immunol. 2005;5:284–295. doi: 10.1038/nri1591. [DOI] [PubMed] [Google Scholar]
- 22.Kosaka Y, Felices M, Berg LJ. Itk and Th2 responses: action but no reaction. Trends Immunol. 2006;27:453–460. doi: 10.1016/j.it.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Miller AT, Wilcox HM, Lai Z, Berg LJ. Signaling through Itk promotes T helper 2 differentiation via negative regulation of T-bet. Immunity. 2004;21:67–80. doi: 10.1016/j.immuni.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 24.Fowell DJ, Shinkai K, Liao XC, Beebe AM, Coffman RL, Littman DR, Locksley RM. Impaired NFATc translocation and failure of Th2 development in Itk-deficient CD4+ T cells. Immunity. 1999;11:399–409. doi: 10.1016/s1074-7613(00)80115-6. [DOI] [PubMed] [Google Scholar]
- 25.Schaeffer EM, Yap GS, Lewis CM, Czar MJ, McVicar DW, Cheever AW, Sher A, Schwartzberg PL. Mutation of Tec family kinases alters T helper cell differentiation. Nat Immunol. 2001;2:1183–1188. doi: 10.1038/ni734. [DOI] [PubMed] [Google Scholar]
- 26.Mueller C, August A. Attenuation of immunological symptoms of allergic asthma in mice lacking the tyrosine kinase ITK. J Immunol. 2003;170:5056–5063. doi: 10.4049/jimmunol.170.10.5056. [DOI] [PubMed] [Google Scholar]
- 27.Kashiwakura J, Suzuki N, Nagafuchi H, Takeno M, Takeba Y, Shimoyama Y, Sakane T. Txk, a nonreceptor tyrosine kinase of the Tec family, is expressed in T helper type 1 cells and regulates interferon gamma production in human T lymphocytes. J Exp Med. 1999;190:1147–1154. doi: 10.1084/jem.190.8.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeba Y, Nagafuchi H, Takeno M, Kashiwakura J, Suzuki N. Txk, a member of nonreceptor tyrosine kinase of Tec family, acts as a Th1 cell-specific transcription factor and regulates IFN-gamma gene transcription. J Immunol. 2002;168:2365–2370. doi: 10.4049/jimmunol.168.5.2365. [DOI] [PubMed] [Google Scholar]
- 29.Schaeffer EM, Debnath J, Yap G, McVicar D, Liao XC, Littman DR, Sher A, Varmus HE, Lenardo MJ, Schwartzberg PL. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science. 1999;284:638–641. doi: 10.1126/science.284.5414.638. [DOI] [PubMed] [Google Scholar]
- 30.Sahu N, Venegas AM, Jankovic D, Mitzner W, Gomez-Rodriguez J, Cannons JL, Sommers C, Love P, Sher A, Schwartzberg PL, August A. Selective expression rather than specific function of Txk and Itk regulate Th1 and Th2 responses. J Immunol. 2008;181:6125–6131. doi: 10.4049/jimmunol.181.9.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thauland TJ, Koguchi Y, Wetzel SA, Dustin ML, Parker DC. Th1 and Th2 cells form morphologically distinct immunological synapses. J Immunol. 2008;181:393–399. doi: 10.4049/jimmunol.181.1.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackman RP, Balamuth F, Bottomly K. CTLA-4 differentially regulates the immunological synapse in CD4 T cell subsets. J Immunol. 2007;178:5543–5551. doi: 10.4049/jimmunol.178.9.5543. [DOI] [PubMed] [Google Scholar]
- 33.Huse M, Quann EJ, Davis MM. Shouts, whispers and the kiss of death: directional secretion in T cells. Nat Immunol. 2008;9:1105–1111. doi: 10.1038/ni.f.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kupfer A, Singer SJ. The specific interaction of helper T cells and antigen-presenting B cells. IV. Membrane and cytoskeletal reorganizations in the bound T cell as a function of antigen dose. J Exp Med. 1989;170:1697–1713. doi: 10.1084/jem.170.5.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poo WJ, Conrad L, Janeway CA., Jr Receptor-directed focusing of lymphokine release by helper T cells. Nature. 1988;332:378–380. doi: 10.1038/332378a0. [DOI] [PubMed] [Google Scholar]
- 36.Morales-Tirado V, Johannson S, Hanson E, Howell A, Zhang J, Siminovitch KA, Fowell DJ. Cutting edge: selective requirement for the Wiskott-Aldrich syndrome protein in cytokine, but not chemokine, secretion by CD4+ T cells. J Immunol. 2004;173:726–730. doi: 10.4049/jimmunol.173.2.726. [DOI] [PubMed] [Google Scholar]
- 37.Huse M, Lillemeier BF, Kuhns MS, Chen DS, Davis MM. T cells use two directionally distinct pathways for cytokine secretion. Nat Immunol. 2006;7:247–255. doi: 10.1038/ni1304. [DOI] [PubMed] [Google Scholar]
- 38.Marsland BJ, Kopf M. T-cell fate and function: PKC-theta and beyond. Trends Immunol. 2008;29:179–185. doi: 10.1016/j.it.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 39.Ferrara TJ, Mueller C, Sahu N, Ben-Jebria A, August A. Reduced airway hyperresponsiveness and tracheal responses during allergic asthma in mice lacking tyrosine kinase inducible T-cell kinase. J Allergy Clin Immunol. 2006;117:780–786. doi: 10.1016/j.jaci.2005.12.1330. [DOI] [PubMed] [Google Scholar]
- 40.Grogan JL, Wang ZE, Stanley S, Harmon B, Loots GG, Rubin EM, Locksley RM. Basal chromatin modification at the IL-4 gene in helper T cells. J Immunol. 2003;171:6672–6679. doi: 10.4049/jimmunol.171.12.6672. [DOI] [PubMed] [Google Scholar]
- 41.Asmal M, Colgan J, Naef F, Yu B, Lee Y, Magnasco M, Luban J. Production of ribosome components in effector CD4+ T cells is accelerated by TCR stimulation and coordinated by ERK-MAPK. Immunity. 2003;19:535–548. doi: 10.1016/s1074-7613(03)00268-1. [DOI] [PubMed] [Google Scholar]
- 42.Scheu S, Stetson DB, Reinhardt RL, Leber JH, Mohrs M, Locksley RM. Activation of the integrated stress response during T helper cell differentiation. Nat Immunol. 2006;7:644–651. doi: 10.1038/ni1338. [DOI] [PubMed] [Google Scholar]
- 43.Yamashita M, Shinnakasu R, Asou H, Kimura M, Hasegawa A, Hashimoto K, Hatano N, Ogata M, Nakayama T. Ras-ERK MAPK cascade regulates GATA3 stability and Th2 differentiation through ubiquitin-proteasome pathway. J Biol Chem. 2005;280:29409–29419. doi: 10.1074/jbc.M502333200. [DOI] [PubMed] [Google Scholar]
- 44.Guo L, Urban JF, Zhu J, Paul WE. Elevating calcium in Th2 cells activates multiple pathways to induce IL-4 transcription and mRNA stabilization. J Immunol. 2008;181:3984–3993. doi: 10.4049/jimmunol.181.6.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohrs K, Wakil AE, Killeen N, Locksley RM, Mohrs M. A two-step process for cytokine production revealed by IL-4 dual-reporter mice. Immunity. 2005;23:419–429. doi: 10.1016/j.immuni.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katzman SD, Fowell DJ. Pathogen-imposed skewing of mouse chemokine and cytokine expression at the infected tissue site. J Clin Invest. 2008;118:801–811. doi: 10.1172/JCI33174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 48.Lohning M, Stroehmann A, Coyle AJ, Grogan JL, Lin S, Gutierrez-Ramos JC, Levinson D, Radbruch A, Kamradt T. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc Natl Acad Sci U S A. 1998;95:6930–6935. doi: 10.1073/pnas.95.12.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu D, Chan WL, Leung BP, Huang F, Wheeler R, Piedrafita D, Robinson JH, Liew FY. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J Exp Med. 1998;187:787–794. doi: 10.1084/jem.187.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He R, Oyoshi MK, Garibyan L, Kumar L, Ziegler SF, Geha RS. TSLP acts on infiltrating effector T cells to drive allergic skin inflammation. Proc Natl Acad Sci U S A. 2008;105:11875–11880. doi: 10.1073/pnas.0801532105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fang L, Adkins B, Deyev V, Podack ER. Essential role of TNF receptor superfamily 25 (TNFRSF25) in the development of allergic lung inflammation. J Exp Med. 2008;205:1037–1048. doi: 10.1084/jem.20072528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Imanishi T, Hara H, Suzuki S, Suzuki N, Akira S, Saito T. Cutting edge: TLR2 directly triggers Th1 effector functions. J Immunol. 2007;178:6715–6719. doi: 10.4049/jimmunol.178.11.6715. [DOI] [PubMed] [Google Scholar]
- 53.Rakasz E, Blum AM, Metwali A, Elliott DE, Li J, Ballas ZK, Qadir K, Lynch R, Weinstock JV. Localization and regulation of IFN-gamma production within the granulomas of murine schistosomiasis in IL-4-deficient and control mice. J Immunol. 1998;160:4994–4999. [PubMed] [Google Scholar]
- 54.Metwali A, Blum A, Elliott DE, Weinstock JV. Interleukin-4 receptor alpha chain and STAT6 signaling inhibit gamma interferon but not Th2 cytokine expression within schistosome granulomas. Infect Immun. 2002;70:5651–5658. doi: 10.1128/IAI.70.10.5651-5658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wurtz O, Bajenoff M, Guerder S. IL-4-mediated inhibition of IFN-gamma production by CD4+ T cells proceeds by several developmentally regulated mechanisms. Int Immunol. 2004;16:501–508. doi: 10.1093/intimm/dxh050. [DOI] [PubMed] [Google Scholar]
- 56.Hessel EM, Chu M, Lizcano JO, Chang B, Herman N, Kell SA, Wills-Karp M, Coffman RL. Immunostimulatory oligonucleotides block allergic airway inflammation by inhibiting Th2 cell activation and IgE-mediated cytokine induction. J Exp Med. 2005;202:1563–1573. doi: 10.1084/jem.20050631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kato A, Favoreto S, Jr, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol. 2007;179:1080–1087. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kato A, Schleimer RP. Beyond inflammation: airway epithelial cells are at the interface of innate and adaptive immunity. Curr Opin Immunol. 2007;19:711–720. doi: 10.1016/j.coi.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ansel KM, Lee DU, Rao A. An epigenetic view of helper T cell differentiation. Nat Immunol. 2003;4:616–623. doi: 10.1038/ni0703-616. [DOI] [PubMed] [Google Scholar]
- 60.Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, Martin B, Wilhelm C, Stockinger B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008 doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- 61.Gerosa F, Nisii C, Righetti S, Micciolo R, Marchesini M, Cazzadori A, Trinchieri G. CD4(+) T cell clones producing both interferon-gamma and interleukin-10 predominate in bronchoalveolar lavages of active pulmonary tuberculosis patients. Clin Immunol. 1999;92:224–234. doi: 10.1006/clim.1999.4752. [DOI] [PubMed] [Google Scholar]
- 62.Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4(+)CD25(−)Foxp3(−) Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J Exp Med. 2007;204:285–297. doi: 10.1084/jem.20061886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nylen S, Maurya R, Eidsmo L, Manandhar KD, Sundar S, Sacks D. Splenic accumulation of IL-10 mRNA in T cells distinct from CD4+CD25+ (Foxp3) regulatory T cells in human visceral leishmaniasis. J Exp Med. 2007;204:805–817. doi: 10.1084/jem.20061141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Niesner U, Albrecht I, Janke M, Doebis C, Loddenkemper C, Lexberg MH, Eulenburg K, Kreher S, Koeck J, Baumgrass R, Bonhagen K, Kamradt T, Enghard P, Humrich JY, Rutz S, Schulze-Topphoff U, Aktas O, Bartfeld S, Radbruch H, Hegazy AN, Lohning M, Baumgart DC, Duchmann R, Rudwaleit M, Haupl T, Gitelman I, Krenn V, Gruen J, Sieper J, Zeitz M, Wiedenmann B, Zipp F, Hamann A, Janitz M, Scheffold A, Burmester GR, Chang HD, Radbruch A. Autoregulation of Th1-mediated inflammation by twist1. J Exp Med. 2008;205:1889–1901. doi: 10.1084/jem.20072468. [DOI] [PMC free article] [PubMed] [Google Scholar]