

Abstract
Daidzein (1) is a natural estrogenic isoflavone. We report here that 1 can be transformed into antiestrogenic ligands by simple alkyl substitutions of the 7-hydroxyl hydrogen. To test the effect of such structural modifications on the hormonal activities of the resulting compounds, a series of daidzein analogues have been designed and synthesized. When MCF-7 cells were treated with the analogues, those resulting from hydrogen substitution by isopropyl (3d), isobutyl (3f), cyclopentyl (3g), and pyrano- (2), inhibited cell proliferation, estrogen-induced transcriptional activity, and estrogen receptor (ER) regulated progesterone receptor (PgR) gene expression. However, methyl (3a) and ethyl (3b) substitutions of the hydroxyl proton only led to moderate reduction of the estrogenic activities. These results demonstrated the structural requirements for the transformation of daidzein from an ER agonist to an antagonist. The most effective analogue, 2 was found to reduce in vivo estrogen stimulated MCF-7 cell tumorigenesis using a xenograft mouse model.
Keywords: Daidzein analogues, antiestrogen, isoflavones, breast cancer, phytoestrogen, SERM, estrogen receptor, xenograft model
1. Introduction
Daidzein (1) is one of many known isoflavones that naturally occur in plants such as soybeans and kudzu vines (Peruraria lobata).1 Like other constitutive isoflavones found in soy tissues (e.g., genistein),2 daidzein has been shown to act as a variety of biologically active agents ranging from antioxidant, estrogen at doses lower than 20 μM, and breast cancer cell growth and proliferation promoter.3,4,5. The estrogenic effect, however, can be reversed when daidzein concentration exceeds 20 μM where it has been found to inhibit the growth of different cancer cells.4,6,7 For example, daidzein was found to antagonize the inhibitory effects of tamoxifen on pS2 expression8 and proliferation9 of breast tumor cells. The diverse functionality of daidzein is also reflected in its glucosyl derivative, 7-O-glucosyl-4′-hydroxyisoflavone (daidzin) which acts as an anti-alcohol-addiction agent,10 presumably through binding to the enzyme aldehyde dehydrogenase.
The exact mechanism underlying the cellular inhibitory effects of high dose daidzein and its closely related analogue genistein is not completely understood. Previous studies suggest their role as weak agonists competing with estradiol for ER binding5. Additional studies have proposed several possible signaling pathways that may be responsible for these effects, such as binding to the epidermal growth factor receptor kinase11 and interfering with transforming growth factor signaling.12
Interestingly, in soy, daidzein is also a key intermediate in the biosynthesis of the anti-estrogenic glyceollins in response to bacterial infection.13,14 This naturally occurring transformation of estrogenic to anti-estrogenic function prompted us to examine the structural features that distinguish the estrogenic activity of daidzein from the anti-estrogenic activity of the glyceollins. As previously published, the biosynthesis of glyceollin I from the daidzein precursor leads to the incorporation of 1) a 7,8-phenol ring of the chromanone structure where a six-membered ring is formed via an ether linkage13,14 and 2) a central moiety where a freely rotating biphenyl bond in daidzein becomes a rigid pterocarpan structure in glyceollin I. Both or one of the structural modifications could be responsible for the functional reversal of ER activity. Molecular modeling using the crystal structure of ERα indicates that daidzein can fit in the binding pocket of ERα when the receptor is optimized with both 17β-estradiol (E2) and 4-hydroxytamoxifen (4-OH-TAM), whereas glyceollins can only fit the pocket when the crystal structure of ERα is optimized with 4-OH-TAM. The modeling results15 suggest that the structural features in the 7-phenolic position may be the critical one responsible for the antiestrogenic effects of glyceollin I.
In this study we explored the potential antiestrogenic properties of daidzein analogues by modifying the 7-hydroxyl functional group while keeping the daidzein skeleton intact. We started with an analogue that is structurally identical with glyceollin I on the 7 and 8 ring substitutions but without the pterocarpan moiety. Using molecular modeling as a guiding tool, we designed and synthesized an additional 8 daidzein analogues that vary in the 7-O substitution. We also evaluated the effect of daidzein and daidzein analogues on estrogen responsive element (ERE) transcriptional activity, ERα binding affinity, MCF-7 breast cancer cells colonial survival, PgR gene expression and ERα docking. Finally, we conducted in vivo studies of the anti-estrogenic effects of the most potent analogue on MCF-7 cell tumorigenesis using a xenograft mouse model. The structure activity relationship studies on these daidzein analogues reveal the progressive loss of an ER agonist activity concurrent to the acquisition of ER antagonistic activity with the substitution of the 7-OH hydrogen with varying hydrophobic groups.
2. Results and Discussion
2.1. Chemistry
The synthetic route of 3-(4-hydroxyphenyl)-8,8-dimethyl-8H-pyrano[2,3-f]chromen-4-one (2) is diagrammed in Scheme 1. Briefly, following a literature method,16 a condensation reaction between 1 and an unsaturated aldehyde masked as its acetal in refluxing p-xylene in the presence of picoline as base brought about the desired ring closure in position 8 of 1 to give the target compound 2 as the major product. The minor byproduct formed by substitution and ring closure on daidzein's 4′ and 5′ positions was negligible and did not present a problem in the isolation and purification of 2.
Scheme 1.

Synthesis of 2 from daidzein.
This strategy, however, did not work as hoped for all other daidzein analogues from 3a to 3h (Scheme 2). The main problems appeared to be associated with the much higher proportion of di-substitution on both hydroxyl groups. The challenge was overcome by adopting a synthetic strategy17 that used the appropriate alkyl halide in a DMSO suspension of potassium carbonate. The facile reactions proceeded at room temperature giving the desired compounds as the main products with varying amounts of byproducts from substitution on the other hydroxyl group (4-OH). Notably, in some cases, it was necessary to carefully control reaction time and substrate ratio in order to achieve the desired outcome of majority 7-O substitution. In fact, for several compounds (e.g., 3f and 3g), isolation of the target 7-O-substituted product from their 4′-O-substituted byproducts was rather difficult and separation by semi-preparative HPLC was necessary to afford greater than 98% purity of these analogues.
Scheme 2.

Synthesis of daidzein analogues 3a – 3h.
2.2 Biological Activities of Daidzein Analogues
Effects of daidzein analogues on ER transcriptional activation in MCF-7 cells
We have previously described the estrogenic and anti-estrogenic activities of individual flavonoids and phytochemicals using a luciferase based ERE reporter assay.2, 5, 14 These studies have demonstrated the potent estrogenic activities of certain isoflavones such as genistein or daidzein, as well as the ability of glyceollins to antagonize estrogen-stimulated ERE activity15. To test how structural modifications on the 7-hydroxyl group of daidzein affect estrogenic properties, the ER transcriptional activity induced by the daidzein analogues were measured and compared to daidzein using the ERE based luciferase reporter gene assay. As shown in Figure 1, the estrogenic activities of synthetic daidzein analogues were plotted as dose response curves when MCF-7 cells were treated with increasing concentrations of the test compounds. At 1 μM concentration where daidzein induced maximal estrogenic effect in MCF-7 cells, the relative estrogenic potencies of the analogues are seen to decrease in roughly the following order: daidzein > 3a > 3f > 3e > 3b ≅ 3c > 3d > 2 > 3g > 3h. While this order varied slightly as analogue concentrations were increased further to 25 μM (Table 1), several trends have emerged in the dose-dependent data. First, the parent compound daidzein showed more potent estrogenic activity than any of its derivatives when hydrogen was replaced with alkyl groups with varying chain lengths and configurations. Second, shorter alkyl groups appeared to have less effect on the reduction of ER agonist activity than their longer, bulkier counterparts. For example, at 25 μM concentration, 3b (ethyl-), 3a (methyl-), and 3c (n-propyl-) daidzein analogues induced ERE transcriptional activity at 80.4, 70.9, and 38.4 %, respectively, retaining greater estrogenic activities than other analogues with longer chain or bulkier substituting groups. Finally, the analogue showing little or no estrogen activity, 2, resembled the naturally occurring glyceollin I15 in the chromanone moiety, suggesting that the unique ring substitution on the 7O- and 8-chromanone ring was responsible for the diminished estrogenic activity of the modified daidzein.
Figure 1. Effects of daidzein and daidzein analogues on ERE transcriptional activity in MCF-7 cells.

MCF-7 cells were transiently transfected with pGL2-ERE2x-TK-luciferase plasmid. After a 6 h transfection, cells were treated with compounds (DMSO, daidzein and daidzein analogues) and incubated overnight. Data are represented as relative light units (RLUs) normalized to E2 (100 ± SEM). The values are the means and the SEM of triplicates from a single experiment and representative for at least three independent experiments. a, significant difference from daidzein, p<0.05, b, significant difference from daidzein, p < 0.01, c, significant difference from daidzein, p < 0.001.
Table 1.
Summary of biological activities of 7-O-substituted daidzein derivatives*
| Analogue Structure | Name | Max % Agonist |
Max % Inhibition |
ERα BINDING MAX % |
|---|---|---|---|---|
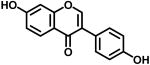 |
Daidzein | 108.5% @ 1μM | 17.2% @ 10μM | 112 |
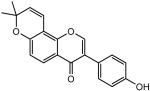 |
2 | 17.7% @ 10μM | 100% @ 25μM 54.6 @ 10μM |
44.1 |
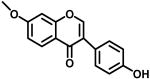 |
3a | 68.5% @ 25μM | 33.2% @ 25μM | 82.7 |
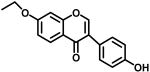 |
3b | 79% @ 25μM | 18.8% @ 25μM | 66.6 |
 |
3c | 33.4% @ 10μM | 41.1% @ 25μM | 44.9 |
 |
3d | 30.6% @ 10μM | 68.1% @ 25μM 33.4% @ 10μM |
42.9 |
 |
3e | 16.5% @ 10μM | 49.8% @ 25μM | 81.9 |
 |
3f | 22.0% @ 1μM | 91.3% @ 25μM 60.5 @ 10μM |
38.3 |
 |
3g | 5.4% @ 1μM | 89.8% @ 25μM 54.1 @ 10μM |
19.2 |
| 3h | 0.8% @ 0.1μM | 46.4% @ 25μM | 88.5 |
Data are represented as relative agonistic and antagonists activities for individual compounds with double normalization in which E2 (0.1 nM) was set at 100% with vehicle (DMSO) set at 0%.
Antagonist Activities of Daidzein Analogues on ER transcriptional activity
To evaluate the antiestrogenic activities of daidzein analogues, MCF-7 cells were treated with the synthesized compounds at increasing concentrations in the presence of 0.1 nM E2. The luciferase activities of the treated cells were then measured by the ERE reporter gene assay. Figure 2 shows the dose response curves of MCF-7 cells treated with E2 plus daidzein and its synthetic analogues where the luciferase activity is normalized to 100 for cells treated with 0.1 nM E2 alone. The inhibitory effect of some analogues on E2 became more pronounced as their concentration was increased to 10 μM. At 25 μM (Table 1), the relative antiestrogenic potencies of the analogues were observed to increase in the following order: daidzein < 3b < 3a < 3c < 3h < 3e < 3d < 3g < 3f < 2. The analogues that reduced estradiol-dependent luciferase activity by greater than 60% at 25 μM included 2, 3f, 3g, and 3d, of which 2 was the most effective antiestrogen, completely blocking ERE activity at 25 μM. In contrast, analogues 3a, 3b, 3c, 3h, and 3e decreased E2 induced transcriptional activity by less than 47% at the highest ligand concentration. Compared with estrogenic activity assay results, the analogues showing strong antiestrogenic activity exhibited little or no estrogen activity. For example, 2, 3f, and 3g all induced negligible ERE activity, but they behaved as potent estrogen inhibitors (Figure 1 and Figure 2A). Treatment with fulvestrant (ICI) or 4-OH-TAM alone and in combination with estrogen caused a decrease in ERE transcriptional activity over 90% and 80%, respectively (Figure 2B). On the other hand, the strongest estrogenic analogues, 3a, 3b, and 3c, failed to inhibit 50% ERE activity in the presence of E2 at all concentrations tested (Figure 2A). Taken together, the alkyl substituted daidzein analogues can be viewed in two groups in terms of their ER mediated transcriptional activities. The first group (3a, 3b, and 3c) where the 7-OH of daidzein is alkoxylated by the methyl, ethyl, and n-propyl group, respectively, has retained much of the estrogenic function of daidzein. The second group, consisting of 2, 3g, 3f, and to a lesser extent 3d, has been transformed into potent antiestrogens. This complete reversal from estrogenic to antiestrogenic activity in certain daidzein analogues highlights the importance of the structure of 7-O substitution in determining the ligand's interaction with the estrogen receptor. It is also interesting to note the significant difference in ERE activity between the i-propyl (3d) and n-propyl (3c) substituted analogue, suggesting that a branched alkyl group may provide a better binding mechanism toward the ER.
Figure 2. Effects of daidzein and daidzein analogues on E2 stimulation on ERE transcriptional activity in MCF-7 cells.


MCF-7 cells were transiently transfected with pGL2-ERE2x-TK-luciferase plasmid. After a 6 h transfection, cells were treated with (A) compounds (DMSO, E2, daidzein and daidzein analogues plus E2) or (B) with compounds (DMSO, E2, ICI, ICI + E2, 4-OH-TAM, 4-OH-TAM + E2) and incubated overnight. Data are represented as relative light units (RLUs) normalized to E2 (100 ± SEM). The values are the means and the SEM of triplicates from a single experiment and representative for at least three independent experiments.
a, significant difference from E2, p<0.05, b, significant difference from E2, p < 0.01, c, significant difference from E2 p < 0.001, and d, significant difference from DMSO p < 0.001.
Relative affinity of daidzein analogues for ERα
In an attempt to understand the biological activity of the synthesized daidzein analogues, we examined the ability of each daidzein analogues to bind to ERα using a competitive binding assay with fluorescent detection (Figure 3). Analysis of the competition binding curve yielded IC50 values, representing the concentration of unlabeled ligand required to displace 50% of the tracer from the ERα. Unlabeled E2 was used as a reference and its displacement of 50% tracer E2 bound to ERα occurred at 1.5 nM. Compared to daidzein, which has an IC50 value of 0.45 μM, none of the analogues displayed higher binding affinities towards the estrogen receptor. The maximal %bound to ERα for the analogues are summarized in Table 1. At 10 μM ligand concentration, four analogues showed over 50% bound ERα, i.e., 3a, 3b, 3e, and 3h while the rest of analogues appeared to have lower binding affinities towards the ER. The binding data are partially consistent with the observation that structural modifications on the 7-O position of daidzein reduced more estrogenic potency of the analogues to various degrees at lower concentrations (10 μM) than at higher doses (25 μM). However, the reason for higher binding affinities exhibited by 3h and 3e which are neither estrogenic nor antiestrogenic is unclear. The binding curves suggest that at higher concentrations (e.g., 25 μM as used in the luciferase reporter assay), the degree of binding to ER by several analogues may rapidly approach that of daidzein. This is also consistent with the fact that both estrogenic (3a, 3b, and 3c) and antiestrogenic (2, 3f, 3g) activities of the analogues reached maximal levels at 25 μM (Figures 1 and 2). For example, 3a reached 38% ERE activity at 10 μM; at 25 μM, its ERE activity increased dramatically to 70.9% (Figure 1). The binding curve for 3a confirmed this rapid increase in estrogenic activity as its %bound value appear to approach that of daidzein. Notably, for antiestrogenic compounds, binding affinities to ERα did not appear to correlate well with their ability to inhibit E2 induced ERE activity.
Figure 3. Relative binding affinity of daidzein and daidzein analogues for ERα.
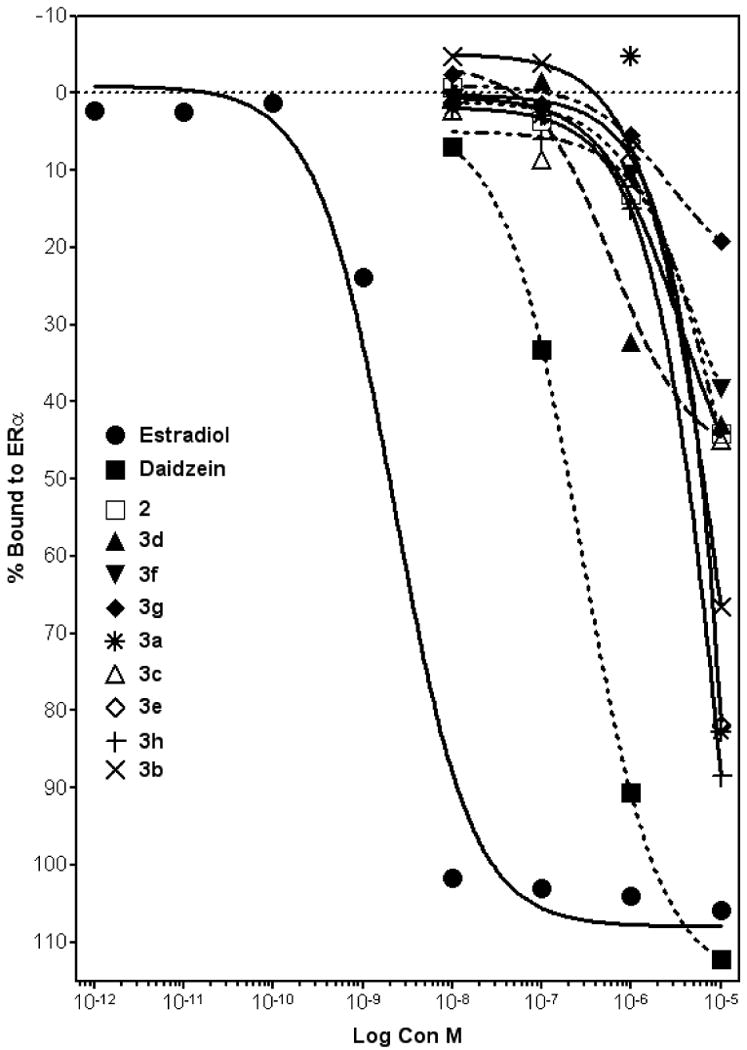
Increasing concentrations of daidzein analogues were added to the ER complex and compared to E2. Data points and error bars represent the mean ± SEM of at least three experiments (n = 3) for each concentration tested. (p < 0.05).
Effect of Daidzein Analogues on Estrogen-stimulated clonogenic proliferation
To further evaluate the estrogenic or antiestrogenic activities of the daidzein analogues determined from the ERE transcriptional assay, we selected two analogues, 3a and 2, for further test by a clonogenic assay for their ability to inhibit the estradiol-induced proliferation of MCF-7 cells. As discussed earlier, 3a behaved as an estrogen with a slightly lower potency than daidzein, whereas 2 acted as an antiestrogen that was able to inhibit 100% ER mediated transcriptional activity. In the colony assay MCF-7 cells were treated with increasing concentrations of daidzein, 2, 3a, and 4-OH-TAM in the presence or absence of 0.1 nM E2. Results are illustrated in Figure 4 where estrogen-dependent cell growth was normalized to 100 for the control (DMSO) (Fig 4A) or for E2 alone (Fig 4B). The addition of 0.1 nM E2 to the culture media resulted in an approximately 3-fold increase in the clonogenicity of the MCF-7 cells compared to DMSO treated controls. At 1 μM, daidzein also promoted the clonogenicity of MCF-7 cells by over 3-fold, while 3a increased cell proliferation by 43%, confirming that 3a is a weaker estrogen than daidzein, a direct consequence of the replacement of 7-hydroxy group by 7-methoxy group. In contrast, 2 inhibited the clonogenicity by 15% at the same concentration, demonstrating its antiestrogenic property at 1 μM concentration. When concentration was raised to 10 μM, daidzein and 3a continued to increase the clonogenicity of MCF-7 cells by 2.5 times and 24%, respectively. Analogue 2, however, completely blocked cell clonogenicity at 10 μM, similar to treatment with 4-OH-TAM at 100 nM. Figure 4B illustrates the effect of daidzein, analogues 2 and 3a, and 4-OH-TAM on cell clonogenicity in the presence of 0.1 nM E2. Even in the presence of 0.1 nM estradiol, 2 at 10 μM was able to inhibit the clonogenicity of MCF-7 cells completely, similar to 4-OH TAM at 100 nM. Thus, the colony assay results clearly demonstrated the divergence of hormonal activities in daidzein analogues by simple modifications of the 7-OH group of daidzein.
Figure 4. Effects of daidzein, 2 and 3a on E2 stimulation on colony formation on MCF-7 Cells.
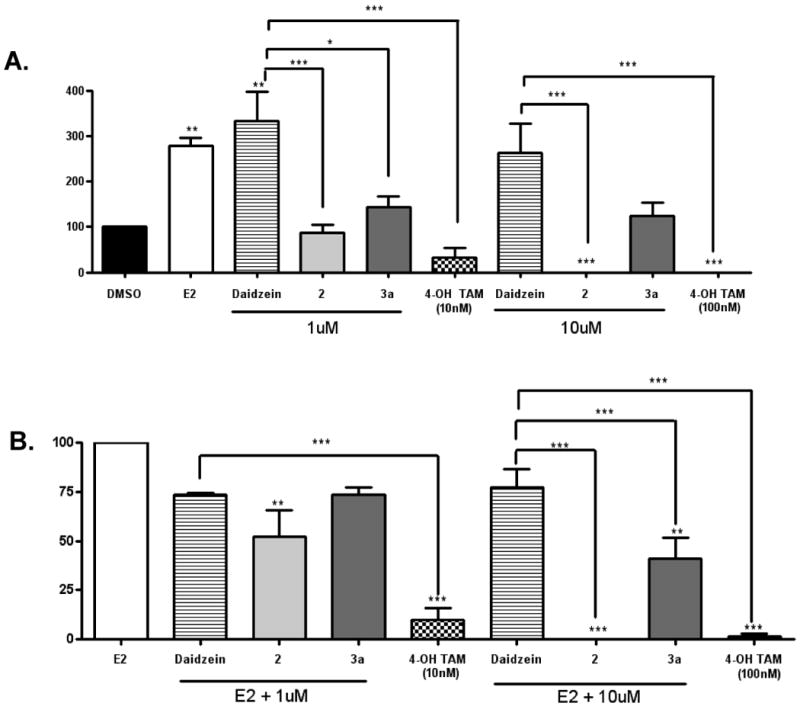
MCF-7 cells were placed in phenol red-free DMEM supplemented with 5% dextran-coated charcoal-treated FBS for 48 h before plating. MCF-7 cells (1000) were plated in six well plates. Forty eight hours later, cells were treated with DMSO (vehicle), E2, daidzein, daidzein analogues (at 1 μM and 10 μM) and 4-OH-TAM (at 10 nM and 100 nM) (A) and E2, E2 + daidzein, E2 + daidzein analogues (at 1 μM and 10 μM) and E2 + 4-OH-TAM (at 10 nM and 100 nM) (B). Colonies of > 50 cells were counted as positive. Results are normalized to percentage of clonogenic survival from DMSO control cells. The values are the means and the SEM of triplicates from a single experiment and representative for at least three independent experiments. Significant difference from vehicle control (A) and E2 (B), *p < 0.05, **p < 0.01 and *** p < 0.001; Tukey test.
Regulation of ER-mediated PgR Gene Expression by Daidzein Analogues
To further examine the differing hormonal activities of daidzein analogues we monitored ER-mediated gene expression of progesterone receptor (PgR) by quantitative real time RT-PCR in MCF-7 cells treated with daidzein, 2 (an antiestrogenic analogue), and 3a (an estrogenic analogue) (Figure 5). Progesterone receptor (PgR) is an estrogen-responsive gene, whose expression has been shown to indicate a responsive estrogen receptor pathway. The expression of PgR mRNA in MCF-7 cells that were treated with DMSO (control) and daidzein, 2 and 3a either in the presence or absence of E2 were determined. When treated with ligands only, daidzein and 3a induced a 3-fold increase in PgR expression compared with control, while a 5fold increase was induced by E2. However, 2 caused a decrease in PgR expression compared with DMSO, demonstrating its non-estrogenic nature. When MCF-7 cells were treated with the ligands in the presence of 0.1 nM E2, both daidzein and 3a were able to promote the level of PgR expression by 5- and 4-fold, respectively. In contrast, 2 inhibited the E2 induced PgR expression by reducing its upregulation from 5-fold to 3-fold. Recently we have demonstrated the ability of the known anti-estrogen fulvestrant (100 nM) to suppress E2-stimulated PgR expression by ∼75% in our MCF-7 cell system.15 In comparison, analogue 2 resulted in a ∼40% suppression of E2-stimulated PgR expression (Figure 5).
Figure 5. Effects of daidzein, 2 and 3a on PgR expression.
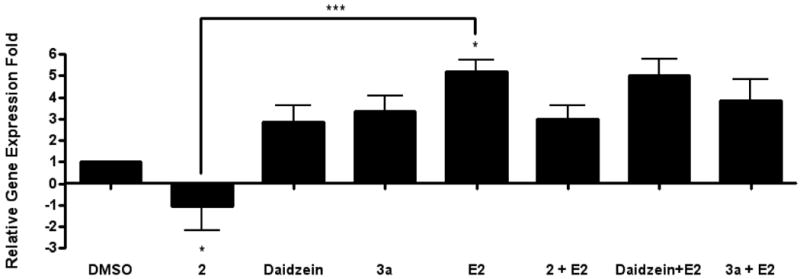
Total RNA was isolated from MCF-7 cells, reverse-transcribed into cDNA and subjected to realtime RT-PCR analysis for quantitation. Treatment on MCF-7 cells was as follows: DMSO (vehicle), daidzein, 2, 3a, E2, daidzein + E2, 2 +E2, 3a + E2 (10 μM and 10 μM + E2). Results are expressed as the mean unit ± S.E.M. (***p < 0.05; **p < 0.01; *p < 0.001) with significant differences from vehicle control.
In vivo Anti-estrogenic effects of Analogue 2 on MCF-7 cell tumorigenesis
Due to the anti-estrogenic effects of analog 2 on MCF-7 cells observed in our in vitro biological assays, this compound was selected for further testing of estrogen stimulated MCF-7 cell tumorigenesis using a xenograft model. Ovariectomized female immunocompromised mice were injected with MCF-7 cells and supplemented with exogenous estrogen pellets. Once tumors were palpable animals were treated with analogue 2, tamoxifen, or ICI and tumor volume compared to vehicle control animals. At day 37 post cell injection a decrease in tumor volume was observed in all treatment groups compared to control (351.17 ± 43.88). Compound 2 treatment resulted in reduced tumor volumes similar to tamoxifen treated animals (241.53 ± 68.29 and 178.12 ± 41.18 mm3 respectively), while treatment with the pure anti-estrogen ICI 182,780 was most effective in attenuating estrogen-induced tumorigenesis (130.95 ± 23.49). These results confirm that the anti-estrogenic properties of compound 2 observed in vitro extend to in vivo conditions as well.
Molecular Modeling
The estrogenic compound 3a and antiestrogenic compound 2, along with the parent compound daidzein were subjected to docking studies. To understand the nature of their interactions with ERα, two protein conformations were selected as template coordinates for the docking studies. It has been well established that the binding mode of ligand to the estrogen receptor causes structural perturbations in and around the ligand binding pocket resulting in two protein conformations that contribute to the estrogenic or antiestrogenic effects.18, 19, 20 Two such representative structures – 3ERD (ERα bound to the agonist diethylstilbestrol) and 3ERT (ERα bound to the antagonist 4-OH-TAM) were taken as protein templates. The compounds were docked to the template protein structures using the Surflex docking program. The Surflex scoring21 and the binding postures for the compounds daidzein and 3a were comparable for the estrogenic and antiestrogenic protein conformations with a slight preference to the estrogenic conformation. The binding modes of daidzein and 3a for the estrogenic protein conformation are represented in Figure 7A and 7B. Daidzein forms hydrogen bonds with GLU353, ARG394, GLY521 and HIS524 (Figure 7D). 3a forms hydrogen bonds with LEU387, ARG394 and HIS524 (Figure 7E), resulting in fewer hydrogen bonding interactions than daidzein which could account for its weaker estrogenic activity. Compound 2 docked to the antiestrogenic conformation of the protein with a significantly better Surflex score than when docked to the estrogenic conformation of the protein which implies that it can have antiestrogenic property. The binding posture of 2 (Figures 7C and 7F) for the antiestrogenic protein conformation was similar to that of other ERα antagonists tamoxifen and raloxifene. These docking studies further confirm the nature of estrogenic and antiestrogenic activity exhibited by the compounds 3a and 2.
Figure 7. Molecular modeling of daidzein analogues 2 and 3a.
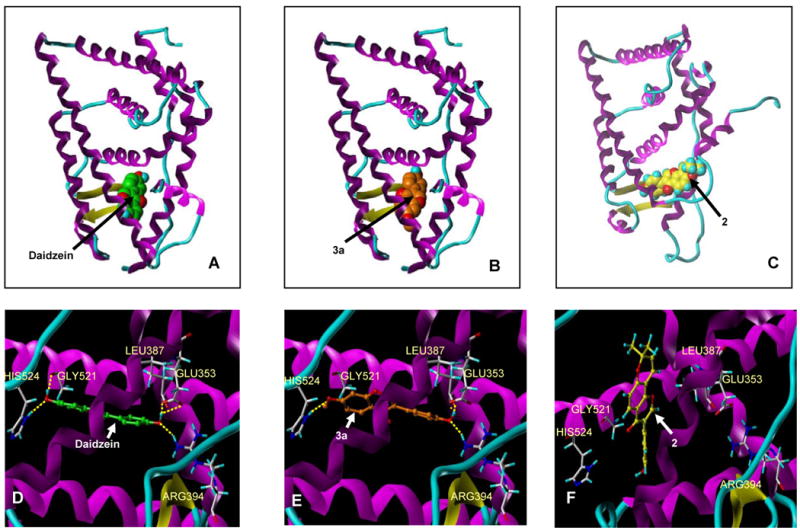
Binding modes of daidzein (A), 2 (B) to estrogenic conformer of ERα and 2 (C) to antiestrogenic conformer of ERα, wherein, the protein is depicted as ribbon/tube model colored by secondary structure and the compounds are shown as spacefill models. D, E and F depict the hydrogen bond interactions made by the compounds daidzein, 3a and 2 with the protein as a ribbon/tube model colored by secondary structure and the compounds as ball & stick models.
3. Conclusion
Previous studies in our laboratory demonstrated the diverse estrogenic and anti-estrogenic activity of naturally occurring flavonoid phytochemicals. Through these studies we identified glyceollin I, a naturally occurring phytoalexin in activated soy, behaved as a potent antiestrogen.15 However, daidzein, the precursor in the biological synthesis of glyceollin I was strongly estrogenic. The contrasting hormonal activity of daidzein and glyceollin I provided the rationale to design and synthesize a series of daidzein analogues that have modified structures on the 7-hydroxyl position of daidzein. In particular, one such analogue, 2 was designed to answer the central question: what is the structural requirement that is responsible for the regulation of the agonistic and antagonistic activities of isoflavone phytochemcials. 2 has retained the daidzein skeleton with the addition of a chromanone moiety. Results from ERE luciferase assay, colony assay, and PgR expression revealed that 2, in contrast to daidzein, acted predominantly as an antiestrogen with limited agonist activity. The in vitro antiestrogenic potency of 2 was found comparable to glyceollin I,15 strongly suggesting that substitution on the chromanone structure, not the presence of pterocarpan skeleton, is responsible for the loss of estrogenic and acquisition of antiestrogenic activity from daidzein to glyceollin I. To further test the in vivo efficacy of 2 as an ER antagonist, nude mice xenografted with E2-dependent MCF-7 cells was treated with the analogue 2, resulting in significant tumor regression.
By synthesizing and testing additional daidzein analogues we have further demonstrated the effect of varying 7O-substitution groups on estrogenic or antiestrogenic activities of the compounds. Short chain alkyl substitutions (3a, 3b, 3c) weakened the estrogenic potency of daidzein but did not render the compounds antiestrogenic; longer chain alkyl substitutions (3e, 3h) appeared to further diminish the estrogenic activity. On the other hand, 3d, 3f, and 3g where branched alkyl or cycloalkyl substitutions were introduced, exhibited significant antiestrogenic property. Molecular modeling study of representative daidzein analogues using the crystal structure of ERα revealed that daidzein and 3a fitted well in the ligand binding pocket of ER in its estrogen bound configuration. The antiestrogenic 2 was found to dock better in ER in its antiestrogen-bound configuration. These results demonstrated the structural requirements for the transformation of daidzein from an estrogen to an antiestrogen. The most effective compound, 2 may serve as a useful novel antiestrogen for breast cancer treatment and prevention.
4. Experimental
4.1. Chemistry
All reagents and solvents were purchased from either Aldrich Chemical Co. (WI, USA) or Acros organics (NY, USA) and were used as received. All organic solvents used were of reagent grade quality and were used without further purification. Daidzein (I) was synthesized by Tyger Scientific Inc. (NJ, USA). 1H spectra were recorded at 400 MHz on a Varian Unity-400 spectrometer (Varian Inc, Palo Alto, CA), using DMSO-d6 as solvent and as internal standard (δ 2.49). GC-MS analyses were performed on an Agilent Technologies 5975C inert MSD mass spectrometer. Melting points were determined with a Mel-temp II point apparatus and are uncorrected. Crude synthetic products were purified by the following methods: chromatography on Silica Gel (60–100 mesh, Fisher Scientific) column and recrystallization from acetone. Analytical thin layer chromatography (TLC) was performed on 250 μ fluorescent plates Whatman, Germany) and visualized by using UV light. For all products, the purity was ascertained to be greater than 95% by the HPLC method using a Shimadzu (Columbia, MD) 2010 HPLC-UV/MS system with a C-18 reverse phase column.
4.1.1. 3-(4-Hydroxyphenyl)-8,8-dimethyl-8H-pyrano[2,3-f]chromen-4-one (2)
According to the literature method16, to a solution of 1 (1.9 g, 7.2 mmol) in 360 mL of xylene was added 1,1-diethoxy-3-methyl-2-butene (2.4 g, 14.7 mmol) in 15 mL of xylene and 3-picoline (3.7 mmol in 43 mL xylene). The reaction mixture was refluxed at 130 °C for 18 hours. The solvent was evaporated under vacuum and the residue was chromatographed over silica gel using hexanes as eluant. Final product was recrystallized from acetone to give 0.34 g of 2 as colorless needle crystalline, with an overall yield of 15%. mp: 262-264 °C (Dec.). GC-MS (m/z 320 M+, m/z 305, m/z 254, m/z 187, m/z 152, m/z 118). 1HNMR (DMSO-d6, 400 MHz, δ): 1.45 (6H, s), 5.94 (1H, d, J=10.0 Hz), 6.79 (1H, d, J=10.0 Hz), 6.80 (2H, d, J=8.8Hz), 6.92 (1H, d, J=8.4Hz), 7.38 (2H, d, J=8.8Hz), 7.88 (1H, d, J=8.4 Hz), 8.36 (1H, s), 9.57 (1H, s, D2O exchangeable).
4.1.2. 3-(4-Hydroxyphenyl)-7-methoxychromen-4-one (3a)
Following a literature method17, to a solution of 1 (1.27g, 5 mmol) in 10 mL of DMSO was added anhydrous K2CO3 (0.9 g, 6.5 mmol) and iodomethane (0.71 g, 5.0 mmol). The reaction mixture was stirred at room temperature for 2 h and then poured into ice water. The resulting mixture was extracted with ethyl acetate. The organic phases were combined, washed with brine, dried over sodium sulfate, filtered, and concentrated under vacuum. The residue was chromatographed over silica gel using hexanes/ethyl acetate (4:1) as eluant. The product was recrystallized from acetone to provide 0.36g of 3a as a white powder in 27% isolated yield. mp: 210-211. (lit17: 210-211). GC-MS (m/z 268 M+, m/z 225, m/z 151, m/z118). 1HNMR (DMSO-d6, 400 MHz, δ): 3.91 (3H, s), 6.81 (2H, d, J=8.8 Hz), 7.08 (1H, dd, J=8.8 Hz, J=2.4 Hz), 7.16 (1H, d, J=2.4 Hz), 7.40 (2H, d, J=8.8 Hz), 8.03 (1H, d, J=8.8 Hz), 8.38 (1H, s), 9.58 (1H, s, D2O exchangeable).
4.1.3. 3-(4-Hydroxyphenyl)-7-ethoxychromen-4-one (3b)
Following the procedure used to prepare 3a, reaction of 1 with bromoethane provided 0.44g of 3b as a pale yellow solid in 31% yield. mp: 131-132 °C. (lit22: 131-133 °C). GC-MS (m/z 282, M+, m/z 253, m/z 225, m/z 165, m/z 137, m/z 118). 1HNMR (DMSO-d6, 400 MHz, δ): 1.38 (3H, t, J=6.8 Hz), 4.19 (2H, q, J=6.8Hz), 6.82 (2H, d, J=8.8 Hz), 7.06 (1H, dd, J=8.8Hz, J=2.4 Hz), 7.14 (1H, d, J=2.4 Hz), 7.40 (2H, d, J=8.8 Hz), 8.02 (1H, d, J=8.8 Hz), 8.37 (1H, s), 9.57 (1H, s, D2O exchangeable).
4.1.4. 3-(4-Hydroxyphenyl)-7-propoxychromen-4-one (3c)
According to the procedure used to prepare 3a, reaction of 1 with 1-bromopropane provided 0.57g of 3c in 38% yield as a white powder. mp: 173-175 °C (Dec.). GC-MS (m/z 296 M+, m/z 253, m/z225, m/z137, m/z118) 1HNMR (DMSO-d6, 400 MHz, δ): 0.99 (3H, t, J=7.0Hz), 1.77 (2H, sextet, J=7.2 Hz), 4.08 (2H, t, J=7.0Hz), 6.81 (2H, d, J=8.4Hz), 7.06 (1H, dd, J=8.8 Hz, J=2.4Hz), 7.13 (1H, d, J=2.4 Hz), 7.39 (2H, d, J=8.4 Hz), 8.01 (1H, d, J=8.8 Hz), 8.35 (1H, s), 9.56 (1H, s, D2O exchangeable).
4.1.5. 3-(4-Hydroxyphenyl)-7-isopropoxychromen-4-one (3d)
According to the procedure used to prepare 3a, reaction of 1 with 2-bromopropane provided 0.26g of 3d in 18% yield as a white powder. mp: 179-180 °C (Dec.). GC-MS (m/z 296, M+, m/z 253, m/z 225, m/z137, m/z 118). 1HNMR (DMSO-d6, 400 MHz, δ): 1.32 (6H, d, J=6.4 Hz), 4.82 (sept, 1H, J=6.0 Hz), 6.796 (2H, d, J=8.4 Hz), 7.03 (1H, dd, J=8.8 Hz, J=2.4 Hz), 7.13 (1H, d, J=2.4 Hz), 7.38 (d, 2H, J=8.4 Hz), 8.00 (1H, d, J=8.8 Hz), 8.35 (1H, s), 9.56 (1H, s, D2O exchangeable).
4.1.6. 7-Butoxy-3-(4-hydroxyphenyl) chromen-4-one (3e)
Using the procedure used to prepare 3a, reaction of 1 with 1-bromobutane provided 0.66g of 3e as a white powder in 43% yield. mp: 162-164 °C. GC-MS (m/z 310, M+, m/z 254, m/z 253, m/z 226, m/z 225, m/z 197, m/z 137, m/z 118). 1HNMR (DMSO-d6, 400 MHz, δ): 0.94 (3H, t, J=7.2 Hz), 1.45 (2H, sextet, J=7.2 Hz),1.737 (2H, quint, J=7.2 Hz), 4.12 (2H, t, J=7.0 Hz), 6.80 (d, 2H, J=8.4Hz), 7.06 (1H, dd, J=8.8Hz, J = 2.4Hz), 7.13 (1H, d, J=2.4 Hz), 7.39 (2H, d, J=8.4 Hz), 8.00 (1H, d, J=8.8 Hz), 8.35 (1H, s), 9.57 (1H, s, D2O exchangeable).
4.1.7. 3-(4-Hydroxyphenyl)-7-isobutoxychromen-4-one (3f)
By the same procedure used to prepare 3a, reaction of 1 with 2-bromobutane provided 0.39g of 3f in 25% yield as a white powder. mp: 176-178 °C. GC-MS (m/z 310 M+, m/z 253, m/z 137, m/z 118). 1HNMR (DMSO-d6, 400 MHz, δ): 0.990 (6H, d, J=6.4Hz), 2.05 (1H, nonet, J=6.40 Hz), 3.89 (2H, d, J=6.4Hz), 6.792 (2H, d, J=8.8Hz), 7.061 (1H, dd, J=8.8 Hz, J=2.4 Hz),7.119 (1H, d, J=2.4 Hz), 7.381 (2H,d, J=8.8Hz), 8.001(1H, d, J=8.8 Hz), 8.350 (1H, s), 9.57 (1H, s, D2O exchangeable).
4.1.8. 7-Cyclopentyloxy-3-(4-hydroxyphenyl)chromen-4-one (3g)
According to the procedure used to prepare 3a, reaction of 1 with bromocyclopentane provided 0.35g of 3g as a colorless needle crystalline in 22% yield. mp: 204-206 °C. GC-MS (m/z 322 M+, m/z 254, m/z 225, m/z 137, m/z 118). 1HNMR (DMSO-d6, 400 MHz, δ): 1.61 (2H, m), 1.74 (4H, m),1.99 (2H, m;), 5.01 (1H, m), 6.81 (2H, d, J=8.4 Hz),7.02 (1H, d, J=8.8Hz, J=2.0Hz), 7.09 (1H, d, J=2.0Hz), 7.39 (2H, d, J=8.4 Hz), 8.01 (1H, d, J=8.8 Hz), 8.35 (1H, s), 9.57 (1H, s, D2O exchangeable).
4.1.9. 7-Hexyloxy-3-(4-hydroxyphenyl)chromen-4-one (3h)
According to the procedure used to prepare 3a, reaction of 1 with 1-bromohexane provided 0.68g of 3h as a colorless crystalline in 40% yield. mp: 147-149 °C (lit23: 145-148 °C). GC-MS (m/z 338 M+, m/z 254, m/z 226, m/z 137, m/z 118). 1HNMR (DMSO-d6, 400 MHz, δ): 0.87 (3H, t, J=7.2 Hz), 1.31 (4H, m), 1.42 (2H, quintet, J=7.2Hz), 1.75 (2H, quintet, J=7.2 Hz), 4.11 (2H, t, J=7.0 Hz), 6.80 (2H, d, J=8.4 Hz), 7.05 (1H, dd, J=8.8 Hz, J=2.4 Hz), 7.13 (1H, d, J=2.4 Hz), 7.39 (2H, d, J=8.4 Hz), 8.00 (1H, d, J=8.8 Hz), 8.352 (1H, s), 9.57 (1H, s, D2O exchangeable).
4.2 Biological Assays
Cell Culture
Human cancer cell lines derived from breast, MCF-7 (ER-positive cells) were cultured in 75 cm2 culture flasks in Dulbecco's Modified Eagle's Medium (DMEM) (Invitrogen, Co.) supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Inc., Gaithersburg, MD), basic minimum MEM essential (50×, Invitrogen Co.) and MEM non-essential (100×, Invitrogen, Co.) amino acids, sodium pyruvate (100×, Invitrogen Co.), antimycotic-antibiotic (10,000 U/mL penicillin G sodium; 10,000 μg/mL streptomycin sulfate; 25 μg/mL amphotericin B as Fungizone®), and human recombinant insulin (4 mg/mL. Invitrogen Co). The culture flasks were maintained in a tissue culture incubator in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. For estrogen studies, cells were washed with PBS 3 times and grown in phenol red-free DMEM supplemented with 5% dextran-coated charcoal-treated FBS (5% CS-FBS) for 72 hours before plating for each particular experiment.
ERE-Luciferase assay
As previously described,15,24 MCF-7 cells, grown for 2 days in 5%-CS-FBS containing phenol-red free DMEM, were plated in 24 well plates at a density of 5×105 cells/well in the same media and allowed to attach overnight. After 18 hours, cells were transfected with 300 μg pGL2-ERE2X-TK-luciferase plasmid (Panomics) for 6 hours according to the manufacturers protocol using Effectene (Qiagen) and treated with vehicle DMSO, E2, various concentrations of daidzein analogues (all with and without estrogen), ICI and 4-OH-TAM overnight. Media was removed and cells were lysed with reporter lysis buffer. Relative Light Units (RLUs) were measured in an Opticomp II luminometer (MGM Laboratories) using luciferase reagent (Promega).
ERα Binding Assays
Receptor binding determinations of daidzein analogues were achieved using the method of Bolger25 et al as was applied by Burow14 et al. In this method, recombinant ER is in equilibrium with a fluorescent ligand (ES2) and a concentration of the competitor (daidzein analogues). The relative displacement of the ES2 is measured as a change in polarization anisotropy. Serial dilutions of competitors (daidzein analogues and estradiol) were prepared from DMSO stock solutions in screening buffer at the desired concentrations. The ER and ES2 were combined with each competitor aliquot to a final concentration of 2 nM ER and 3 nM ES2, respectively. In addition, both a no binding control (ER + ES2, equivalent to 0% competitor inhibition) and a 100% binding control (only free ES2, no ER, equivalent to 100% competitor inhibition) were prepared. All competitor and controls were prepared in duplicate within a binding experiment. After 2-hour incubation at room temperature, the anisotropy values for each sample and control were measured using the Beacon 2000. Anisotropy values were converted to percent inhibition using the following formula: I% = (A0 -A)/(A0 -A100) × 100, where I% is the percent inhibition, A0 is 0% inhibition, A100 is 100% inhibition, and A represents the observed value. This conversion to percent inhibition makes the data more intuitive and normalizes the experiment-to-experiment differences in the range of anisotropy values. The percent inhibition versus competitor concentration curves were analyzed by nonlinear least-squares curve fitting (Prism 5.0a, GraphPad Software, San Diego California USA, www.graphpad.com) to yield IC50 values (the concentration of competitor needed to displace half of the bound ligand). To compare binding affinities of the test compounds to those reported in the literature, IC50 values were converted to relative binding affinities (RBA) using E2 as a standard. The E2 RBA was set equal to 100 RBA = (IC50/IC50 of E2) × 100.
Colony Formation Assay
Similar to our previous studies15, 24, cells were cultured in 5% FBS-DMEM and media was changed to phenol red-free 5% CS-DMEM for 48 hours prior to assay. MCF-7 cells were seeded at a density of 1 × 103 cells/well in a 6 well plate containing phenol red-free 5% CS-DMEM. The cells were allowed to attach overnight and treated on the following day with DMSO vehicle, 1 nM E2, the individual daidzein analogues (with and without estrogen) and 4-OH-TAM. Cells were incubated at 37° C, 5% CO2 in a humidified incubator. Media was replaced every 7 days and treated with appropriate drug for 3 weeks. After 3 weeks the media were removed and the cells were fixed with formaldehyde and dried overnight. The cells were then washed and stained with crystal violet and dried. The colonies were counted.
Real Time RT-PCR
Total cellular RNA was extracted using the RNeasy® mini column (Qiagen), following the manufacturer's instructions. Briefly, a maximum of 1 × 107 cells are disrupted in buffer containing guanidine isothiocyanate and homogenized. Ethanol is added to the lysate, creating conditions that promote selective binding of RNA to the RNeasy silica-gel membrane. The sample is then applied to the RNeasy mini-column. Total RNA binds to the membrane, and high-quality RNA is then eluted in RNase-free water. All binding, wash and elution steps were performed by centrifugation in a microcentrifuge. The concentration of RNA was determined using an ultraviolet spectrophotometer. Reverse transcription (RT) was carried out using the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen). One microgram of total RNA was reverse transcribed to cDNA following the manufacturer's instructions. For each RT, a blank was prepared using all the reagents except the RNA sample (for which an equivalent volume of DEPC treated water was substituted). This blank was used as a non-template control in the real-time PCR experiments. The level of PgR transcripts was determined using a real-time quantitative PCR. Primers for PCR were designed to span intron/exon junctions to minimize amplification of residual genomic DNA. The primer sequence for PgR is (5′-TACCCGCCCTATCTCAACTACC-3′; 5′-TGCTTCATCCCCACAG-ATTAAACA-3′). PCR mix contained optimal concentrations of primers, cDNA and SYBR Green PCR Master Mix (Bio-Rad). Quantification and relative gene expression were calculated with internal controls. The ratio between these values obtained provided the relative gene expression levels.
In Vivo Assay of Anti-Estrogenic Activities of Analogue 2
Xenograft experiments were conducted using previously published protocols (Salvo et al 2006, Rhodes et al 2010). Briefly, 4-6 week old female ovariectomized NU/Nu mice were obtained from Charles River Laboratories (Wilmington, MA). The animals were allowed a period of adaptation in a sterile and pathogen-free environment with phytoestrogen-free food and water ad libitum. MCF-7 cells were harvested in the exponential growth phase using a PBS/EDTA solution and washed. Mice were injected bilaterally in the mammary fat pad (MFP) with 5×10ˆ6 viable cells suspended in 50ul sterile PBS mixed with 100ul Matrigel (reduced factor; BD Biosciences, Bedford, MA). 17β-estradiol pellets (0.72mg, 60-day release; Innovative Research of America, Sarasota, FL) were implanted subcutaneously in the lateral area of the neck using a precision trochar (10 gauge) at the time of cell injection. All procedures in animals were carried out under anesthesia using a mix of isofluorane and oxygen delivered by mask. Tumors were allowed to form and at day 15 post cell injection mice were randomized into groups of 5 mice each. Mice were treated daily with intra-peritoneal injections of either vehicle (1:5 DMSO/PBS) or analog 2 (50mg/kg/animal) for 21 days. Tamoxifen animals were implanted with a slow release tamoxifen pellet () in the lateral area of the neck on the opposite side from the estrogen pellet. ICI animals were given a onetime intra-peritoneal injection of ICI 182,780 (5mg in castor oil) on day 15 post cell injection. Tumor size was measured 3 times weekly using digital calipers. The volume of the tumor was calculated using the following formula: 4/3 πLSˆ2 (L=larger radius; S= shorter radius). All procedures involving these animals were conducted in compliance with State and Federal laws, standards of the U.S. Department of Health and Human Services, and guidelines established by Tulane University Animal Care and Use Committee. The facilities and laboratory animals program of Tulane University are accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care.
Molecular modeling
The coordinates for the estrogen receptor alpha (ERα) bound to the agonist diethylstilbestrol (3ERD.pdb)18 and bound to the antagonist 4-OH-TAM (3ERT.pdb)21 were taken from the Protein Data Bank (http://www.rcsb.org). Oxygen atoms representing water were removed and hydrogen atoms were added to the template proteins using Amber99 force field. The representative compounds daidzein, 3a and 2 were utilized for the docking studies. The 3D structures of the molecules were built using the Tripos SYBYL 8.1 Program (Tripos, St. Louis, MO). Initial geometric optimizations of the ligands were carried out using the standard MMFF94 force field, with a 0.001 kcal/mol energy gradient convergence criterion and a distance-dependent dielectric constant employing Gasteiger and Marsili charges. Additional geometric optimizations were performed using the semi-empirical method molecular orbital package (MOPAC). The compounds were docked into the binding pockets of 3ERD and 3ERT using the Surflex docking program. Surflex21 is a fully automatic flexible molecular docking algorithm that combines the scoring function from the Hammerhead docking system26 with a search engine that relies on a surface-based molecular similarity method as a means to rapidly generate suitable putative poses for molecular fragments. A protomol27 representing the binding site was first generated using the position of the native ligand of the crystal structures. This protomol consists of the preferred locations of various molecular probes (CH4, C=O, N–H), which are then used by the docking engine to search for the best three-dimensional morphological similarity between the protomol and the ligand to dock. A proto-thresh value of 0.5 and a proto-bloat value of 0 were used to generate a compact protomol to which the compounds were docked. The Surflex scores and visual inspection of docking results were considered for obtaining the optimal binding posture. The resulting protein-ligand complexes were then subjected to staged minimization using 200 steps of Steepest descent and 1000 steps of Conjugate gradient methods (with Amber99 force fields).
Statistical analysis
Data were summarized as the Mean ± Standard Error of the Mean (SEM) using the Graph Pad Prism V.4 software program. Analysis of variance models were employed to compare relative ERE transcriptional reporter assays between controls versus treatment. A Tukey post-test was performed to compare differences between groups where a p value < 0.05 was considered significant.
Figure 6. Anti-estrogenic effects of analogue 2 on MCF-7 cell tumorigenesis in vivo.
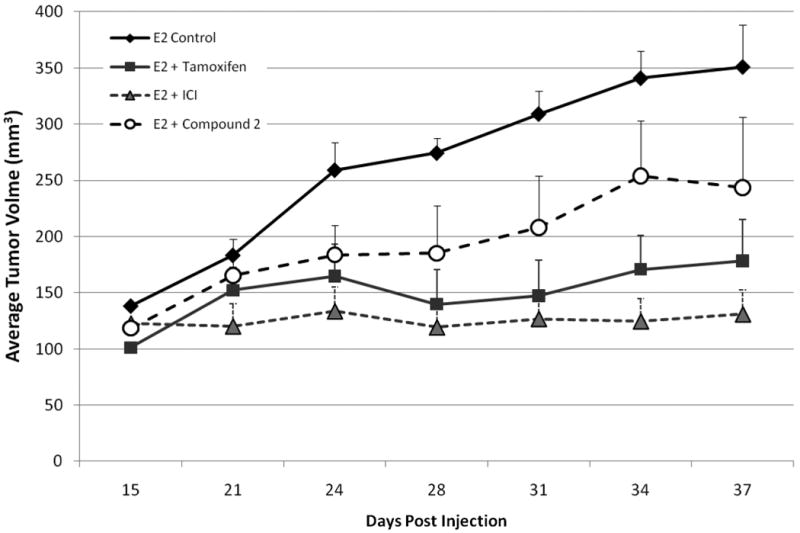
4-6 week old ovariectomized female Nu/Nu mice were injected bilaterally in the MFP with 5×106 MCF-7 cells in matrigel (reduced factor). All animals were implanted with a 17β-estradiol pellet (0.72mg, 60-day release) subcutaneously in the lateral area of the neck at the time of cell injection. Tumors were allowed to form and at day 15 post cell injection mice were randomized into groups (n=5). Animals were treated daily with i.p. injections of either vehicle (1:5 DMSO/PBS) or analog 2 (50mg/kg/animal). Tamoxifen animals were implanted with a slow release tamoxifen pellet () in the lateral area of the neck (opposite side from E2 pellet). ICI animals were given a onetime i.p. injection of ICI 182,780 (5mg in castor oil). Tumor size was measured 3 times weekly using digital calipers. Data represented as mean tumor volume + SEM.
Acknowledgments
This study was supported by a cooperative agreement with the U.S. Department of Agriculture Grant 58-6435-7-019; Department of Defense Grant W81XWH-04-1-0557; the Louisiana Cancer Research Consortium (LCRC); Office of Naval Research Grant N00014-99-1-0763, and National Center for Research Resources RCMI program through Grant 1G12RR026260-01.
Abbreviations
- E2
17β-estradiol
- ER
estrogen receptor
- ERE
estrogen responsive element
- ICI (ICI182,780)
fulvestrant
- 4-OH-TAM
4-hydroxytamoxifen
- PgR
progesterone receptor
- 3ERT
human estrogen receptor alpha ligand-binding domain in complex with 4-hydroxytamoxifen
- 3ERD
ERα bound to the agonist diethylstilbestrol
- MOPAC
molecular orbital package
References
- 1.Rooke N, Li DJ, Li J, Keung WM. The mitochondrial monoamine oxidase–aldehyde dehydrogenase pathway: a potential site of action of daidzin. J Med Chem. 2000;43:4169–4179. doi: 10.1021/jm990614i. [DOI] [PubMed] [Google Scholar]
- 2.Boué SM, Tilghman SL, Elliott S, Zimmerman MC, Williams KY, Payton-Stewart F, Miraflor AP, Howell MH, Shih BY, Carter-Wientjes CH, Segar C, Beckman BS, Wiese TE, Cleveland TE, McLachlan JA, Burow ME. Identification of the potent phytoestrogen glycinol in elicited soybean (Glycine max) Endocrinology. 2009;150:2446–2453. doi: 10.1210/en.2008-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Lemos ML. Effects of soy phytoestrogens genistein and daidzein on breast cancer growth. Ann Pharmacother. 2001;35:1118–1121. doi: 10.1345/aph.10257. [DOI] [PubMed] [Google Scholar]
- 4.Kohen F, Gayer B, Kulik T, Frydman V, Nevo N, Katzburg S, Limor R, Sharon O, Stern N, Somjen D. Synthesis and evaluation of the antiproliferative activities of derivatives of carboxyalkyl isoflavones linked to N-t-Boc-hexylenediamine. J Med Chem. 2007;50:6405–6410. doi: 10.1021/jm070727z. [DOI] [PubMed] [Google Scholar]
- 5.Collins-Burow BM, Burow ME, Duong BN, McLachlan JA. Estrogenic and antiestrogenic activities of flavonoid phytochemicals through estrogen receptor binding-dependent and -independent mechanisms. Nutr Cancer. 2000;38:229–244. doi: 10.1207/S15327914NC382_13. [DOI] [PubMed] [Google Scholar]
- 6.Lo FH, Mak NK, Leung KN. Studies on the anti-tumor activities of the soy isoflavone daidzein on murine neuroblastoma cells. Biomed Pharmacother. 2007;61:591–595. doi: 10.1016/j.biopha.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 7.Valachovicova T, Slivova V, Bergman H, Shuherk J, Sliva D. Soy isoflavones suppress invasiveness of breast cancer cells by the inhibition of NF-kappaB/AP-1-dependent and -independent pathways. Int J Oncol. 2004;25:1389–1395. [PubMed] [Google Scholar]
- 8.Sathyamoorthy N, Wang TT. Differential effects of dietary phyto-oestrogens daidzein and equol on human breast cancer MCF-7 cells. Eur J Cancer. 1997;33:2384–2389. doi: 10.1016/s0959-8049(97)00303-1. [DOI] [PubMed] [Google Scholar]
- 9.Zava DT, Duwe G. Estrogenic and antiproliferative properties of genistein and other flavonoids in human breast cancer cells in vitro. Nutr Cancer. 1997;27:31–40. doi: 10.1080/01635589709514498. [DOI] [PubMed] [Google Scholar]
- 10.Lowe ED, Gao GY, Johnson LN, Keung WM. Structure of daidzin, a naturally occurring anti-alcohol-addiction agent, in complex with human mitochondrial aldehyde dehydrogenase. J Med Chem. 2008;51:4482–4487. doi: 10.1021/jm800488j. [DOI] [PubMed] [Google Scholar]
- 11.Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem. 1987;262:5592–5595. [PubMed] [Google Scholar]
- 12.Kim H, Peterson TG, Barnes S. Mechanisms of action of the soy isoflavone genistein: emerging role for its effects via transforming growth factor beta signaling pathways. Am J Clin Nutr. 1998;68:1418S–1425S. doi: 10.1093/ajcn/68.6.1418S. [DOI] [PubMed] [Google Scholar]
- 13.Graham TL, Kim JE, Graham MY. Role of constitutive isoflavone conjugates in the accumulation of glyceollin in soybean infected with Phytophthora megasperma. Mol Plant Microbe Interact. 1990;3:157–166. [Google Scholar]
- 14.Burow ME, Boue SM, Collins-Burow BM, Melnik LI, Duong BN, Carter-Wientjes CH, Li S, Wiese TE, Cleveland TE, McLachlan JA. Phytochemical glyceollins, isolated from soy, mediate antihormonal effects through estrogen receptor α and β. J Clin Endocrinol Metab. 2001;86:1750–1758. doi: 10.1210/jcem.86.4.7430. [DOI] [PubMed] [Google Scholar]
- 15.Zimmermann MC, Tilghman SL, Boué SM, Salvo VA, Elliott S, Williams KY, Skripnikova EV, Ashe H, Payton-Stewart F, Vanhoy-Rhodes L, Fonseca JP, Corbitt C, Collins-Burow BM, Howell MH, Lacey M, Shih BY, Carter-Wientjes C, Cleveland TE, McLachlan JA, Wiese TE, Beckman BS, Burow ME. Glyceollin I, a novel antiestrogenic phytoalexin isolated from activated soy. J Pharmacol Exp Ther. 2009;332:35–45. doi: 10.1124/jpet.109.160382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khupse RS, Erhardt PW. Total syntheses of racemic, natural (-) and unnatural (+) glyceollin I. Org Lett. 2008;10:5007–5010. doi: 10.1021/ol802112r. [DOI] [PubMed] [Google Scholar]
- 17.Zhang ZT, Wang QY, He Y, Yu KB. Synthesis and crystal structure of 7-ethoxyl-4′-hydroxyisoflavone. J Chem Crystal. 2005;35:89–95. [Google Scholar]
- 18.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 19.Kong EH, Pike AC, Hubbard RE. Structure and mechanism of the oestrogen receptor. Biochem Soc Trans. 2003;31:56–59. doi: 10.1042/bst0310056. [DOI] [PubMed] [Google Scholar]
- 20.Nettles KW, Bruning JB, Gil G, Nowak J, Sharma SK, Hahm JB, Kulp K, Hochberg RB, Zhou H, Katzenellenbogen JA, Katzenellenbogen BS, Kim Y, Joachmiak A, Greene GL. NFkappaB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat Chem Biol. 2008;4:241–247. doi: 10.1038/nchembio.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 22.Gao GY, Li DJ, Keung WM. Synthesis of daidzin analogues as potential agents for alcohol abuse. Bioorg Med Chem. 2003;11:4069–4081. doi: 10.1016/s0968-0896(03)00397-3. [DOI] [PubMed] [Google Scholar]
- 23.Gao GY, Li DJ, Keung WM. Synthesis of potential antidipsotropic isoflavones: inhibitors of the mitochondrial monoamine oxidase –aldehyde dehydrogenase pathway. J Med Chem. 2001;44:3320–3328. doi: 10.1021/jm0101390. [DOI] [PubMed] [Google Scholar]
- 24.Boue SM, Cleveland TE, Carter-Wientjes C, Shih BY, Bhatnagar D, McLachlan JA, Burow ME. Phytoalexin-enriched functional foods. J Agric Food Chem. 2009;57:2614–2622. doi: 10.1021/jf8040403. [DOI] [PubMed] [Google Scholar]
- 25.Bolger R, Nestich S, Wiese T, Ervin K, Checovich W. Rapid screening of environmental chemicals for estrogen receptor binding capacity. Environ Health Perspect. 1998;106:551–557. doi: 10.1289/ehp.98106551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Welch W, Ruppert J, Jain AN. Hammerhead: fast, fully automated docking of flexible ligands to protein binding sites. Chem Biol. 1996;3:449–62. doi: 10.1016/s1074-5521(96)90093-9. [DOI] [PubMed] [Google Scholar]
- 27.Ruppert J, Welch W, Jain AN. Automatic identification and representation of protein binding sites for molecular docking. Protein Sci. 1997;6:524–33. doi: 10.1002/pro.5560060302. [DOI] [PMC free article] [PubMed] [Google Scholar]