

Abstract
Because the liver is central in the maintenance of glucose homeostasis and energy storage, knowledge of the physiology as well as physiopathology of hepatic energy metabolism is a prerequisite to our understanding of whole body metabolism. Hepatic fuel metabolism changes considerably depending on physiological circumstances (fed vs fasted state). In consequence, hepatic carbohydrate, lipid and protein synthesis/utilization are tightly regulated according to needs. Fatty liver and hepatic insulin resistance (both frequently associated with the metabolic syndrome) or increased hepatic glucose production (as observed in type 2 diabetes) resulted of alterations in substrates oxidation/storage balance in the liver. Because AMPK is considered as a cellular energy sensor, it is important to gain understanding of the mechanism by which hepatic AMPK coordinates hepatic energy metabolism. AMPK has been implicated as a key regulator of physiological energy dynamics by limiting anabolic pathways (to prevent further ATP consumption) and by facilitating catabolic pathways (to increase ATP generation). Activation of hepatic AMPK leads to increased fatty acid oxidation and simultaneously inhibition of hepatic lipogenesis, cholesterol synthesis and glucose production. In addition to a short-term effect on specific enzymes, AMPK also modulates the transcription of genes involved in lipogenesis and mitochondrial biogenesis. The identification of AMPK targets in hepatic metabolism should be useful in developing treatments to reverse metabolic abnormalities of type 2 diabetes and the metabolic syndrome.
Keywords: AMP-Activated Protein Kinases, chemistry, genetics, metabolism, Aminoimidazole Carboxamide, analogs & derivatives, metabolism, Animals, Dyslipidemias, drug therapy, metabolism, physiopathology, Energy Metabolism, physiology, Fatty Liver, drug therapy, metabolism, physiopathology, Gluconeogenesis, physiology, Glucose, metabolism, Homeostasis, Humans, Hypoglycemic Agents, metabolism, Lipid Metabolism, Liver, cytology, enzymology, Liver Cirrhosis, drug therapy, metabolism, physiopathology, Mitochondria, metabolism, Protein Conformation, Protein Subunits, chemistry, genetics, metabolism, Ribonucleotides, metabolism
Keywords: MP-activated protein kinase, energy metabolism, hepatic glucose production, fatty liver, liver, therapeutic, type 2 diabetes
Introduction
The ability to store nutrients is vital to survival for all organisms in periods of acute or prolonged shortage of nutrient supply. In mammals, the liver is a major organ responsible for maintaining both short-term and long-term whole-body energy needs by its capacity to induce a number of changes in terms of stored and released energy in the body under widely changing physiological conditions. Alterations in energy intake induce a number of metabolic and hormonal changes in the body. The liver functions as a mediator between dietary, as well as endogenous, sources of energy and the extrahepatic organs that continuously need energy, primarily the brain. Metabolically, the liver is the most versatile organ of the whole body with a metabolism cycling daily between the fed and fasted states. In the fed (anabolic) state, digestion yields simple sugars that are converted to pyruvate (via glycolysis), which is either oxidized to provide energy or channelled into pathways for synthesis of fatty acids (via lipogenesis) for storage. Excess glucose is also stored as glycogen (the total hepatic glucose store is only sufficient for a few hours use), providing a short-term source of carbohydrates for emergency use. The coordinated regulation of these processes allows for efficient utilization of dietary carbohydrate. Hepatic fuel metabolism changes considerably under the different nutritional situations. In the fasted (catabolic) state, the liver becomes a glucose producer (breakdown of stored hepatic glycogen and synthesis of glucose from lactate, amino acids, glycerol, and pyruvate), lipogenesis is slowed down, and fatty acid oxidation and ketogenesis are activated to supply energy and metabolic fuels for extrahepatic organs. Because the liver is the metabolic factory of the body, alterations in liver function clearly affect whole-body metabolism and energy homeostasis and underlie the development of metabolic diseases, including type 2 diabetes and the metabolic syndrome. Hence, understanding the mechanisms regulating hepatic energy metabolism is a prerequisite for developing new pharmacological strategies aimed at treating these metabolic diseases.
Over the last few years, a growing body of evidence indicates that AMP-activated protein kinase (AMPK), a serine threonine kinase comprising a catalytic α subunit and regulatory β and γ subunits, represents a point of convergence of regulatory signals monitoring systemic and cellular energy status. AMPK functions as a fuel sensor in most tissues and organs, including liver, skeletal muscle, heart, hypothalamus and adipose tissue, where it inhibits anabolic pathways and stimulates catabolic pathways in response to limited energy availability, thus simultaneously sparing limited energy resources and acquiring extra energy. AMPK acts first by directly affecting enzymes activities, involved in carbohydrate, lipid and protein biosynthesis, and second by longer-term transcriptional control of key players of these metabolic pathways. AMPK emerges as a major regulator of glucose and lipid metabolism and represents an attractive target for therapeutic intervention in the treatment of hepatic disorders. Important progress has recently been made in the comprehension of the pathophysiological role of AMPK in hepatic energy metabolism and this review provides a general overview of AMPK regulation, its activators and its functions in the liver.
1. Structure and regulation of AMPK in the liver
AMPK exists as a heterotrimeric complex consisting of a catalytic subunit α and two regulatory subunits β and γ involved in heterotrimer formation and ligand sensing. The conventional serine/threonine kinase activity of AMPK is supported by the α subunit which is characterized by the presence of a threonine residue within the activation loop of the kinase domain. The C-terminal region of α subunit is required for the association with the regulatory β and γ subunits. The β subunit contains a C-terminal region acting as scaffold binding α and γ subunits and a central region that allowed AMPK complex to bind glycogen but this has been recently disputed in the liver (Parker et al., 2007). The γ subunit contains four tandem repeats known as cystathionine β-synthase (CBS) motifs which bind AMP or ATP molecules in a competitive manner. Recent structural studies revealed that two sites on the γ subunit bind either AMP or Mg.ATP, a third site contains a tightly bound AMP that does not exchange (Xiao et al., 2007). Isoforms of all three subunits encoding by different genes (α1, α2, β1, β2, γ1, γ2, γ3) have been identified in mammals that allow for the generation of 12 different heterotrimeric combinations. These heterotrimeric AMPK complexes show relative tissue specificity. AMPKα1 and α2-containing complexes account each for about half of total AMPK activity in rodent liver (Cheung et al., 2000) but AMPKα1-containing complexes are predominant in human hepatocytes (B. Guigas, unpublished results). Expression of the β1 and γ1 regulatory subunits predominates in rodent liver (Cheung et al., 2000). To the best of our knowledge, no selective association between catalytic α1 and α2 and regulatory β and γ subunits or differences in the activity of the various hepatic AMPK complexes combination have been reported. Nevertheless, it should be noted that AMPK complexes distribution can be regulated intracellularly with AMPKα2-containing complexes present in both nucleus and cytoplasm raising the possibility of direct regulation of gene transcription (Salt et al., 1998) and with AMPKα1-containing complexes localized in the cytoplasm but also at the plasma membrane (Evans et al., 2005, Hallows et al., 2003). Changes in the subcellular localization of AMPKα2 in response to specific stimuli appear to be conserved from yeast to mammals via a mechanism involving the interaction with the regulatory β subunits (Suzuki et al., 2007, Vincent et al., 2001). In mammalian muscle cells, AMPKα2 bound to the β2 subunit translocates to the nucleus in a manner dependent on a nuclear localization signal that is present in AMPKα2 but not in AMPKα1 subunit. AMPKα2 bound to the β1 subunit is anchored into the cytoplasm at the outer mitochondrial membrane through the myristoylation of β1 subunit. These data suggest that activation of AMPK complexes may elicit distinct metabolic effects in tissues and cells depending on the expression of the different α- and β-subunit isoforms and illustrate the complexity of the molecular mechanisms by which energy metabolism can be regulated by AMPK.
Regulation of AMPK activity involves both direct allosteric activation and reversible phosphorylation. Activation of AMPK requires phosphorylation on Thr-172 within the catalytic subunit and three upstream kinases have been identified corresponding to the tumor suppressor LKB1 kinase, CaMKKβ (Cα2+/calmodulin-dependent protein kinase kinase β) and possibly TAK1 (TGFβ-activated kinase-1, a member of the mitogen-activated protein kinase kinase family). LKB1 is mainly involved in Thr-172 phosphorylation following change in AMP:ATP ratio (Hawley et al., 2003, Shaw et al., 2004, Woods et al., 2003). It has become evident in the last years that LKB1 plays a crucial role in activating AMPK to control glucose and lipid metabolism in the liver (Shaw et al., 2005, Imai et al., 2006). It has been suggested that LKB1 may be constitutively active (Lizcano et al., 2004, Sakamoto et al., 2004) but recent studies indicates that cytosolic localization and activity of LKB1 can be governed by LKB1 acetylation status in the liver (Lan et al., 2008). CaMKKβ is viewed as an alternate upstream kinase that could also phosphorylate Thr-172 and activate AMPK in intact cells by an AMP-independent manner in response to increased intracellular Ca2+ concentrations (Hawley et al., 2005, Hurley et al., 2005, Woods et al., 2005). However, CaMKKβ is highly expressed in neural tissue and the role for Ca2+-mediated AMPK activation in the liver remains to be investigated. The third potential upstream kinase is TAK1, which activates the S. cerevisiae homologue of AMPK, the SNF1 complex, when expressed in the yeast (Momcilovic et al., 2006), and could also phosphorylate Thr-172 and activate AMPK in mouse embryonic fibroblasts (Xie et al., 2006) but its role as an upstream kinase remains controversial (Goransson et al., 2007). In addition to phosphorylation, AMPK is allosterically activated by AMP which binds to the regulatory γ subunit. Binding of AMP to AMPK induces a conformational change in the kinase domain that protects AMPK from dephosphorylation of Thr-172 (Riek et al., 2008), probably catalysed by a form of protein phosphatase-2C (Sanders et al., 2007). The combination of the allosteric and phosphorylation effects causes >1000-fold increase in kinase activity (Suter et al., 2006) allowing to respond to small changes in cellular energy status in a highly sensitive manner.
2. Activators of AMPK in the liver
AMPK is activated in response to a variety of metabolic stresses that typically, but not exclusively, change the cellular AMP:ATP ratio, either by increasing ATP consumption (activation of biosynthetic pathways) or reducing ATP production following hypoxia, glucose deprivation and inhibition of mitochondrial oxidative phosphorylation with metabolic poisons (arsenite, oligomycin, dinitrophenol, azide and antimycin A) (Towler and Hardie, 2007). AMPK plays a central role in the metabolic adaptation to acute and chronic nutritional stresses. For instance, AMPK is activated in the liver by the metabolic challenges imposed by either a 24-h fast (Munday et al., 1991, Witters et al., 1994) and dietary energy restriction (Jiang et al., 2008). However, in other studies, fasting and caloric restriction did not activate liver AMPK (Gonzalez et al., 2004, To et al., 2007). Interestingly, it has been noted that the metabolic demands during muscular work results in a decrease in energy status of the liver (Camacho et al., 2006). Indeed, increased AMPK activation has been demonstrated following short-term exercise in rat liver (Carlson and Winder, 1999, Park et al., 2002). Long-term exercise also induced significant increase in AMPK phosphorylation as well as AMPKα1-and α2-subunit mRNA levels in the liver, suggesting a role for hepatic AMPK in long-term exercise-induced hepatic adaptations (Takekoshi et al., 2006).
In the liver, the transition from the fasted to the fed state is also associated with physiological changes in energy dynamics. The reversal of the metabolic response to starvation includes alterations in enzyme phosphorylation states and changes in the concentration of key regulatory molecules. It has been reported that AMPK coordinates the changes in the activity and/or expression of a number of enzymes of lipid metabolism during refeeding (Munday et al., 1991, Gonzalez et al., 2004, Assifi et al., 2005, Dentin et al., 2005). Refeeding causes a 40% decrease in the activity of AMPKα1 within 1 h, with additional decrease in both AMPKα1 and AMPKα2 activities occurring between 1 and 24 h (Assifi et al., 2005). It is noteworthy that LKB1 activity could also be modulated during the starvation-refeeding transition in association with its acetylation and intracellular localization (Lan et al., 2008). The modification of AMPK activity during the starved-fed transition is compatible with earlier studies that linked these changes to increase in plasma insulin [reported to decrease AMPK activity in isolated hepatocytes (Witters and Kemp, 1992)], and decrease in glucagon [showed to activate hepatic AMPK (Sim and Hardie, 1988)], possibly by a protein kinase A-induced phosphorylation and activation of LKB1 (Kimball et al., 2004). In addition, changes in circulating levels of various hormones and adipokines during refeeding, may directly affect AMPK activity in the hepatocytes and could also contribute to the fasted-to-fed transition from catabolism to anabolism in the liver. It has been reported that hepatic AMPK can be regulated by ghrelin (Barazzoni et al., 2005, Kola et al., 2005), endocannabinoids (Kola et al., 2005), glucocorticoids (Christ-Crain et al., 2008), resistin (Banerjee et al., 2004) and adiponectin (Yamauchi et al., 2002) (Table 1).
Table 1.
Pharmacological and natural modulators of AMPK activity in the liver
| Molecule/Compound | References | |
|---|---|---|
| Activation | A-769662 | (Goransson et al., 2007) |
| AICAR | (Corton et al., 1995) | |
| Metformin | (Zhou et al., 2001) | |
| Thiazolidinediones | (Fryer et al., 2002, Saha et al., 2004) | |
| Adiponectin | (Yamauchi et al., 2002) | |
| Glucocorticoids | (Christ-Crain et al., 2008) | |
| Betaine | (Song et al., 2007) | |
| Berberine | (Lee et al., 2006) | |
| Bitter melon | (Tan et al., 2008) | |
| Combretastatin A4 | (Zhang et al., 2008) | |
| EGCG (epigallocatechin gallate) | (Collins et al., 2007) | |
| Galegine | (Mooney et al., 2008) | |
| Ginseng | (Park et al., 2008) | |
| Resveratrol | (Zang et al., 2006) | |
| Oltipraz | (Bae et al., 2007) | |
| Inhibition | Ghrelin | (Barazzoni et al., 2005, Kola et al., 2005) |
| Endocannabinoids | (Kola et al., 2005) | |
| Resistin | (Banerjee et al., 2004) |
As well as responding to metabolic stresses, hepatic AMPK activity is modulated by various pharmacological and natural drugs including [AICAR] (5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside) (Corton et al., 1995), compound A-769662 (Cool et al., 2006), polyphenols (Zang et al., 2006) and two major classes of existing antidiabetic drugs biguanides (metformin and phenformin) (Zhou et al., 2001) and thiazolidinediones (TZDs) (Saha et al., 2004).
AICAR is a cell-permeable nucleoside which could be metabolically converted to 5-aminoimidazole-4-carboxamide ribotide (AICA ribotide or ZMP) by adenosine kinase. ZMP shares some structural similarities with 5′-AMP and can mimic all of the allosteric effects of 5′-AMP on the AMPK system (Corton et al., 1995). During the last decade, AICAR has been extensively used both in vitro and in vivo to activate hepatic AMPK (Viollet et al., 2006) because it was generally assumed that activation of AMPK by AICAR does not affect cellular levels of AMP, ADP or ATP (Corton et al., 1995). However, this view has been recently challenged, showing that treatment of hepatocytes with AICAR concentrations above 200 μM depleted intracellular ATP levels (Guigas et al., 2006, Mukhtar et al., 2008). Importantly, AMPK-independent effects of AICAR in the control of hepatic glucose uptake (Guigas et al., 2006), phosphatidylcholine synthesis (Jacobs et al., 2007) and autophagic proteolysis (Meley et al., 2006) probably associated with its effect on ATP depletion where also recently reported. Detrimental effect of AICAR is likely to be due to an AMPK-independent inhibition of mitochondrial oxidative phosphorylation induced by a concomitant effect of ZMP on the mitochondrial respiratory chain complex 1 and a drop of adenine nucleotides and inorganic phosphate following its phosphorylation (Guigas et al., 2007). Furthermore, we established that ZTP accumulation induced uncoupling of mitochondrial oxidative phosphorylation, an effect that could worsen the change in cellular energetic by decreasing the yield of ATP synthesis (Guigas et al., 2007). In addition, it should be noted another important caveat in the use of AICAR because ZMP accumulation does affect certain other AMP-regulated enzymes such as glucokinase and fructose 1,6-bisphosphatase causing inhibition of glycolysis and gluconeogenesis in hepatocytes (Vincent et al., 1992, Vincent et al., 1991). Therefore, like all pharmacological approaches, results of experiments using AICAR must be interpreted with caution and it remains to be clearly determined whether all AICAR effects described are mediated by AMPK. In addition, it has been demonstrated that uptake of AICAR into cells, mediated by the adenosine transport system, is blocked by a number of protein kinase inhibitors, thus preventing ZMP accumulation and subsequent AMPK activation (Fryer et al., 2002, Guigas, unpublished results).
Recently, a new class of AMPK activators have been identified after the screening of a chemical library using partially purified AMPK from rat liver. The non nucleoside thienopyridone A-592017 emerged from the initial screen, and after optimization the more potent one, A-769662, was developed. Specificity of this new compound has been tested on a panel of 76 protein kinases and the majority of kinases were not significantly affected at 10 μM (Goransson et al., 2007) suggesting that A-769662 is a new specific activator of AMPK. Unlike other AMPK activators, A-769662 directly activates native AMPK complex purified from rat liver in cell-free essays by mimicking both effects of AMP on allosteric activation and inhibition of dephosphorylation of AMPK complex (Goransson et al., 2007, Sanders et al., 2007). However, A-769662 and AMP binding sites on AMPK complex are unlikely to be identical. Firstly, in the presence of saturating AMP concentration, A-769662 stimulated further AMPK activity (Cool et al., 2006, Goransson et al., 2007). Second, A-769662 allosterically activates AMPK complexes harboring a mutation in the γ1 subunit that abolishes allosteric activation by AMP (Sanders et al., 2007). Interestingly, an AMPK complex lacking the glycogen binding domain of the β subunit or containing a mutation of Ser-108 to alanine (an autophosphorylation site within the glycogen binding domain of the β1 subunit) completely abolished the allosteric effect of A-769662, while only partially reducing AMP activation (Sanders et al., 2007). It has been reported that AMPK activation by A-769662 was independent of the upstream kinase utilized (Goransson et al., 2007, Sanders et al., 2007). Importantly, neither change in adenine nucleotide levels (Cool et al., 2006) nor alterations in mitochondrial oxidative phosphorylation (Guigas et al., 2008) following treatment with A-769662 have been detected in hepatocytes. Addition of A-769662 to primary mouse hepatocytes stimulates AMPK activity and phosphorylation of its known downstream targets but was completely abolished in hepatocytes lacking AMPKα1 and α2 catalytic subunits (Goransson et al., 2007). Short-term in vivo treatment with this compound recapitulates many of the effects expected for an hepatic activation of AMPK (Foretz et al., 2005) as it is mainly targeted to the liver (Cool et al., 2006).
Polyphenols, including resveratrol and epigallocatechin-3-gallate (EGCG), have been recently identified as potent activators for AMPK in vitro and in vivo (Baur et al., 2006, Collins et al., 2007, Zang et al., 2006). Some of the beneficial metabolic actions of polyphenols are mediated by their ability to activate sirtuin 1 (SIRT1), a mammalian ortholog of Sir2 (silent information regulator 2), an NAD-dependent deacetylase that acts as a master metabolic sensor of NAD+ and modulates cellular metabolism and life span (Baur et al., 2006). It has been demonstrated that SIRT1 activation by resveratrol functions as an upstream regulator in the LKB1/AMPK signaling axis (Hou et al., 2008). The ability of resveratrol to stimulate AMPK was mimicked by overexpression of SIRT1 and abolished by knockdown or pharmacological inhibition of SIRT1. Furthermore, AMPK activation by resveratrol-activated SIRT1 is mediated by the upstream kinase, LKB1, but not CaMKKβ. The mechanism by which SIRT1 activation by polyphenols leads to LKB1-dependent AMPK activation relies on the direct deacetylation of LKB1 by SIRT1 (Lan et al., 2008). Indeed, activation of SIRT1 deacetylates LKB1, which in turn increases its cytoplasmic localization, its association with the LKB1 activator STRAD and its ability to activate AMPK. Nevertheless, it should be noted that AMPK activation by polyphenols may involve distinct regulators. It has been shown that the EGCG-induced increase in AMPK phosphorylation is mediated by CaMKK activation through production of ROS (Collins et al., 2007). Furthermore, the SIRT1 regulation of LKB1/AMPK signaling appears to be tissue-specific as resveratrol-stimulated AMPK activation in neurons was independent of SIRT1 (Dasgupta and Milbrandt, 2007).
The molecular pathway of AMPK activation by the antidiabetic drug metformin has been unclear (Zhou et al., 2001) but the direct inhibition of the respiratory chain complex 1 by the biguanide constitutes the more convincing molecular mechanism, thus, leading to reduced cellular ATP and increased AMP (El-Mir et al., 2000, Brunmair et al., 2004, Owen et al., 2000). This is supported by the fact that metformin treatment in primary hepatocytes resulted in a decrease in ATP content (Guigas et al., 2006). It should be noted that metformin could also exert AMPK-independent effects in the liver, probably due to its effect on cellular ATP levels (Guigas et al., 2006). It has been reported that TZDs acutely activate AMPK by a mechanism independent of PPARγ-regulated gene transcription, which appears to be associated with change in cellular energy state and could potentially increase AMPK activity (Brunmair et al., 2004, Saha et al., 2004).
3. Regulation of hepatic lipid metabolism
AMPK phosphorylates and inactivates a number of metabolic enzymes involved in lipid metabolism in order to maintain liver energy status. AMPK coordinates the changes in the hepatic lipid metabolism and, so, regulates the partitioning of fatty acids between oxidative and biosynthetic pathways. 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase) and acetyl CoA carboxylase (ACC), key enzymes in cholesterol and fatty acid synthesis respectively, were the first enzymes shown to be phosphorylated and inactivated by AMPK (Corton et al., 1994, Henin et al., 1995). Although the action of AMPK is achieved by rapid and direct phosphorylation of these enzymes, long-term effects have also been clearly demonstrated on gene expression. AMPK activation by AICAR or by the use of adenovirus-mediated overexpression of a constitutively active form of the α2 catalytic subunit (AMPKα2-CA) inhibits glycolytic and lipogenic gene expression (Foretz et al., 2005, Leclerc et al., 2001, Woods et al., 2000, Foretz et al., 1998, Leclerc et al., 1998), preserving glucose for ATP-producing pathways rather than for lipid synthesis. Of note, AMPK activation reduces expression of sterol regulatory element-binding protein-1c (SREBP1c) (Zhou et al., 2001, Foretz et al., 2005) and carbohydrate response element–binding protein (ChREBP) (Foretz et al., 2005, Kawaguchi et al., 2002), transcription factors playing a key role in the transcriptional regulation of lipogenic and glycolytic genes by insulin and glucose, respectively. In addition, it has been also reported that AMPK directly phosphorylates ChREBP and modulates its DNA binding activity (Kawaguchi et al., 2002). Polyunsaturated fatty acids (PUFAs) are known to repress glycolytic and lipogenic gene expression and raised the question about a role of AMPK in mediating the effect of PUFAs on gene transcription. To address this point, we investigated the effect of PUFAs in mice lacking AMPKα1/α2 catalytic subunits in the liver (AMPKα1α2LS−/−). In the absence of hepatic AMPK, PUFAs continue to inhibit glycolytic and lipogenic gene expression indicating the existence of AMPK-independent mechanism(s) (Viollet et al., 2006).
In the cholesterol synthesis pathway, AMPK blocks the conversion of HMG-CoA to mevalonate. One could expect detrimental effect on cholesterol homeostasis when AMPK activity is altered. In total and liver-specific AMPKα2 KO mice, plasma levels for total and HDL cholesterol are not statistically different compared to controls but have a tendency to be higher (Andreelli et al., 2006, Viollet et al., 2003). This suggested that remaining α1 subunit activity in AMPKα2 KO mice is sufficient to control hepatic cholesterol synthesis and that HMG-CoA reductase is a target for both catalytic isoforms of AMPK.
ACC is an important rate-controlling enzyme for the synthesis of malonyl-CoA, which is both a critical precursor for biosynthesis of fatty acids and a potent inhibitor of mitochondrial fatty acid oxidation via the allosteric regulation of carnitine palmitoyltransferase-1 (CPT-1) which catalyzes the entry of long-chain fatty acyl-CoA into mitochondria. Inhibition of ACC by AMPK leads to a fall in malonyl-CoA content and a subsequent decrease in fatty acid synthesis concomitantly with an increase in β-oxidation (Brusq et al., 2006, Velasco et al., 1997). Exposure of hepatocytes to AICAR also produces a strong stimulation of long-chain fatty acid oxidation as a result of an increase in the rate of ketogenesis and an inhibition of triacylglycerol synthesis via phosphorylation of sn-glycerol-3-phosphate acyltransferase (GPAT) (Muoio et al., 1999). In addition, overexpression of AMPKα2-CA in the liver increases plasma ketone bodies levels, a surrogate marker for hepatic β-oxidation (Foretz et al., 2005). Interestingly, AMPK-induced ACC phosphorylation is impaired in hepatocytes deleted of both catalytic subunit, contributing to increase intracellular malonyl CoA levels and triglyceride (TG) accumulation in the liver (B. Viollet, M. Foretz, unpublished results). Thus, these results indicate that AMPK regulates cellular lipid metabolism in large part through stimulation of fatty acid oxidation. However, recent studies showed that AMPK also stimulates a previously unrecognized pathway that involves constitutive exocytosis of lipoproteins (Puljak et al., 2008). This alternative pathway appears to be quantitatively important for intracellular lipid homeostasis and may function in parallel with fatty acid oxidation to regulate intracellular lipid content.
4. Regulation of hepatic mitochondrial biogenesis and function
As reported recently, AMPK has been also involved in the control of mitochondrial biogenesis in the liver, a role already evidenced in skeletal muscles (Reznick and Shulman, 2006). First evidences were brought by treatment with resveratrol, a polyphenol constituent of red wine, which increases mitochondrial number in liver in association with AMPK activation (Baur et al., 2006). Second, AMPKα1α2LS−/− mice have reduced mitochondrial biogenesis as suggested by decreased transcript and protein expression of key mitochondrial constituents such as peroxisome proliferator-activated receptor-γ coactivator-1 α (PGC-1α), cytochrome c oxidase I (COX I), COX IV and cytochrome c genes (Guigas et al., 2007) (Figure 1). Interestingly, both mitochondrial respiration and basal level of ATP were also significantly lower in hepatocytes isolated from AMPKα1α2LS−/− mice compared with control mice (Guigas et al., 2006). This result emphasizes the importance of AMPK in the regulation of cellular energy homeostasis via the control of adaptive mitochondrial function. Thus, diminished AMPK activity may be an important contributing factor in the reduced mitochondrial function and dysregulated intracellular lipid metabolism associated with hepatic insulin resistance. The exact mechanisms by which deletion of AMPK in the liver affects PGC-1α expression and mitochondrial biogenesis remain to be elucidated but could be due to a lack of protein-protein interaction, as suggested in skeletal muscle (Jager et al., 2007).
Figure 1. Alterations in mitochondrial biogenesis in hepatocytes from AMPKα1α2LS−/− mice.
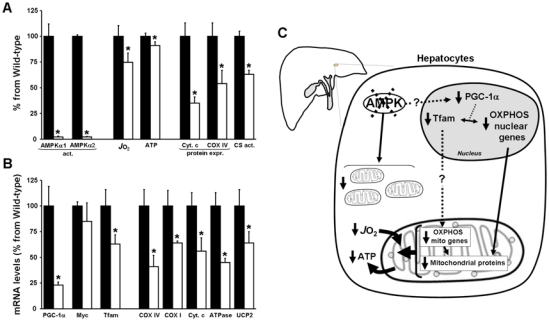
(A) Hepatocytes from wild-type (black bars) or AMPKα1α2LS−/− mice (open bars) were isolated and AMPKα1 and α2 activities, oxygen consumption rate (JO2), intracellular ATP concentrations, cytochrome c (Cyt. C) and cytochrome oxidase (COX IV) contents, and citrate synthase activity (CS) were measured. (B) In separate experiments, RNA was extracted from freeze-clamped livers of wild-type (black bars) and AMPKα1α2LS−/− (open bars) mice starved for 24h and quantitative real-time PCR was performed on cDNA. The mRNA contents for the indicated gene were normalized for β-actin and expressed relative to that in wild-type mice. (C) The results of all experiments indicating that AMPK is an important regulator of mitochondrial biogenesis in the liver are summarized in a simplified scheme. Deletion of AMPK • subunits in hepatocytes leads to reduced mitochondrial biogenesis as suggested by decreased transcript and protein expression of key mitochondrial constituents such as peroxisome proliferator-activated receptor-γ coactivator-1 α (PGC-1α) and mitochondrial transcription factor A (Tfam) which control the expression of nuclear and mitochondrial genes involved in mitochondrial oxidative phosphorylation (OXPHOS). Accordingly, mitochondrial respiration and basal level of ATP were significantly decreased. These data emphasize the importance of AMPK in the control of adaptive mitochondrial function. *p<0.05 compared with wild-type mice.
5. Regulation of hepatic glucose production
Recent results from various animal models confirm the physiological importance of hepatic AMPK for whole-body glucose homeostasis. It has been first shown that systemic infusion of AICAR in normal and insulin-resistant obese rats leads to the inhibition of hepatic glucose production (HGP) (Bergeron et al., 2001). Then, several studies reported that AICAR treatment improve blood glucose levels and glucose tolerance in animal models of type 2 diabetes. However, the relative roles played by skeletal muscle and liver in mediating the hypoglycemic effect of AICAR remain unclear. In muscle-specific AMPK dominant-negative mice, AICAR has a weaker hypoglycemic effect than in control animals (Mu et al., 2001), suggesting that this compound lowers glycemia at least partly by increasing muscle glucose uptake. We demonstrated that AMPKα1α2LS−/− mice display an impaired hypoglycemic response to AICAR, indicating that the liver as well participate to the hypoglycemic effect of AICAR (Figure 2). It has been also reported that short-term hepatic expression of AMPK-CA leads to mild hypoglycemia in normal mice (Foretz et al., 2005, Viana et al., 2006) and abolishes hyperglycemia in diabetic ob/ob and STZ-induced diabetic mice (Foretz et al., 2005). The hypoglycemic effect of AMPK activation is consistent with the abolition of HGP, as suggested by the down-regulation of gluconeogenic gene expression (e.g., phosphoenolpyruvate carboxykinase [PEPCK] and glucose-6-phosphatase [G6Pase]) and inhibition of glucose production in hepatic cells expressing AMPK-CA or treated with AICAR (Lochhead et al., 2000, Foretz et al., 2005, Viana et al., 2006). Using transcriptional profiling of hepatocyte cell lines treated with AICAR, the dual specificity phosphatase Dusp4 and the immediate early transcription factor Egr1 have been identified as transcriptional targets of AMPK that are necessary for its ability to fully repress glucose production (Berasi et al., 2006). Egr1 and Dusp4 act sequentially to mediate the inhibitory effect of AMPK on hepatic gluconeogenesis: increase in Egr1 protein is accompanied with increased binding to the Dusp4 promoter leading to Dusp4 expression and inhibition of PEPCK and G6Pase gene transcription (Berasi et al., 2006).
Figure 2. The hypoglycemic effect of AICAR is dependent partially on hepatic AMPK.
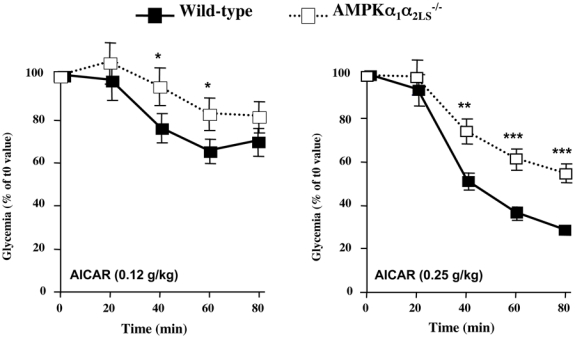
AMPKα1α2LS−/− mice deleted for both AMPK catalytic subunit in the liver are resistant to hypoglycemic effect of AICAR. AICAR tolerance test was performed on overnight fasted AMPKα1α2LS−/− mice injected intraperitonealy with AICAR at 0.12 g/kg and 0.25 g/kg and tail blood was collected at 0, 20, 40, 60 and 80 min for determination of glucose concentration using a glucometer. *p<0.005, **p<0.001, ***p<0.0001 vs control mice by unpaired, two-tailed Student t test.
The importance of AMPK in the control of glucose output by the liver was recently highlighted by the potent effects of circulating adipocyte-derived hormones on whole-body glucose metabolism. A physiological link has been established between circulating resistin levels and hepatic AMPK activity in the maintenance of blood glucose (Banerjee et al., 2004). Low blood glucose levels and reduced HGP in mice lacking resistin are likely related, at least in part, to activation of AMPK and decreased expression of gluconeogenic enzymes in the liver. Interestingly, adiponectin signaling opposes that of resistin in the control of glucose homeostasis and activation of AMPK is though to underlie the ability of this adipocyte hormone to reduce HGP (Yamauchi et al., 2002). According to this result, adiponectin failed to regulate HGP in liver-specific AMPKα2 KO mice (Andreelli et al., 2006). AdipoR1 and AdipoR2 serve as adiponectin receptors and functional differences in adiponectin-signaling pathways in the liver have been recently demonstrated with AdipoR1 being tightly linked to activation of AMPK pathways whereas AdipoR2 being more associated with the activation of peroxisome proliferator-activated receptor-α (PPAR-α) pathways (Yamauchi et al., 2007, Bjursell et al., 2007).
Recently, the transcriptional co-activator transducer of regulated CREB activity 2 (TORC2) has been identified as a pivotal component of the gluconeogenic program (Koo et al., 2005). TORC2 mediates CREB-dependent transcription of PGC1α and its subsequent gluconeogenic targets PEPCK and G6Pase. TORC2 is regulated by multiple signaling pathways in response to changes in glucagon and insulin levels or intracellular energy status. Phosphorylation on Ser171 confers binding of the protein 14-3-3 and sequestration of TORC2 out of the nucleus, resulting in inhibition of gluconeogenic gene expression. The kinase responsible for phosphorylating Ser171 of TORC2 was initially identified as salt-inducible kinase 2 (SIK2), an AMPK-related protein kinase (Screaton et al., 2004). Recently, activation of AMPK was also found to phosphorylate TORC2 and regulate cytoplasmic translocation of TORC2 in primary hepatocyte cultures (Koo et al., 2005). Interestingly, in the absence of LKB1 in the liver, the expression of some of the key gluconeogenic genes are enhanced and the antidiabetic drug metformin no longer reduced blood glucose levels, demonstrating that hepatic LKB1/AMPK axis is required to maintain blood glucose levels (Shaw et al., 2005). LKB1 phosphorylates and activates a number of kinases including AMPK and SIK family members (Lizcano et al., 2004), suggesting that genetic deletion of LKB1 in liver will result in the loss of multiple metabolic checkpoints in the regulation of TORC2. In LKB1-deficient liver, AMPK was almost completely inactive, TORC2 was predominantly nuclear and fasting blood glucose levels were highly increased concomitantly with PEPCK and G6Pase gene expression (Shaw et al., 2005). The reduction of TORC2 protein levels by injection of adenoviruses bearing small hairpin RNA for TORC2 in mice lacking LKB1 in the liver resulted in decreased PGC-1α protein levels accompanied by a significant decrease in fasting blood glucose levels, suggesting that TORC2 is a critical downstream target of LKB1-dependent kinases in the control of gluconeogenesis (Shaw et al., 2005). In addition, a recent study has shown that metformin regulates hepatic gluconeogenesis through AMPK-dependent regulation of the orphan nuclear receptor small heterodimer partner (SHP) (Kim et al., 2008). AMPK activation caused upregulation of SHP gene expression and this in turn, inhibits PEPCK and G6Pase gene expression. It has been proposed that metformin could regulate the expression of hepatic gluconeogenic genes via both a short-term pathways, involving the protein stabilization or phosphorylation of AMPK-targeted transcription factors and coactivators, such as TORC2 (Shaw et al., 2005), HNF4α (Leclerc et al., 2001, Hong et al., 2003) and FoxO1 (Barthel et al., 2002) and a long-term effect exerted via the induction of SHP gene expression (Kim et al., 2008) (Figure 3). Lastly, it should be noted that AMPK has been also involved in the repression of gluconeogenic genes in response to ECGC treatment via a LKB1-independent pathway relying on CaMKK activation through production of ROS (Collins et al., 2007).
Figure 3. AMPK controls gluconeogenic gene expression by promoting TORC2 phosphorylation.
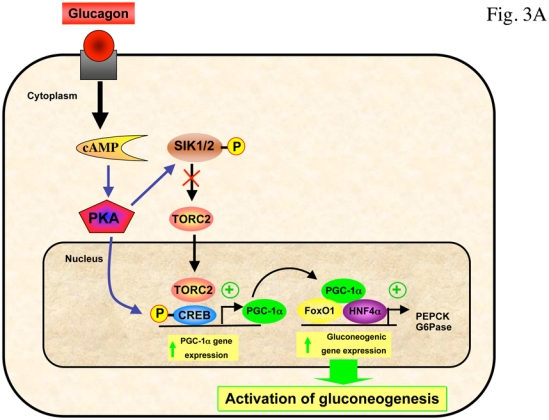
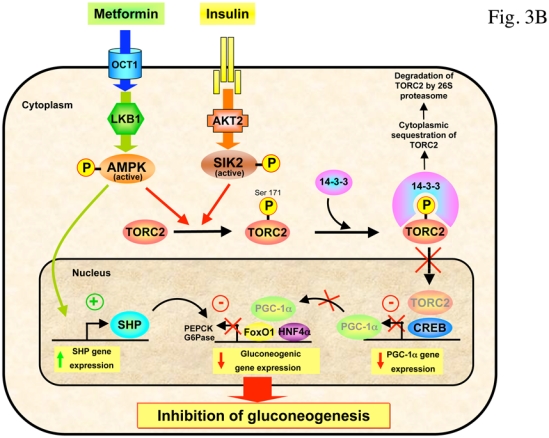
(A) In response to fasting, glucagon promotes dephosphorylation of TORC2 on Ser171 through the PKA-dependent inhibition of SIK, leading to its nuclear translocation and association with CREB transcription factor, driving the expression of the PGC1α co-activator. Expression of the coactivator PGC-1α drives the transcription of key gluconeogenic enzymes such as PEPCK and G6Pase in association with the transcription factor HNF4a and the forkhead family activator FoxO1. (B) Phosphorylation of TORC2 (Ser171) is controlled by insulin through AKT2-dependent activation of SIK2 and by AMPK. Phosphorylated TORC2 is sequestered in the cytoplasm via a phosphorylation-dependent interaction with 14-3-3 proteins and degraded by 26S proteasome to inhibit gluconeogenic program. The antidiabetic drug metformin inhibits gluconeogenic gene expression through the LKB1/AMPK/TORC2 axis. TORC2 represents a central point in the control of hepatic gluconeogenesis that senses both hormone and energy status. AMPK activation also caused upregulation of SHP gene expression and this in turn, inhibits PEPCK and G6Pase gene expression.
6. Management of glucose homeostasis and dyslipidemia
The growing evidence that AMPK regulates the coordination of anabolic and catabolic metabolic processes represents an attractive therapeutic target for intervention in many conditions of disordered energy balance, including obesity, type 2 diabetes, and the metabolic syndrome (Winder and Hardie, 1999). Support for this idea came from in vivo treatment with AICAR of various animal models of insulin resistance, causing improvement of metabolic disturbances, at least partly by lowering blood glucose levels and increasing insulin sensitivity (Bergeron et al., 2001, Buhl et al., 2002, Iglesias et al., 2002, Song et al., 2002, Pold et al., 2005, Fiedler et al., 2001). Chronic treatment with AICAR was also shown to reduce plasma TG levels and adiposity in obese animals (Bergeron et al., 2001, Buhl et al., 2002, Song et al., 2002). Furthermore, similar beneficial metabolic effects have been also obtained by direct AMPK activation in the liver by overexpression of AMPK-CA or by the use of the AMPK activator A-769662, leading to decreased plasma glucose, plasma TG levels and adiposity in diabetic, obese and high fat fed mice (Cool et al., 2006, Foretz et al., 2005, Yang et al., 2008). These results suggest that direct targeting of the liver is crucial for improving glucose and lipid metabolism, thereby preventing and treating type 2 diabetes. Accordingly, it appears that the major effect of intravenous AICAR infusion in type 2 diabetic patients was on the liver with inhibition of hepatic glucose output and decreased blood glucose levels (Boon et al., 2008). However, as liver samples were not collected in this study, one can only speculate on the role of hepatic AMPK in this therapeutic effect of AICAR. Indeed, inhibition of hepatic glucose output could have resulted from AMPK-independent effects, such as inhibition of fructose-1,6-bisphosphatase by ZMP accumulation in the liver [(Vincent et al., 1991) and see above] Furthermore, AICAR administration could also stimulate hepatic fatty acid oxidation and/or inhibit whole body lipolysis, thereby reducing plasma non-esterified fatty acids (NEFA) concentration (Boon et al., 2008). Unexpectedly, the effect of AICAR infusion on skeletal muscle glucose uptake was not evident and no changes in AMPK activity were detected in skeletal muscle. Similarly, in another study performed in healthy humans, AMPK activity was not modified in skeletal muscle after AICAR infusion (Cuthbertson et al., 2007),reinforcing the role of the liver in the action of AICAR.
Several reports indicate that metformin and TZDs can reduce risk for type 2 diabetes in people with glucose intolerance (Knowler et al., 2002). Interestingly, these two drugs have been reported to activate AMPK which is now considered as a new target for the management of insulin-resistant state. Consistent with these data, AMPK now appears to be activated by a multitude of natural products such as polyphenols and traditional Chinese medicine able to reduce blood glucose levels in obese and diabetic animal models (table 1). Pathophysiology and management of type 2 diabetes are complex. Increased concentrations of NEFA and inflammatory cytokines (e.g., tumor necrosis factor α [TNFα] and interleukin 6 [IL-6]) released by expanded visceral adipose tissue are crucial mechanisms involved in the alteration of insulin signaling cascade (Stumvoll et al., 2005). Adiponectin is another important key of the pathophysiology of type 2 diabetes. Adiponectin has favourable effects on insulin resistance, hepatic steatosis and inflammation (Kadowaki et al., 2006). It is now well established that circulating levels of adiponectin are decreased in individuals with obesity and insulin resistance, suggesting that its deficiency may have a causal role in the etiopathogenesis of these diseases and their consequences. Thus, adiponectin replacement or restoration of endogenous secretion in humans may represent a promising approach to prevent and/or treat obesity, insulin resistance and the metabolic syndrome. The chronic effects of adiponectin on insulin resistance were investigated in vivo by generating adiponectin transgenic mice. Globular adiponectin transgenic ob/ob mice showed partial amelioration of insulin resistance and diabetes (Yamauchi et al., 2003) and full length adiponectin showed suppression of insulin-mediated endogenous glucose production (Yamauchi et al., 2002). Interestingly, it has been shown that metabolic and insulin-sensitizing effects of TZDs are in part mediated through increase in adiponectin availability. Indeed, TZDs metabolic effects are abolished in lipoatrophic mice suggesting that adipose tissue is an essential key of TZDs action (Chao et al., 2000). In addition, TZDs can markedly enhance the expression and secretion of adiponectin in vitro and in vivo through the activation of its promoter and also by blocking the suppressive effect of TNFα on the production of adiponectin. Even if the intracellular adiponectin signaling is not yet completely described, it is clearly demonstrated that adiponectin activates hepatic AMPK (Yamauchi et al., 2002). In consequence, TZDs probably activate AMPK by an indirect mechanism, through increase of adiponectin levels. This is suggested by the reduction of AMPK activation by rosiglitazone in adiponectin KO mice (Nawrocki et al., 2006). A possible similar mechanism of action could concern AICAR (but not metformin) which increase the expression of adiponectin in human adipose tissue as TZDs (Lihn et al., 2004).
7. Management of fatty liver and hepatic fibrosis
Nonalcoholic fatty liver disease (NAFLD), the most common chronic liver disease in western countries, is considered as the hepatic manifestation of the metabolic syndrome (Angulo, 2002). NAFLD is highly associated with obesity and insulin resistance and represent a broad spectrum of liver abnormalities ranging from simple hepatic steatosis (accumulation of TG inside hepatocytes) to a more severe form, nonalcoholic steatohepatitis (NASH), which is associated with hepatocyte damage, chronic inflammation, and fibrosis, and may progress to cirrhosis and liver failure. Studies in humans and various animal models have suggested that efforts to enhance insulin sensitivity might improve fatty liver disease, a situation frequently observed in patients with metabolic syndrome. The efficacy of metformin as a treatment for fatty liver disease has been confirmed in obese, ob/ob mice, which develop hyperinsulinemia, insulin resistance and fatty livers (Lin et al., 2000). Recent studies suggest that activation of AMPK accounts for the lipid-lowering effect of metformin in cultured hepatocytes (Zang et al., 2004). Similarly, adiponectin restores insulin sensitivity and decreases hepatic steatosis by lowering TG content in the liver of obese mice (Xu et al., 2003, Yamauchi et al., 2001). The action of adiponectin is linked to an activation of hepatic AMPK, ultimately leading to decreased fatty acid biosynthesis and increased mitochondrial fatty acid oxidation (Yamauchi et al., 2002). The role of AMPK has been confirmed by the decrease in liver TG content in lean and obese rodents during AICAR infusion (Bergeron et al., 2001) and treatment with direct AMPK activator A-769662 (Cool et al., 2006). In addition, it has been recently demonstrated that resveratrol improves insulin sensitivity and protects against lipid accumulation in the liver of diabetic and high-fat fed animals concomitantly with activation of hepatic AMPK (Baur et al., 2006, Shang et al., 2008). These effects have been correlated to increased mitochondrial number and SIRT1-mediated PCG-1α deacetylation, and decreased expression of lipogenic genes in the liver. Similarly, the beneficial effect of betaine (trimethylglycine), a naturally occurring metabolite of choline, on high-sucrose diet-induced hepatic steatosis in mice is associated with increased activation of hepatic AMPK (Song et al., 2007). Promising therapeutic effects of betaine supplementation on human NAFLD have been reported in a pilot clinical studies (Abdelmalek et al., 2001) but the use of betaine has been also described earlier in the treatment of alcoholic fatty liver disease (AFLD) (Barak et al., 1997). Although, the underlying causes of NAFLD and AFLD are clearly different, there are similarities in the disturbances of hepatic metabolism. This is supported by reports showing that treatement with adiponectin alleviated alcoholic and non-alcoholic fatty liver disease in mice, partly due to enhanced hepatic fatty acid oxidation and decreased fatty acid synthesis (Xu et al., 2003). Interestingly, chronic ethanol ingestion causes the impairment of AMPK-mediated regulation of fatty acid metabolism and may have an important role in the development of alcoholic fatty liver (You et al., 2004, Garcia-Villafranca et al., 2008). Activation of AMPK by AICAR or metformin largely blocked the ability of ethanol to increase levels of SREBP1c protein and expression of SREBP1c-regulated lipogenic enzymes and also appears to protect the liver from fatty changes associated with chronic alcohol use (You et al., 2004, Tomita et al., 2005). Very recently, treatment with resveratrol has been also shown to prevent the development of alcoholic liver steatosis through the SIRT1-AMPK signaling system associated with increased circulating adiponectin levels and enhanced expression of hepatic AdipoR1 and R2 receptors (Ajmo et al., 2008).
It is now established that hepatic stellate cells (HSCs) play a crucial role in the fibrotic response during the progression of NASH (Bataller and Brenner, 2005, Friedman, 2004). Stimuli such as liver injury activate and transdifferentiate HSCs from vitamin A-storing pericytes to myofibroblast-like cells. Once activated, human HSCs become proliferative, proinflammatory and profibrogenic through increased responsiveness to several soluble mediators (Friedman, 2004). Despite the clear role of insulin resistance in the progression of fibrosis, the molecular mechanisms involved in these conditions are still unclear. Adiponectin levels which have been directly correlated with insulin sensitivity are closely and inversely associated with degree of hepatic steatosis, necroinflammation, and fibrosis in NAFLD (Targher et al., 2006). Recent studies have demonstrated that in rat HSCs, adiponectin inhibits proliferation, migration, and expression of fibrogenic genes, and it may induce apoptosis of activated cells (Ding et al., 2005). Furthermore, in vivo administration of adiponectin prevents proliferation of activated HSCs and reduces the development of fibrosis and liver damage during experimental steatohepatitis (Xu et al., 2003, Kamada et al., 2003). In addition to adiponectin, AICAR and metformin significantly inhibited proliferation and migration of human HSCs in a dose-dependent manner (Caligiuri et al., 2008). The beneficial effect of these compounds is linked to suppression of platelet-derived growth factor (PDGF) expression in HSCs and subsequent inhibition of type I procollagen secretion (Caligiuri et al., 2008, Adachi and Brenner, 2008). Activation of AMPK by adiponectin plays a pivotal role in this molecular pathway since the dose-dependent PDGF suppression is abrogated in the presence of dominant-negative AMPK (Adachi and Brenner, 2008) or by the knock-down of AMPK (Caligiuri et al., 2008). Additional mechanisms, such as short-term inhibition of PDGF-mediated phosphorylation of ribosomal S6 kinase (p70S6K) and 4E binding protein-1 (4EBP1) (downstream effectors of the mammalian target of rapamycin complex 1 (mTORC1) pathway) by AICAR have been demonstrated (Caligiuri et al., 2008). AICAR and metformin could also inhibit HSCs proliferation via suppression of ROS production and subsequent inhibition of AKT pathway (Adachi and Brenner, 2008). Taken together, data from recent studies provide evidence that AMPK and adiponectin inhibit HSCs proliferation and hepatic fibrosis via multiple molecular mechanisms and suggest that use of drugs activating hepatic AMPK may have an additional rationale in their antifibrogenic properties.
Conclusion
Hepatic metabolism is highly sensitive to the variations in nutritional and hormonal signals reflecting changes in whole body energy demands. The AMPK system plays a major role in the regulation of hepatic glucose and lipid metabolism through its acute effect on energy metabolism and long-term effects on gene expression pattern in the liver. Activation of hepatic AMPK causes a switch from anabolic to catabolic state and entails metabolic consequences that are beneficial for diabetic patients, such as inhibition of HGP and restoration of blood glucose levels. AMPK is a key player in regulating energy balance at both cellular and whole-body levels, placing it as an ideal therapeutic target for the treatment of altered energy metabolism that occur in conditions like insulin resistance, type 2 diabetes and the metabolic syndrome. A large number of AMPK activators have been employed with promising results in animal models with obesity or type 2 diabetes and also in preliminary studies with diabetic patients. These encouraging results have provided the rationale for the development of new pharmacological but also nutritional AMPK activators. However, the widespread cellular functions of AMPK make its selective targeting in therapeutics a difficult one, with simultaneous advantageous and deleterious consequences being possible. One of the major caveats in the use of AMPK activators is their possible role in the regulation of food intake. Stimulation of AMPK expressed in specific nuclei of the hypothalamus has been shown to increase food intake (Minokoshi et al., 2004). The ideal AMPK activators will be administrated by the oral route, will activate AMPK at low concentration and be effective in specific target tissue, such as the liver but not the hypothalamus, and will also have minimal off-target and/or side effects. Finally, it is now becoming apparent that distinct expression patterns of AMPK isoforms exist between rodent and human liver. This finding will have significant implications for the understanding of the physiological role of AMPK complexes across species and for the design of new therapeutic agents targeting human AMPK complexes.
Acknowledgments
The authors were supported by the EXGENESIS Integrated Project (LSHM-CT- 2004-005272) funded by the European Commission, Programme Nationale de Recherche sur le Diabète, Agence Nationale de la Recherche, Association de Langue Française pour l’Etude du Diabète et des Maladies Métaboliques and Institut Benjamin Delessert.
References
- Abdelmalek MF, Angulo P, Jorgensen RA, Sylvestre PB, Lindor KD. Betaine, a promising new agent for patients with nonalcoholic steatohepatitis: results of a pilot study. Am J Gastroenterol. 2001;96:2711–7. doi: 10.1111/j.1572-0241.2001.04129.x. [DOI] [PubMed] [Google Scholar]
- Adachi M, Brenner DA. High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate-activated protein kinase. Hepatology. 2008;47:677–85. doi: 10.1002/hep.21991. [DOI] [PubMed] [Google Scholar]
- Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol Alleviates Alcoholic Fatty Liver in Mice. Am J Physiol Gastrointest Liver Physiol. 2008 doi: 10.1152/ajpgi.90358.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreelli F, Foretz M, Knauf C, Cani PD, Perrin C, Iglesias MA, Pillot B, Bado A, Tronche F, Mithieux G, Vaulont S, Burcelin R, Viollet B. Liver adenosine monophosphate-activated kinase-alpha2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology. 2006;147:2432–41. doi: 10.1210/en.2005-0898. [DOI] [PubMed] [Google Scholar]
- Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–31. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- Assifi MM, Suchankova G, Constant S, Prentki M, Saha AK, Ruderman NB. AMP-activated protein kinase and coordination of hepatic fatty acid metabolism of starved/carbohydrate-refed rats. Am J Physiol Endocrinol Metab. 2005;289:E794–800. doi: 10.1152/ajpendo.00144.2005. [DOI] [PubMed] [Google Scholar]
- Bae EJ, Yang YM, Kim JW, Kim SG. Identification of a novel class of dithiolethiones that prevent hepatic insulin resistance via the adenosine monophosphate-activated protein kinase-p70 ribosomal S6 kinase-1 pathway. Hepatology. 2007;46:730–9. doi: 10.1002/hep.21769. [DOI] [PubMed] [Google Scholar]
- Banerjee RR, Rangwala SM, Shapiro JS, Rich AS, Rhoades B, Qi Y, Wang J, Rajala MW, Pocai A, Scherer PE, Steppan CM, Ahima RS, Obici S, Rossetti L, Lazar MA. Regulation of fasted blood glucose by resistin. Science. 2004;303:1195–8. doi: 10.1126/science.1092341. [DOI] [PubMed] [Google Scholar]
- Barak AJ, Beckenhauer HC, Badakhsh S, Tuma DJ. The effect of betaine in reversing alcoholic steatosis. Alcohol Clin Exp Res. 1997;21:1100–2. [PubMed] [Google Scholar]
- Barazzoni R, Bosutti A, Stebel M, Cattin MR, Roder E, Visintin L, Cattin L, Biolo G, Zanetti M, Guarnieri G. Ghrelin regulates mitochondrial-lipid metabolism gene expression and tissue fat distribution in liver and skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E228–35. doi: 10.1152/ajpendo.00115.2004. [DOI] [PubMed] [Google Scholar]
- Barthel A, Schmoll D, Kruger KD, Roth RA, Joost HG. Regulation of the forkhead transcription factor FKHR (FOXO1a) by glucose starvation and AICAR, an activator of AMP-activated protein kinase. Endocrinology. 2002;143:3183–6. doi: 10.1210/endo.143.8.8792. [DOI] [PubMed] [Google Scholar]
- Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–42. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berasi SP, Huard C, Li D, Shih HH, Sun Y, Zhong W, Paulsen JE, Brown EL, Gimeno RE, Martinez RV. Inhibition of gluconeogenesis through transcriptional activation of EGR1 and DUSP4 by AMP-activated kinase. J Biol Chem. 2006;281:27167–77. doi: 10.1074/jbc.M602416200. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Previs SF, Cline GW, Perret P, Russell RR, 3rd, Young LH, Shulman GI. Effect of 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes. 2001;50:1076–82. doi: 10.2337/diabetes.50.5.1076. [DOI] [PubMed] [Google Scholar]
- Bjursell M, Ahnmark A, Bohlooly YM, William-Olsson L, Rhedin M, Peng XR, Ploj K, Gerdin AK, Arnerup G, Elmgren A, Berg AL, Oscarsson J, Linden D. Opposing effects of adiponectin receptors 1 and 2 on energy metabolism. Diabetes. 2007;56:583–93. doi: 10.2337/db06-1432. [DOI] [PubMed] [Google Scholar]
- Boon H, Bosselaar M, Praet SF, Blaak EE, Saris WH, Wagenmakers AJ, McGee SL, Tack CJ, Smits P, Hargreaves M, van Loon LJ. Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia. 2008;51:1893–900. doi: 10.1007/s00125-008-1108-7. [DOI] [PubMed] [Google Scholar]
- Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, Roden M, Gnaiger E, Nohl H, Waldhausl W, Furnsinn C. Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes. 2004;53:1052–9. doi: 10.2337/diabetes.53.4.1052. [DOI] [PubMed] [Google Scholar]
- Brusq JM, Ancellin N, Grondin P, Guillard R, Martin S, Saintillan Y, Issandou M. Inhibition of lipid synthesis through activation of AMP kinase: an additional mechanism for the hypolipidemic effects of berberine. J Lipid Res. 2006;47:1281–8. doi: 10.1194/jlr.M600020-JLR200. [DOI] [PubMed] [Google Scholar]
- Buhl ES, Jessen N, Pold R, Ledet T, Flyvbjerg A, Pedersen SB, Pedersen O, Schmitz O, Lund S. Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes. 2002;51:2199–206. doi: 10.2337/diabetes.51.7.2199. [DOI] [PubMed] [Google Scholar]
- Caligiuri A, Bertolani C, Guerra CT, Aleffi S, Galastri S, Trappoliere M, Vizzutti F, Gelmini S, Laffi G, Pinzani M, Marra F. Adenosine monophosphate-activated protein kinase modulates the activated phenotype of hepatic stellate cells. Hepatology. 2008;47:668–76. doi: 10.1002/hep.21995. [DOI] [PubMed] [Google Scholar]
- Camacho RC, Donahue EP, James FD, Berglund ED, Wasserman DH. Energy state of the liver during short-term and exhaustive exercise in C57BL/6J mice. Am J Physiol Endocrinol Metab. 2006;290:E405–8. doi: 10.1152/ajpendo.00385.2005. [DOI] [PubMed] [Google Scholar]
- Carlson CL, Winder WW. Liver AMP-activated protein kinase and acetyl-CoA carboxylase during and after exercise. J Appl Physiol. 1999;86:669–74. doi: 10.1152/jappl.1999.86.2.669. [DOI] [PubMed] [Google Scholar]
- Chao L, Marcus-Samuels B, Mason MM, Moitra J, Vinson C, Arioglu E, Gavrilova O, Reitman ML. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J Clin Invest. 2000;106:1221–8. doi: 10.1172/JCI11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. The Biochemical journal. 2000;346(Pt 3):659–69. [PMC free article] [PubMed] [Google Scholar]
- Christ-Crain M, Kola B, Lolli F, Fekete C, Seboek D, Wittmann G, Feltrin D, Igreja SC, Ajodha S, Harvey-White J, Kunos G, Muller B, Pralong F, Aubert G, Arnaldi G, Giacchetti G, et al. AMP-activated protein kinase mediates glucocorticoid-induced metabolic changes: a novel mechanism in Cushing’s syndrome. Faseb J. 2008;22:1672–83. doi: 10.1096/fj.07-094144. [DOI] [PubMed] [Google Scholar]
- Collins QF, Liu HY, Pi J, Liu Z, Quon MJ, Cao W. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, suppresses hepatic gluconeogenesis through 5′-AMP-activated protein kinase. J Biol Chem. 2007;282:30143–9. doi: 10.1074/jbc.M702390200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell metabolism. 2006;3:403–16. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hardie DG. Role of the AMP-activated protein kinase in the cellular stress response. Curr Biol. 1994;4:315–24. doi: 10.1016/s0960-9822(00)00070-1. [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–65. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Cuthbertson DJ, Babraj JA, Mustard KJ, Towler MC, Green KA, Wackerhage H, Leese GP, Baar K, Thomason-Hughes M, Sutherland C, Hardie DG, Rennie MJ. 5-aminoimidazole-4-carboxamide 1-beta-D-ribofuranoside acutely stimulates skeletal muscle 2-deoxyglucose uptake in healthy men. Diabetes. 2007;56:2078–84. doi: 10.2337/db06-1716. [DOI] [PubMed] [Google Scholar]
- Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104:7217–22. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dentin R, Benhamed F, Pegorier JP, Foufelle F, Viollet B, Vaulont S, Girard J, Postic C. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. The Journal of clinical investigation. 2005;115:2843–54. doi: 10.1172/JCI25256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Saxena NK, Lin S, Xu A, Srinivasan S, Anania FA. The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol. 2005;166:1655–69. doi: 10.1016/S0002-9440(10)62476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–8. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- Evans AM, Mustard KJ, Wyatt CN, Peers C, Dipp M, Kumar P, Kinnear NP, Hardie DG. Does AMP-activated protein kinase couple inhibition of mitochondrial oxidative phosphorylation by hypoxia to calcium signaling in O2-sensing cells? The Journal of biological chemistry. 2005;280:41504–11. doi: 10.1074/jbc.M510040200. [DOI] [PubMed] [Google Scholar]
- Fiedler M, Zierath JR, Selen G, Wallberg-Henriksson H, Liang Y, Sakariassen KS. 5-aminoimidazole-4-carboxy-amide-1-beta-D-ribofuranoside treatment ameliorates hyperglycaemia and hyperinsulinaemia but not dyslipidaemia in KKAy-CETP mice. Diabetologia. 2001;44:2180–6. doi: 10.1007/s001250100027. [DOI] [PubMed] [Google Scholar]
- Foretz M, Ancellin N, Andreelli F, Saintillan Y, Grondin P, Kahn A, Thorens B, Vaulont S, Viollet B. Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes. 2005;54:1331–9. doi: 10.2337/diabetes.54.5.1331. [DOI] [PubMed] [Google Scholar]
- Foretz M, Carling D, Guichard C, Ferre P, Foufelle F. AMP-activated protein kinase inhibits the glucose-activated expression of fatty acid synthase gene in rat hepatocytes. J Biol Chem. 1998;273:14767–71. doi: 10.1074/jbc.273.24.14767. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol. 2004;1:98–105. doi: 10.1038/ncpgasthep0055. [DOI] [PubMed] [Google Scholar]
- Fryer LG, Parbu-Patel A, Carling D. Protein kinase inhibitors block the stimulation of the AMP-activated protein kinase by 5-amino-4-imidazolecarboxamide riboside. FEBS Lett. 2002;531:189–92. doi: 10.1016/s0014-5793(02)03501-9. [DOI] [PubMed] [Google Scholar]
- Garcia-Villafranca J, Guillen A, Castro J. Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie. 2008;90:460–6. doi: 10.1016/j.biochi.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Gonzalez AA, Kumar R, Mulligan JD, Davis AJ, Weindruch R, Saupe KW. Metabolic adaptations to fasting and chronic caloric restriction in heart, muscle, and liver do not include changes in AMPK activity. Am J Physiol Endocrinol Metab. 2004;287:E1032–7. doi: 10.1152/ajpendo.00172.2004. [DOI] [PubMed] [Google Scholar]
- Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, Viollet B, Hardie DG, Sakamoto K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. 2007 doi: 10.1074/jbc.M706536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigas B. unpublished results. [Google Scholar]
- Guigas B, Bertrand L, Taleux N, Foretz M, Wiernsperger N, Vertommen D, Andreelli F, Viollet B, Hue L. 5-Aminoimidazole-4-Carboxamide-1-{beta}-D-Ribofuranoside and Metformin Inhibit Hepatic Glucose Phosphorylation by an AMP-Activated Protein Kinase-Independent Effect on Glucokinase Translocation. Diabetes. 2006;55:865–74. doi: 10.2337/diabetes.55.04.06.db05-1178. [DOI] [PubMed] [Google Scholar]
- Guigas B, Sakamato K, Taleux N, Reyna SM, Musi N, Viollet B, Hue L. Beyond AICA riboside: In search of new specific AMP-activated protein kinase activators. IUBMB Life. 2008 doi: 10.1002/iub.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigas B, Taleux N, Foretz M, Detaille D, Andreelli F, Viollet B, Hue L. AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside. Biochem J. 2007;404:499–507. doi: 10.1042/BJ20070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows KR, Kobinger GP, Wilson JM, Witters LA, Foskett JK. Physiological modulation of CFTR activity by AMP-activated protein kinase in polarized T84 cells. American journal of physiology. 2003;284:C1297–308. doi: 10.1152/ajpcell.00227.2002. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Henin N, Vincent MF, Gruber HE, Van den Berghe G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. Faseb J. 1995;9:541–6. doi: 10.1096/fasebj.9.7.7737463. [DOI] [PubMed] [Google Scholar]
- Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem. 2003;278:27495–501. doi: 10.1074/jbc.M304112200. [DOI] [PubMed] [Google Scholar]
- Hou X, Xu S, Maitland-Toolan KA, Sato K, Jiang B, Ido Y, Lan F, Walsh K, Wierzbicki M, Verbeuren TJ, Cohen RA, Zang M. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem. 2008;283:20015–26. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–6. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- Iglesias MA, Ye JM, Frangioudakis G, Saha AK, Tomas E, Ruderman NB, Cooney GJ, Kraegen EW. AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes. 2002;51:2886–94. doi: 10.2337/diabetes.51.10.2886. [DOI] [PubMed] [Google Scholar]
- Imai K, Inukai K, Ikegami Y, Awata T, Katayama S. LKB1, an upstream AMPK kinase, regulates glucose and lipid metabolism in cultured liver and muscle cells. Biochem Biophys Res Commun. 2006;351:595–601. doi: 10.1016/j.bbrc.2006.10.056. [DOI] [PubMed] [Google Scholar]
- Jacobs RL, Lingrell S, Dyck JR, Vance DE. Inhibition of hepatic phosphatidylcholine synthesis by 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside is independent of AMP-activated protein kinase activation. J Biol Chem. 2007;282:4516–23. doi: 10.1074/jbc.M605702200. [DOI] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–22. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Zhu Z, Thompson HJ. Dietary energy restriction modulates the activity of AMP-activated protein kinase, Akt, and mammalian target of rapamycin inmammary carcinomas, mammary gland, and liver. Cancer Res. 2008;68:5492–9. doi: 10.1158/0008-5472.CAN-07-6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–92. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y, Tamura S, Kiso S, Matsumoto H, Saji Y, Yoshida Y, Fukui K, Maeda N, Nishizawa H, Nagaretani H, Okamoto Y, Kihara S, Miyagawa J, Shinomura Y, Funahashi T, Matsuzawa Y. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology. 2003;125:1796–807. doi: 10.1053/j.gastro.2003.08.029. [DOI] [PubMed] [Google Scholar]
- Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J Biol Chem. 2002;277:3829–35. doi: 10.1074/jbc.M107895200. [DOI] [PubMed] [Google Scholar]
- Kim YD, Park KG, Lee YS, Park YY, Kim DK, Nedumaran B, Jang WG, Cho WJ, Ha J, Lee IK, Lee CH, Choi HS. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes. 2008;57:306–14. doi: 10.2337/db07-0381. [DOI] [PubMed] [Google Scholar]
- Kimball SR, Siegfried BA, Jefferson LS. Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J Biol Chem. 2004;279:54103–9. doi: 10.1074/jbc.M410755200. [DOI] [PubMed] [Google Scholar]
- Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, Nathan DM. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, Williams LM, Hawley SA, Hardie DG, Grossman AB, Korbonits M. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. The Journal of biological chemistry. 2005;280:25196–201. doi: 10.1074/jbc.C500175200. [DOI] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, Takemori H, Montminy M. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–11. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization and activity of LKB1; possible role in AMP-activated protein kinase activation. J Biol Chem. 2008 doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc I, Kahn A, Doiron B. The 5′-AMP-activated protein kinase inhibits the transcriptional stimulation by glucose in liver cells, acting through the glucose response complex. FEBS Lett. 1998;431:180–4. doi: 10.1016/s0014-5793(98)00745-5. [DOI] [PubMed] [Google Scholar]
- Leclerc I, Lenzner C, Gourdon L, Vaulont S, Kahn A, Viollet B. Hepatocyte nuclear factor-4alpha involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes. 2001;50:1515–21. doi: 10.2337/diabetes.50.7.1515. [DOI] [PubMed] [Google Scholar]
- Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, Ye JM, Lee CH, Oh WK, Kim CT, Hohnen-Behrens C, Gosby A, Kraegen EW, James DE, Kim JB. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes. 2006;55:2256–64. doi: 10.2337/db06-0006. [DOI] [PubMed] [Google Scholar]
- Lihn AS, Jessen N, Pedersen SB, Lund S, Richelsen B. AICAR stimulates adiponectin and inhibits cytokines in adipose tissue. Biochem Biophys Res Commun. 2004;316:853–8. doi: 10.1016/j.bbrc.2004.02.139. [DOI] [PubMed] [Google Scholar]
- Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med. 2000;6:998–1003. doi: 10.1038/79697. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J. 2004;23:833–43. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead PA, Salt IP, Walker KS, Hardie DG, Sutherland C. 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes. 2000;49:896–903. doi: 10.2337/diabetes.49.6.896. [DOI] [PubMed] [Google Scholar]
- Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–9. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–74. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281:25336–43. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- Mooney MH, Fogarty S, Stevenson C, Gallagher AM, Palit P, Hawley SA, Hardie DG, Coxon GD, Waigh RD, Tate RJ, Harvey AL, Furman BL. Mechanisms underlying the metabolic actions of galegine that contribute to weight loss in mice. Br J Pharmacol. 2008;153:1669–77. doi: 10.1038/bjp.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–94. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Mukhtar MH, Payne VA, Arden C, Harbottle A, Khan S, Lange AJ, Agius L. Inhibition of glucokinase translocation by AMP-activated protein kinase is associated with phosphorylation of both GKRP and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Am J Physiol Regul Integr Comp Physiol. 2008;294:R766–74. doi: 10.1152/ajpregu.00593.2007. [DOI] [PubMed] [Google Scholar]
- Munday MR, Milic MR, Takhar S, Holness MJ, Sugden MC. The short-term regulation of hepatic acetyl-CoA carboxylase during starvation and re-feeding in the rat. Biochem J. 1991;280 ( Pt 3):733–7. doi: 10.1042/bj2800733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM, Seefeld K, Witters LA, Coleman RA. AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. The Biochemical journal. 1999;338 ( Pt 3):783–91. [PMC free article] [PubMed] [Google Scholar]
- Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, Rossetti L, Scherer PE. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281:2654–60. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–14. [PMC free article] [PubMed] [Google Scholar]
- Park H, Kaushik VK, Constant S, Prentki M, Przybytkowski E, Ruderman NB, Saha AK. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem. 2002;277:32571–7. doi: 10.1074/jbc.M201692200. [DOI] [PubMed] [Google Scholar]
- Park MW, Ha J, Chung SH. 20(S)-ginsenoside Rg3 enhances glucose-stimulated insulin secretion and activates AMPK. Biol Pharm Bull. 2008;31:748–51. doi: 10.1248/bpb.31.748. [DOI] [PubMed] [Google Scholar]
- Parker GJ, Koay A, Gilbert-Wilson R, Waddington LJ, Stapleton D. AMP-activated protein kinase does not associate with glycogen alpha-particles from rat liver. Biochem Biophys Res Commun. 2007;362:811–5. doi: 10.1016/j.bbrc.2007.08.080. [DOI] [PubMed] [Google Scholar]
- Pold R, Jensen LS, Jessen N, Buhl ES, Schmitz O, Flyvbjerg A, Fujii N, Goodyear LJ, Gotfredsen CF, Brand CL, Lund S. Long-term AICAR administration and exercise prevents diabetes in ZDF rats. Diabetes. 2005;54:928–34. doi: 10.2337/diabetes.54.4.928. [DOI] [PubMed] [Google Scholar]
- Puljak L, Parameswara V, Dolovcak S, Waldrop SL, Emmett D, Esser V, Fitz JG, Kilic G. Evidence for AMPK-dependent regulation of exocytosis of lipoproteins in a model liver cell line. Exp Cell Res. 2008;314:2100–9. doi: 10.1016/j.yexcr.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznick RM, Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. J Physiol. 2006;574:33–9. doi: 10.1113/jphysiol.2006.109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek U, Scholz R, Konarev P, Rufer A, Suter M, Nazabal A, Ringler P, Chami M, Muller SA, Neumann D, Forstner M, Hennig M, Zenobi R, Engel A, Svergun D, Schlattner U, et al. Structural properties of AMP-activated protein kinase: dimerization, molecular shape, and changes upon ligand binding. J Biol Chem. 2008;283:18331–43. doi: 10.1074/jbc.M708379200. [DOI] [PubMed] [Google Scholar]
- Saha AK, Avilucea PR, Ye JM, Assifi MM, Kraegen EW, Ruderman NB. Pioglitazone treatment activates AMP-activated protein kinase in rat liver and adipose tissue in vivo. Biochem Biophys Res Commun. 2004;314:580–5. doi: 10.1016/j.bbrc.2003.12.120. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Goransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. Am J Physiol Endocrinol Metab. 2004;287:E310–7. doi: 10.1152/ajpendo.00074.2004. [DOI] [PubMed] [Google Scholar]
- Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. The Biochemical journal. 1998;334 ( Pt 1):177–87. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. The Biochemical journal. 2007;403:139–48. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Yates JR, 3rd, Takemori H, Okamoto M, Montminy M. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell. 2004;119:61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Shang J, Chen LL, Xiao FX, Sun H, Ding HC, Xiao H. Resveratrol improves non-alcoholic fatty liver disease by activating AMP-activated protein kinase. Acta Pharmacol Sin. 2008;29:698–706. doi: 10.1111/j.1745-7254.2008.00807.x. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science (New York, NY. 2005;310:1642–6. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim AT, Hardie DG. The low activity of acetyl-CoA carboxylase in basal and glucagon-stimulated hepatocytes is due to phosphorylation by the AMP-activated protein kinase and not cyclic AMP-dependent protein kinase. FEBS Lett. 1988;233:294–8. doi: 10.1016/0014-5793(88)80445-9. [DOI] [PubMed] [Google Scholar]
- Song XM, Fiedler M, Galuska D, Ryder JW, Fernstrom M, Chibalin AV, Wallberg-Henriksson H, Zierath JR. 5-Aminoimidazole-4-carboxamide ribonucleoside treatment improves glucose homeostasis in insulin-resistant diabetic (ob/ob) mice. Diabetologia. 2002;45:56–65. doi: 10.1007/s125-002-8245-8. [DOI] [PubMed] [Google Scholar]
- Song Z, Deaciuc I, Zhou Z, Song M, Chen T, Hill D, McClain CJ. Involvement of AMP-activated protein kinase in beneficial effects of betaine on high-sucrose diet-induced hepatic steatosis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G894–902. doi: 10.1152/ajpgi.00133.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365:1333–46. doi: 10.1016/S0140-6736(05)61032-X. [DOI] [PubMed] [Google Scholar]
- Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5’-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem. 2006;281:32207–16. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Okamoto S, Lee S, Saito K, Shiuchi T, Minokoshi Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the alpha2 form of AMP-activated protein kinase. Mol Cell Biol. 2007;27:4317–27. doi: 10.1128/MCB.02222-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekoshi K, Fukuhara M, Quin Z, Nissato S, Isobe K, Kawakami Y, Ohmori H. Long-term exercise stimulates adenosine monophosphate-activated protein kinase activity and subunit expression in rat visceral adipose tissue and liver. Metabolism. 2006;55:1122–8. doi: 10.1016/j.metabol.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Tan MJ, Ye JM, Turner N, Hohnen-Behrens C, Ke CQ, Tang CP, Chen T, Weiss HC, Gesing ER, Rowland A, James DE, Ye Y. Antidiabetic activities of triterpenoids isolated from bitter melon associated with activation of the AMPK pathway. Chem Biol. 2008;15:263–73. doi: 10.1016/j.chembiol.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Targher G, Bertolini L, Rodella S, Zoppini G, Scala L, Zenari L, Falezza G. Associations between plasma adiponectin concentrations and liver histology in patients with nonalcoholic fatty liver disease. Clin Endocrinol (Oxf) 2006;64:679–83. doi: 10.1111/j.1365-2265.2006.02527.x. [DOI] [PubMed] [Google Scholar]
- To K, Yamaza H, Komatsu T, Hayashida T, Hayashi H, Toyama H, Chiba T, Higami Y, Shimokawa I. Down-regulation of AMP-activated protein kinase by calorie restriction in rat liver. Exp Gerontol. 2007;42:1063–71. doi: 10.1016/j.exger.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Tomita K, Tamiya G, Ando S, Kitamura N, Koizumi H, Kato S, Horie Y, Kaneko T, Azuma T, Nagata H, Ishii H, Hibi T. AICAR, an AMPK activator, has protective effects on alcohol-induced fatty liver in rats. Alcohol Clin Exp Res. 2005;29:240S–5S. doi: 10.1097/01.alc.0000191126.11479.69. [DOI] [PubMed] [Google Scholar]
- Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–41. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- Velasco G, Geelen MJ, Guzman M. Control of hepatic fatty acid oxidation by 5′-AMP-activated protein kinase involves a malonyl-CoA-dependent and a malonyl-CoA-independent mechanism. Arch Biochem Biophys. 1997;337:169–75. doi: 10.1006/abbi.1996.9784. [DOI] [PubMed] [Google Scholar]
- Viana AY, Sakoda H, Anai M, Fujishiro M, Ono H, Kushiyama A, Fukushima Y, Sato Y, Oshida Y, Uchijima Y, Kurihara H, Asano T. Role of hepatic AMPK activation in glucose metabolism and dexamethasone-induced regulation of AMPK expression. Diabetes Res Clin Pract. 2006;73:135–42. doi: 10.1016/j.diabres.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Vincent MF, Bontemps F, Van den Berghe G. Inhibition of glycolysis by 5-amino-4-imidazolecarboxamide riboside in isolated rat hepatocytes. Biochem J. 1992;281 ( Pt 1):267–72. doi: 10.1042/bj2810267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent MF, Marangos PJ, Gruber HE, Van den Berghe G. Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes. 1991;40:1259–66. doi: 10.2337/diab.40.10.1259. [DOI] [PubMed] [Google Scholar]
- Vincent O, Townley R, Kuchin S, Carlson M. Subcellular localization of the Snf1 kinase is regulated by specific beta subunits and a novel glucose signaling mechanism. Genes Dev. 2001;15:1104–14. doi: 10.1101/gad.879301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–8. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L, Hue L, Andreelli F. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. The Journal of physiology. 2006;574:41–53. doi: 10.1113/jphysiol.2006.108506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol. 1999;277:E1–10. doi: 10.1152/ajpendo.1999.277.1.E1. [DOI] [PubMed] [Google Scholar]
- Witters LA, Gao G, Kemp BE, Quistorff B. Hepatic 5′-AMP-activated protein kinase: zonal distribution and relationship to acetyl-CoA carboxylase activity in varying nutritional states. Arch Biochem Biophys. 1994;308:413–9. doi: 10.1006/abbi.1994.1058. [DOI] [PubMed] [Google Scholar]
- Witters LA, Kemp BE. Insulin activation of acetyl-CoA carboxylase accompanied by inhibition of the 5′-AMP-activated protein kinase. J Biol Chem. 1992;267:2864–7. [PubMed] [Google Scholar]
- Woods A, Azzout-Marniche D, Foretz M, Stein SC, Lemarchand P, Ferre P, Foufelle F, Carling D. Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase. Mol Cell Biol. 2000;20:6704–11. doi: 10.1128/mcb.20.18.6704-6711.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Cα2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–8. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, Martin SR, Carling D, Gamblin SJ. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- Xie M, Zhang D, Dyck JR, Li Y, Zhang H, Morishima M, Mann DL, Taffet GE, Baldini A, Khoury DS, Schneider MD. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:17378–83. doi: 10.1073/pnas.0604708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–95. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Imai Y, Shimozawa N, Hioki K, Uchida S, Ito Y, Takakuwa K, Matsui J, Takata M, Eto K, Terauchi Y, Komeda K, Tsunoda M, Murakami K, et al. Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem. 2003;278:2461–8. doi: 10.1074/jbc.M209033200. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–6. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, Okada-Iwabu M, Kawamoto S, Kubota N, Kubota T, Ito Y, Kamon J, Tsuchida A, Kumagai K, Kozono H, Hada Y, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nature medicine. 2007;13:332–9. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- Yang J, Maika S, Craddock L, King JA, Liu ZM. Chronic activation of AMP-activated protein kinase-alpha1 in liver leads to decreased adiposity in mice. Biochem Biophys Res Commun. 2008;370:248–53. doi: 10.1016/j.bbrc.2008.03.094. [DOI] [PubMed] [Google Scholar]
- You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, Wierzbicki M, Verbeuren TJ, Cohen RA. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55:2180–91. doi: 10.2337/db05-1188. [DOI] [PubMed] [Google Scholar]
- Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H, Brecher P, Ruderman NB, Cohen RA. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem. 2004;279:47898–905. doi: 10.1074/jbc.M408149200. [DOI] [PubMed] [Google Scholar]
- Zhang F, Sun C, Wu J, He C, Ge X, Huang W, Zou Y, Chen X, Qi W, Zhai Q. Combretastatin A-4 activates AMP-activated protein kinase and improves glucose metabolism in db/db mice. Pharmacol Res. 2008;57:318–23. doi: 10.1016/j.phrs.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]