

Abstract
Synaptotagmin I is the major Ca2+ sensor for membrane fusion during neurotransmitter release. The cytoplasmic domain of synaptotagmin consists of two C2 domains, C2A and C2B. On binding Ca2+, the tips of the two C2 domains rapidly and synchronously penetrate lipid bilayers. We investigated the forces of interaction between synaptotagmin and lipid bilayers using single-molecule force spectroscopy. Glutathione-S-transferase-tagged proteins were attached to an atomic force microscope cantilever via a glutathione-derivatized polyethylene glycol linker. With wild-type C2AB, the force profile for a bilayer containing phosphatidylserine had both Ca2+-dependent and Ca2+-independent components. No force was detected when the bilayer lacked phosphatidylserine, even in the presence of Ca2+. The binding characteristics of C2A and C2B indicated that the two C2 domains cooperate in binding synaptotagmin to the bilayer, and that the relatively weak Ca2+-independent force depends only on C2A. When the lysine residues K189–192 and K326, 327 were mutated to alanine, the strong Ca2+-dependent binding interaction was either absent or greatly reduced. We conclude that synaptotagmin binds to the bilayer via C2A even in absence of Ca2+, and also that positively charged regions of both C2A and C2B are essential for the strong Ca2+-dependent binding of synaptotagmin to the bilayer.
Introduction
It is now widely accepted that synaptotagmin I (syt) is a major Ca2+ sensor at the nerve terminal (reviewed in (1,2)). Recent evidence indicates that it plays a dual role, reducing spontaneous neurotransmitter release in the absence of Ca2+, and then responding to Ca2+ influx across the presynaptic membrane by triggering a burst of synchronous transmitter release (3,4). This dual action of syt is recapitulated in an in vitro assay for membrane fusion (5).
Syt is an integral membrane protein of the synaptic vesicle, comprising a short luminal region, a single transmembrane region, and a cytoplasmic region consisting predominantly of two C2 domains, known as C2A and C2B, connected by a short linker (reviewed in (1,2)). The tips of these C2 domains bind three and two Ca2+ ions, respectively (6,7). On Ca2+ binding, the surface of each C2 domain acquires a significant positive charge, enabling syt to interact with negatively charged phospholipids, such as phosphatidylserine (PS) and phosphatidylinositol 4,5-bisphosphate (7–14). Syt also interacts with soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptors (SNAREs), proteins known to be key players in the process of membrane fusion, in both a Ca2+-dependent and a Ca2+-independent manner (3,15–18). The interactions of syt with both phospholipids and SNAREs appear to be essential for its function as a Ca2+ sensor for neurotransmitter release (19–22).
In response to Ca2+ binding, the tips of the C2 domains rapidly and synchronously penetrate the lipid bilayer (11,23–26), causing perturbation of the bilayer structure (27). In addition to the Ca2+ binding sites, other regions of both C2A and C2B have been shown to be important for the normal function of syt. For instance, a polybasic region of C2B, including lysines 326 and 327, is involved in the recruitment of membrane to this domain, and may be involved in bridging the synaptic vesicle and the plasma membrane during exocytotic membrane fusion (28). Mutation of this region significantly impairs the ability of syt to support Ca2+-stimulated neurotransmitter release in Drosophila (29,30). There is also evidence that C2A is able to bind to lipid bilayers in the absence of Ca2+, leading to the suggestion that C2A might be preassociated with the presynaptic membrane with its Ca2+ binding tip exposed to the cytoplasm, and that on binding Ca2+, the domain rotates to allow the tip to penetrate the membrane (31). Interestingly, there is a positively charged region on C2A, encompassing lysines 189–192 in the rat, that might explain the loose association of this domain with the membrane in the absence of Ca2+. Mutation of this region does not affect evoked neurotransmitter release in Drosophila, but does increase spontaneous release (4).
In this study we set out to measure the forces of interaction between syt and a supported lipid bilayer using single-molecule force spectroscopy. By studying the effects of mutations to key regions, in the context of both the complete cytoplasmic domain (C2AB) and the individual C2 domains, we hoped to be able to dissect the regions of syt that contribute to its membrane binding under various conditions. We show that both C2 domains need to be intact and able to sense Ca2+ in order to generate the maximum binding force, consistent with the known cooperation between these two C2 domains with respect to membrane interaction (11,23). The interaction forces were substantially reduced by mutation of the Ca2+-binding region of either C2 domain. Intriguingly, a significant interaction force persists in C2A even when the Ca2+ binding site has been mutated. Mutation of the polybasic region on either C2 domain almost abolished the Ca2+-dependent binding of syt to the bilayer.
Materials and Methods
Protein expression and purification
The following syt constructs, all in the vectors pGEX-2T or 4T, were used: syt I cytoplasmic domain (wild-type C2AB, residues 96–421); C2AB in which aspartate residues crucial to Ca2+ binding to either C2A (D230,232), C2B (D363,365), or both C2A and C2B (6,7) were neutralized by conversion to asparagines, designated C2AMB, C2ABM, and C2AMBM, respectively; C2AB with mutations of various lysine residues to alanines, designated C2AK189-192AB and C2ABK326,327A; wild-type C2A (residues 96–265), C2AM and C2AK189-192A; and wild-type C2B (residues 248–421) and C2BM. Constructs were kindly provided by Edwin Chapman and Mark Dunning (Department of Physiology, University of Wisconsin-Madison, WI). Glutathione-S-transferase (GST)-tagged proteins were prepared as described previously (27). Where appropriate, bacterial contaminants were removed from recombinant proteins by treatment with 1 M NaCl plus RNase/DNase, as described (32). For control experiments, GST itself was prepared using empty pGEX-2T vector. In all cases, protein purity was checked by sodium dodecyl sulfate-polyacrylamide gel electrophoresis followed by Coomassie Blue staining.
Cantilever modification
Atomic force microscopy (AFM) cantilevers made of silicon nitride (OMCL-TR400PSA; Olympus, Tokyo, Japan) were modified as described previously (33). Cantilevers were treated with 3-aminopropyltriethoxysilane (APTES; Sigma, St. Louis, MO) vapor for 1 h, and incubated with the heterobifunctional cross-linker maleimide-dPEG12-n-hydroxysuccinimide ester (5 mg/mL; Quanta BioDesign, Powell, OH), dissolved in chloroform containing 0.7% triethylamine, for 2 h at room temperature. The polyethylene glycol- (PEG)-modified cantilevers were then incubated with glutathione (10 mM in HEPES-buffered saline (HBS); 100 mM NaCl, 20 mM HEPES, pH 7.2) for 1 h at room temperature. A glutathione-coupled cantilever was immersed in a solution of GST-fusion protein for 1 h at 4°C, washed in HBS, and immediately used in the experiment.
Covalent coupling of GST to mica
Freshly-cleaved mica sheets were treated with APTES for 1 h. The amino-coated mica was then treated with glutaraldehyde (1 mM) for 10 min at room temperature. After washing with HBS, GST solution (10 ng/mL) was added to the mica and incubated for 10 min at room temperature. Free glutaraldehyde on the GST-immobilized mica surface was blocked with 10 mM glycine solution, pH 7.6. After washing with HBS, the substrate was used for the measurement.
Formation of supported lipid bilayers
Supported lipid bilayers were prepared essentially as described previously (27). 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) and brain L-α-phosphatidylserine (PS), sphingomyelin (SM), and cholesterol, obtained from Avanti Polar Lipids (Birmingham, AL) as chloroform stocks, were mixed as appropriate. The chloroform was evaporated under a stream of nitrogen gas and the lipids were rehydrated overnight in water (from a Millipore water purification system) to give a total lipid concentration of 2 mg/mL. The lipid mixture was vortexed to produce large multilamellar vesicles, from which small unilamellar vesicles were prepared by sonication on ice for 10 s. To prepare supported lipid bilayers, 10 μL of the vesicle suspension was added to 20 μL of HBS, containing either 500 μM CaCl2 or 2 mM EGTA. The resulting suspension was deposited onto freshly-cleaved mica (5-mm diameter disks) fixed by epoxy (Aron Alpha type 102; Agar Scientific, Stansted, UK) to 15-mm steel SPM specimen discs (Agar Scientific). After an 8-min adsorption on mica, the sample was rinsed with HBS to remove unadsorbed liposomes, and then transferred to the atomic force microscope.
Topographic AFM imaging
AFM imaging was carried out at room temperature (20°C) using a Digital Instruments Multimode atomic force microscope equipped with a E-scanner and a Nanoscope IIIa controller with an in-line electronics extender module (Veeco/Digital Instruments, Santa Barbara, CA). All images were collected using tapping mode in fluid, and using oxide-sharpened silicon nitride probes (OMCL-400PSA; Olympus). The cantilevers (typically exhibiting a spring constant of 0.02 N/m) were tuned to 10–20% below the peak of the resonance frequency, generally found between 9.0 and 10 kHz. The drive amplitude was set to generate a root mean-square amplitude of 1.2–1.7 V. The microscope was engaged with a 0-nm scan area to allow for tuning. The setpoint was adjusted to the highest setting that allowed imaging with little noise, to minimize the force applied to the sample. Images were captured at a scan rate of 2 Hz (unless otherwise noted), and with 512 scan lines per area. Data analysis was performed using commercially available software (NanoScope III software; Digital Instruments).
Force measurement
Force measurements were performed with a Digital Instruments Multimode atomic force microscope equipped with an E-scanner and a Nanoscope IIIa controller with an in-line electronics extender module (Veeco/Digital Instruments). Protein-derivatized cantilevers with a specified spring constant of 0.02 N/m were used routinely. Force measurements were performed in HBS containing either 2 mM EGTA or 0.5 mM Ca2+. The loading rate was 2000 pN/s and the trigger channel was set for 20 nm. Because the coupled proteins might be damaged by repetitive cycles of binding and retraction, each functionalized cantilever was used for fewer than 140 retractions. The data were analyzed using a Scanning Probe Image Processor (Image Metrology, Lyngby, Denmark). Rupture forces were obtained from force-extension curves. Force curves that could be fitted to the wormlike chain model of the PEG molecule (34) were taken to represent single-molecule pulling events.
A number of control experiments were carried out. For instance, we examined the interaction between a glutathione-derivatized tip (with no attached protein) and supported lipid bilayers composed of 45% DOPC, 30% SM, 15% PS, and 10% cholesterol (mol/mol). We occasionally saw very large forces (1.5 nN) that might indicate an interaction between lipids attached to the tip and the supported bilayer. However, these traces could not be fitted to the wormlike chain model for the PEG molecule, and so could be excluded from any analysis of protein-lipid interactions. We also tested the interaction between GST on the tip and supported lipid bilayers. We occasionally detected small forces of interaction, although we were unable to fit a force peak to the data. In fact, when GST-syt constructs are attached to the tip, the GST will likely be masked by the attached syt; so only the syt and not the GST will interact with the bilayer.
Results
Functionalization of AFM cantilevers
The method for attaching GST-tagged proteins to the AFM cantilever has been described previously (33). Briefly, a 5.3-nm-long heterobifunctional PEG linker is first attached to an amino-functionalized silicon nitride cantilever, and glutathione (GSH) is then attached to a maleimide group at the end of the PEG linker (Fig. 1). Subsequently, GST-tagged proteins can be coupled noncovalently to the GSH. The PEG linker provides the attached protein with both flexibility to interact with its target and freedom from steric hindrance from the surface of the cantilever. Protein-modified cantilevers prepared in this way have already been used successfully to study the interaction between importin-β and the nucleoporin Nup 153 (35).
Figure 1.
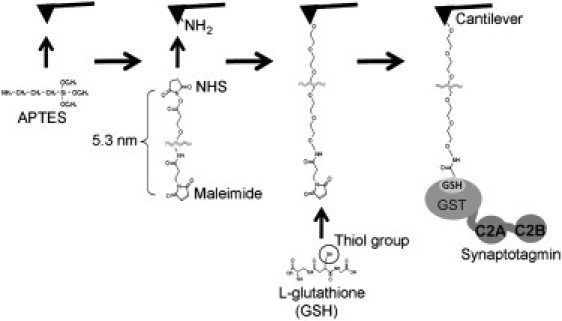
Coupling of GST-tagged proteins to the AFM cantilever. A silicon nitride cantilever is amino-functionalized by incubation with APTES. A 5.3-nm long PEG linker is then attached to the amino-functionalized cantilever through an N-hydroxysuccinimide group. GSH is covalently bound to the free end of the PEG linker through a maleimide group. Finally, a GST-tagged protein, in this case C2AB, is noncovalently attached to the glutathione.
The strength of the glutathione-GST bond was first measured by determination of the rupture force between GSH on the cantilever and GST covalently attached to the mica surface. The mean rupture force was 121 ± 63 (SD) pN (n = 27), in agreement with a previously reported value (33). This provides an upper limit to the force that can be measured between syt and lipid bilayers. As expected, this force was abolished when the buffer solution contained free GSH (100 mM).
AFM imaging of lipid bilayers
In each experiment, before force measurements were made, a topographic image of the supported bilayer was acquired with a nonfunctionalized cantilever. Bilayers typically contained 45% DOPC, 30% SM, 15% PS, and 10% cholesterol (mol/mol). Fig. 2 shows representative images of bilayers in the presence (Fig. 2 A) and absence (Fig. 2 B) of Ca2+. In both cases, the bilayers are featureless, and sections through regions containing both bilayer and bare mica (lower panels) indicate that the bilayer is ∼5.0-nm thick, similar to previously reported values (e.g., (27,36)). This value is larger than the predicted bilayer thickness of ∼4.6 nm (37). The presence of a 1-nm hydration layer between a bilayer and its support has been reported previously (38), and such a hydration layer would account for the discrepancy between expected and observed bilayer thickness in our experiments. Occasionally, features raised ∼1 nm above the level of the bilayer were seen. These have been shown previously (36) to represent SM/cholesterol-rich domains, reminiscent of the rafts seen in biological membranes (37).
Figure 2.
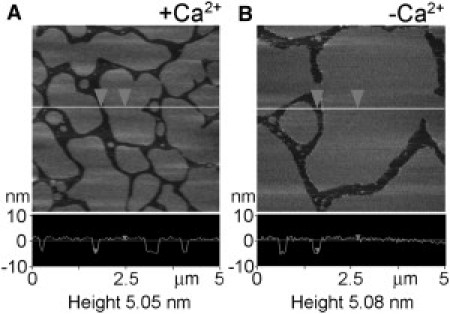
Structure of a supported lipid bilayer. Typical AFM image of a supported lipid bilayer composed of 45% DOPC, 30% SM, 15% PS, and 10% cholesterol either in the presence of Ca2+ (0.5 mM; A) or the absence of Ca2+ (2 mM EGTA; B). (Lower panels) Sections through the bilayers, at the positions indicated (lines). Bilayer surface or the mica support (arrows). The thickness of the bilayer was similar in the presence and the absence of Ca2.
Interaction of wild-type C2AB with lipid bilayers
GST-C2AB was coupled to the cantilever and the rupture force for its interaction with bilayers was measured in the presence and absence of Ca2+. Typical force curves obtained in these two conditions are shown in Fig. 3, A and B, respectively. For each condition, a number of force-extension curves were analyzed, and a frequency distribution of forces was produced. Gaussian functions were fitted to the distributions, and the means of the distributions (along with the percentage of retractions giving force-extension curves) were recorded. In the presence of Ca2+ (0.5 mM), the force distribution had two peaks, at 73 ± 29 (SD) pN and 122 ± 33 pN (Fig. 3 C), and 4.69% of retractions gave force-extension curves. For purposes of comparison, the higher rupture force is similar to the force required to break an antigen-antibody interaction (126 pN (39)), and much greater than the force required to break the interaction between importin-β and the nucleoporin Nup 153 (49 pN (35)). Note also that the mean force required to rupture the glutathione-GST interaction is 121 pN, so the higher force of interaction between C2AB and the bilayer might be an underestimate of the true value.
Figure 3.
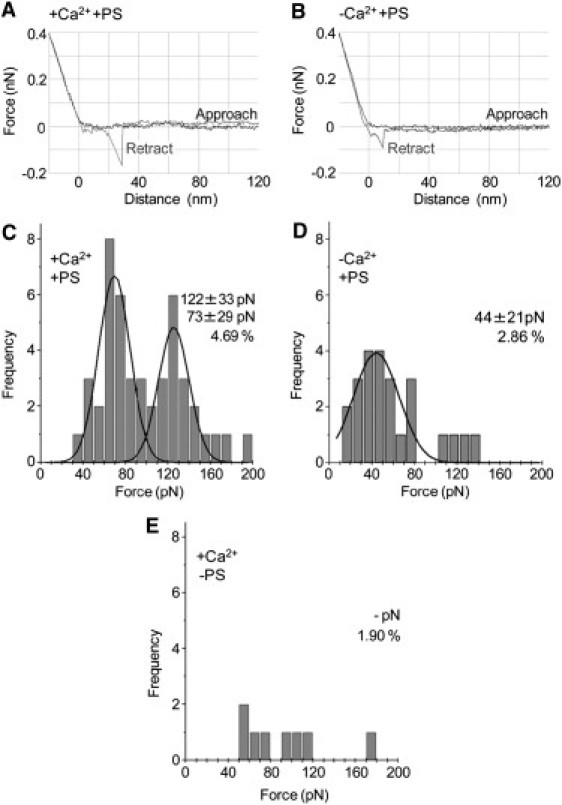
Interaction of wild-type C2AB with PS-containing bilayers in the presence and absence of Ca2+. Typical force curves for the interaction of C2AB with lipid bilayers either in the presence (A) or the absence (B) of Ca2+. The vertical axes indicate cantilever deflections, representing a retraction force from the substrate. The horizontal axes show the distance between the cantilever and the substrate. All force measurements were performed in HBS at room temperature, with a loading rate of 2000 pN/s. To measure single molecule pulling events, only the force peaks that could be fitted to the wormlike chain model of PEG were accepted. (C–E) Frequency distributions of forces for the interaction of C2AB with lipid bilayers either in the presence (C) or absence of Ca2+ (D), and with lipid bilayers composed of 60% DOPC, 30% SM, and 10% cholesterol (i.e., no PS) in the presence of Ca2+ (E). The numbers of accepted force values were 46 (C), 24 (D), and 8 (E). (Curves) Fitted Gaussian functions. The means of the distributions, and the percentage of retractions giving force-extension curves, are indicated.
In the absence of Ca2+ (i.e., plus 2 mM EGTA), only a single low force, at 44 ± 21 pN, was recorded (Fig. 3 D), and now only 2.86% of retractions gave force-extension curves. Hence, C2AB interacts with the lipid bilayer through both Ca2+-dependent and Ca2+-independent forces. When PS was omitted, so that the bilayers contained 60% DOPC, 30% SM, and 10% cholesterol, the percentage of retractions giving force-extension curves fell to 1.90% (Fig. 3 E), and no force peak was detectable, indicating that the interaction between C2AB and the lipid bilayer requires the presence of an acidic phospholipid.
Identification of regions responsible for the Ca2+-dependent and Ca2+-independent interactions with the bilayer
We next examined the behavior of C2AB mutants in which Ca2+ binding to either C2A or C2B (or both) had been disrupted by appropriate aspartate-to-asparagine mutations (D230,232N on C2A and D363,365N on C2B). When the Ca2+ binding site on C2A was mutated (C2AMB), only a single force peak was seen, at 60 ± 22 (Fig. 4 A), in the same range as the lower force seen with the wild-type protein in the presence of Ca2+ (Fig. 3 C). When the Ca2+ binding site on C2B was mutated (C2ABM), again only a single force was seen, at 73 ± 24 pN (Fig. 4 B), identical to the lower force seen with wild-type C2AB (Fig. 3 C). When both Ca2+ binding sites were mutated (C2AMBM), a single, low force of 51 ± 21 pN was seen (Fig. 4 C). The Ca2+-ligand mutations did not have a major effect on the percentage of retractions giving force-extension curves.
Figure 4.
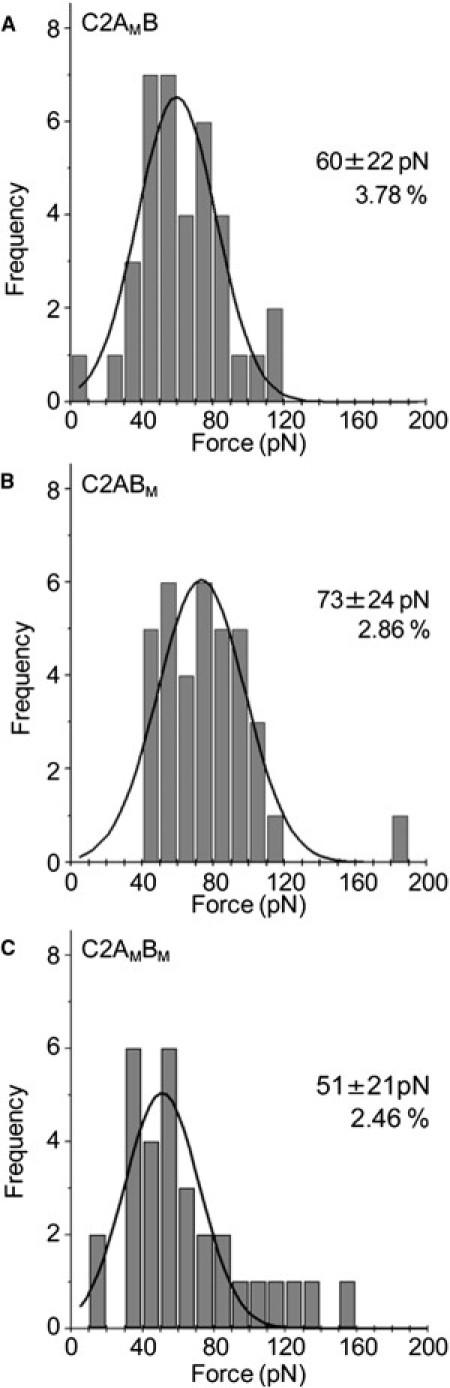
Interaction of C2AB bearing Ca2+ ligand mutations with bilayers in the presence of Ca2+. Force measurements were carried out as described in Fig. 3. Frequency distributions of forces for the interaction of C2AMB (A), C2ABM (B), and C2AMBM (C) with lipid bilayers in the presence of Ca2+ (0.5 mM). The numbers of accepted force values were 37 (A), 36 (B), and 31 (C). The curves indicate the fitted Gaussian functions. The means of the distributions, and the percentage of retractions giving force-extension curves, are indicated.
The results presented so far indicate that both C2 domains need to be intact in order to maximize the higher of the two forces seen with wild-type C2AB. The higher force is reduced when either C2A or C2B lacks the ability to bind Ca2+. However, the measured force remains higher than that seen with C2AB in the absence of Ca2+, or with C2AMBM in the presence of Ca2+, suggesting that some residual Ca2+-dependent binding remains, although it cannot be distinguished from the Ca2+-independent force. This result is consistent with previous reports that C2A and C2B bind cooperatively (and simultaneously) to the bilayer (11). The greater importance of the C2A domain with respect to attachment of C2AB to the bilayer, indicated by the greater impact of mutation of the Ca2+ binding site on C2A on the force of interaction, was also seen in experiments in which bilayers were incubated with wild-type C2AB, C2AMB, and C2ABM, all at 2.5 nM, and then imaged by AFM. Wild-type C2AB formed a dense spread of particles on the bilayer (Fig. 5 A). The binding density was reduced when C2AMB was used (Fig. 5 B), whereas C2ABM formed an array of similar density to that given by wild-type C2AB (Fig. 5 C). The low level of binding of C2AMBM to the bilayer has been reported previously (27).
Figure 5.

AFM images of wild-type and mutant C2AB in association with lipid bilayers. Liposomes composed of DOPC and PS (3:1) were incubated with protein (2.5 nM final concentration) in the presence of Ca2+ (0.5 mM), and supported bilayers were then produced. Free protein was washed away before AFM imaging. (A) Wild-type C2AB. (B) C2AMB. (C) C2ABM.
Next, we measured the forces of interaction between the individual C2 domains and the lipid bilayer. In the presence of Ca2+, wild-type C2A gave two forces, of 54 ± 29 pN and 102 ± 22 pN (Fig. 6 A). In the absence of Ca2+, the higher force disappeared, and only a single low force, of 50 ± 15 pN, was seen (Fig. 6 B). When the Ca2+ binding site on C2A was mutated (C2AM), and binding was measured in the presence of Ca2+, once again only a low force, of 44 ± 18 pN, was seen (Fig. 6 C). In the presence of Ca2+, wild-type C2B showed a binding force of 73 ± 32 pN (Fig. 6 D). In the absence of Ca2+ (Fig. 6 E), no force peak was seen. When C2B's Ca2+ binding site was mutated (C2BM) and binding was measured in the presence of Ca2+, once again there was no detectable force peak (Fig. 6 F). Note that Ca2+ ligand mutations in C2B had a greater effect on the percentage of retractions giving force-extension curves than the corresponding mutations in C2A. These data indicate that the region responsible for the Ca2+-independent force resides in C2A. Incidentally, the absence of detectable force peaks for wild-type C2B in the absence of Ca2+, for C2BM in the presence of Ca2+, and for wild-type C2AB with PS-free bilayers in the presence of Ca2+ (above), strongly indicates that the GST anchor is making no contribution to any of the forces detected in the other experiments.
Figure 6.
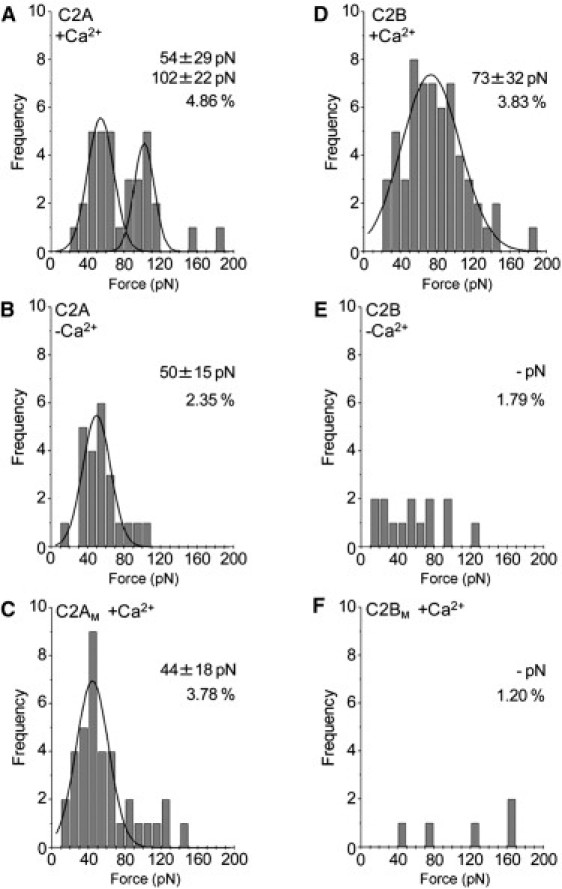
Interaction of wild-type individual C2 domains and C2 domains bearing Ca2+ ligand mutations with bilayers. Force measurements were carried out as described in Fig. 3. (A–C) Frequency distributions of forces for the interaction of C2A with lipid bilayers in the presence (A), and absence (B) of Ca2+, and for C2AM in the presence of Ca2+ (0.5 mM; C). (D–F) Frequency distributions of forces for the interaction of C2B with lipid bilayers in the presence (D) and absence (E) of Ca2+, and for C2BM in the presence of Ca2+ (0.5 mM; F). The numbers of accepted force values were 34 (A), 59 (B), 23 (C), 14 (D), 37 (E), and 5 (F). The curves indicate the fitted Gaussian functions. The means of the distributions, and the percentage of retractions giving force-extension curves, are indicated.
The role of polybasic regions in C2A and C2B in the interactions of syt with the bilayer
As mentioned above, both C2A and C2B have positively charged regions that seem to be important for the function of syt. To examine the role of these regions in the interaction of syt with the bilayer, we measured the interaction forces of C2AB in which lysines 189–192 in C2A and lysines 326 and 327 in C2B had been mutated to alanines. We found that in the presence of Ca2+, C2AK189-192AB gave only a small force, of 54 ± 19 pN; the larger force, normally seen with wild-type C2AB (above), was absent (Fig. 7 A). Similarly, C2ABK326,327A gave only a small force, of 57 ± 23 pN (Fig. 7 B). Because C2A was the domain responsible for the Ca2+-independent binding of syt to the bilayer, and because mutation of lysines 189–192 is known to cause an increase in spontaneous transmitter release (4), we tested the possibility that these residues mediate the Ca2+-independent binding. We found that in the presence of Ca2+ C2AK189-192A gave two force peaks, at 44 ± 25 pN and 89 ± 15 pN (Fig. 7 C). These peaks are similar to those seen with wild-type C2A (Fig. 6 A), although the higher force peak is less well represented. In the absence of Ca2+, C2AK189-192A gave a single low force peak, at 41 ± 23 pN (Fig. 7 D), similar to the behavior of wild-type C2A. Hence, the Ca2+-independent force does not depend on this positively charged region.
Figure 7.
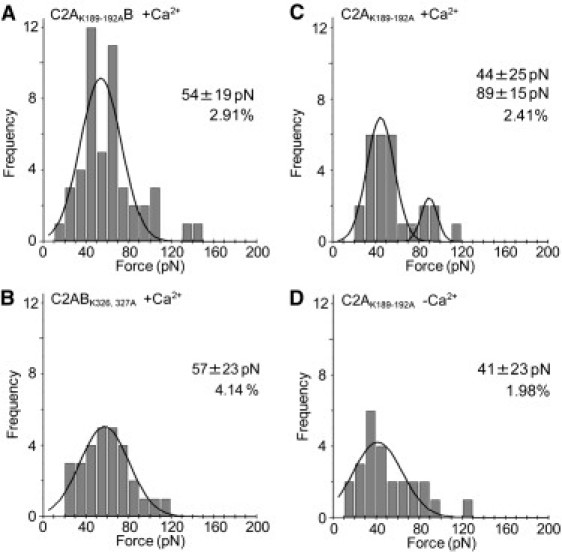
Interaction of C2AB and C2A bearing lysine-alanine mutations with bilayers. Force measurements were carried out as described in Fig. 3. Frequency distributions of forces for the interaction of C2AK189-192A B (A) and C2ABK326,327A (B) in the presence of Ca2+ (0.5 mM), and for C2AK189-192A in the presence (C) and absence (D) of Ca2+. The numbers of accepted force values were 48 (A), 29 (B), 27 (C), and 25 (D). The curves indicate the fitted Gaussian functions. The means of the distributions, and the percentage of retractions giving force-extension curves, are indicated.
Discussion
Our results indicate that syt's two C2 domains cooperate in its binding to lipid bilayers. Many previous studies have addressed the relative contributions of the two C2 domains of syt to its function as a Ca2+ sensor for neurotransmitter release, and a confusing picture has emerged. Some studies have suggested that C2A is dispensable. For example, neutralization of all five acidic residues in the Ca2+ binding site on C2A had no effect on synaptic transmission (40). Further, syt with a C2A domain that could not bind Ca2+ was nevertheless able to rescue exocytosis in syt-null Drosophila (41). On the other hand, a mutation in a Ca2+-binding loop of the C2A domain, R233Q, decreased the Ca2+ binding affinity of syt I and the Ca2+ sensitivity of neurotransmitter release in parallel (42). Further, tryptophan substitutions in the Ca2+ binding loops of C2A (and C2B) that increased Ca2+ binding affinity caused parallel increases in the Ca2+ sensitivity of exocytosis (43). Experiments involving substitution of the Ca2+ ligands in C2B have produced a clearer picture. Essentially, Ca2+ binding to C2B is an absolute requirement for the function of syt in exocytosis (44,45). Overall, these experiments indicate that both C2 domains of syt are involved in its functional role, with C2B being the major partner.
In the in vitro models of membrane fusion, disruption of Ca2+ binding to both C2A and C2B abolished the ability of C2AB to stimulate fusion (21). Interestingly, in a similar assay the ability of C2AB to stimulate fusion required an intact Ca2+ binding site on C2A but not C2B (46). This stimulatory effect required the binding of C2A to PS in the R-SNARE-containing liposomes, whereas C2B apparently interacted with Q-SNAREs in the other liposome population in a Ca2+-independent manner. When full-length syt was anchored in the R-SNARE liposomes, mimicking the in vivo arrangement, it interacted with the Q-SNARE liposomes in a Ca2+-independent manner to accelerate fusion. In the presence of Ca2+, syt could interact with liposomes either in cis or in trans, depending on the location of PS. The former interaction retarded fusion, whereas the latter interaction accelerated it. This study also found that the two C2 domains needed to be linked together in order for C2AB to stimulate fusion; neither C2 domain alone had any effect. Recently, however, it has been shown that C2B can stimulate fusion in response to Ca2+, and also inhibit fusion in the absence of Ca2+, but only at high concentrations and only when the liposomes contain phosphatidylethanolamine (47).
As judged by cosedimentation with liposomes, both C2A and C2B bind individually to lipid bilayers in a Ca2+-dependent manner, with C2A showing the greater Ca2+ affinity (14) and the greater resistance to increasing salt concentration (11). Interestingly, the binding of C2B to lipid bilayers can be enhanced by tethering it to C2A, even when the ability of C2A to bind Ca2+ is abolished (i.e., as in our C2AMB construct; (11)). The Ca2+-independent binding of C2A to bilayers seen in this study would explain this observation. Binding of syt is accompanied by the dipping of the Ca2+ binding regions of the C2 domains into the lipid bilayer (24–26,48,49). Consistent with our results, the extent of bilayer penetration was greater for C2AB than for either C2A or C2B alone (26). Note also that in our experiments there was little evidence for independent disengagement of the two C2 domains from the bilayer: the vast majority of force-extension curves obtained with C2AB indicated single rupture events. In the absence of Ca2+, it has been reported that only C2B binds to bilayers (50), although there is evidence that C2A is at least partially bilayer-bound in the absence of Ca2+ (31).
The polylysine motif on C2B is known to be required for efficient Ca2+-triggered neurotransmitter release (29,30,51,52), consistent with our finding that it is required to support Ca2+-dependent binding of C2AB to the bilayer. The nature of the interaction of the polylysine motif with the bilayer is still unclear. For instance, it has been shown that it is required for the ability of C2B to aggregate liposomes (28); however, it does not penetrate the bilayer (26). It is possible that it makes only a peripheral interaction with the bilayer surface, which, although too weak to detect in our experiments, might assist in orientating the C2B domain in a position favoring Ca2+-triggered membrane penetration of its Ca2+-binding loops. An analogous polylysine motif on C2A has recently been linked with syt's ability to suppress exocytosis in the absence of Ca2+ (4). We expected that mutation of this region (as in C2AK189-192A) might have abolished the Ca2+-independent component of binding; however, this was not the case. Hence, either another specific region or (perhaps more likely) a more diffuse region must mediate the Ca2+-independent binding.
Various experimental approaches have been used to study the interaction of syt with lipid bilayers, and different results have been generated using the same constructs in different assays. For example, it was found that C2B, immobilized as a GST fusion protein, binds radiolabeled liposomes only weakly in pull-down assays, whereas C2B efficiently cosediments with liposomes (32). The question is: Which of the assays most closely reflects the situation in vivo? At the synapse, syt is anchored to the synaptic vesicle, with the C2A domain closer to the vesicle membrane than C2B. This is a very similar arrangement to the one in our experiments, where the AFM cantilever tip substitutes for the vesicle membrane. Further, our experiments involve transient presentation of the syt to the bilayer, similar to the situation in vivo. Based on our results, we suggest that in the nerve terminal syt is docked onto bilayers in the absence of Ca2+ through the Ca2+-independent interaction of C2A, as illustrated in Fig. 8. The positively charged regions on C2A and C2B would help to align the protein on the bilayer so that Ca2+-dependent membrane binding may occur. When Ca2+ enters the terminal, both C2 domains would then engage the bilayer and (in the case of C2B) the t-SNAREs in the plasma membrane, thereby triggering a synchronous burst of neurotransmitter release.
Figure 8.
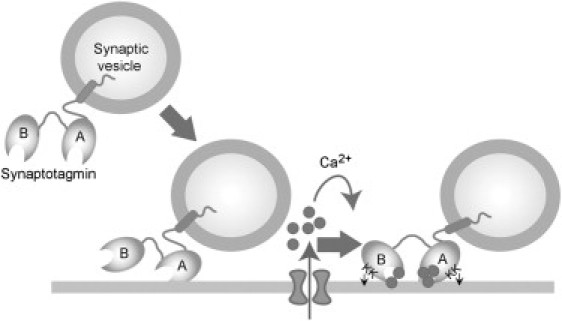
Model of the interaction of syt with the plasma membrane during Ca2+-stimulated exocytosis. In the absence of Ca2+, syt is docked onto bilayers through the Ca2+-independent interaction of C2A. The positively charged regions on C2A and C2B help to align the protein on the bilayer so that Ca2+-dependent membrane binding can occur. When Ca2+ enters the terminal, both C2 domains engage the bilayer, triggering a synchronous burst of neurotransmitter release.
Acknowledgments
We thank Edwin Chapman and Mark Dunning (Department of Physiology, University of Wisconsin-Madison, Madison, WI) for providing syt constructs.
This work was supported by a Biotechnology and Biological Sciences Research Council-Japan Partnering Award (R.M.H. and J.M.E.), a Japan Society for the Promotion of Science (JSPS), Japan-UK Bilateral Joint Project Award (K.T.), a Grant-in-Aid for JSPS Fellows (H.T.), and a Japan Society for the Promotion of Science Grant-in-Aid for Basic Research (K.T.).
Footnotes
Victor Shahin's present address is Institute of Physiology II, Westfälische Wilhelms-Universität Münster, Robert-Koch-Strasse 27b, 48149 Münster, Germany.
References
- 1.Südhof T.C. The synaptic vesicle cycle. Annu. Rev. Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 2.Chapman E.R. How does synaptotagmin trigger neurotransmitter release? Annu. Rev. Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- 3.Pang Z.P., Sun J., Südhof T.C. Genetic analysis of synaptotagmin 2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J. 2006;25:2039–2050. doi: 10.1038/sj.emboj.7601103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mace K.E., Biela L.M., Reist N.E. Synaptotagmin I stabilizes synaptic vesicles via its C2A polylysine motif. Genesis. 2009;47:337–345. doi: 10.1002/dvg.20502. [DOI] [PubMed] [Google Scholar]
- 5.Chicka M.C., Hui E., Chapman E.R. Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+ Nat. Struct. Mol. Biol. 2008;15:827–835. doi: 10.1038/nsmb.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ubach J., Zhang X., Rizo J. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J. 1998;17:3921–3930. doi: 10.1093/emboj/17.14.3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez I., Araç D., Rizo J. Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron. 2001;32:1057–1069. doi: 10.1016/s0896-6273(01)00548-7. [DOI] [PubMed] [Google Scholar]
- 8.Brose N., Petrenko A.G., Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- 9.Davletov B.A., Südhof T.C. A single C2 domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid binding. J. Biol. Chem. 1993;268:26386–26390. [PubMed] [Google Scholar]
- 10.Chapman E.R., Jahn R. Calcium-dependent interaction of the cytoplasmic region of synaptotagmin with membranes. Autonomous function of a single C2-homologous domain. J. Biol. Chem. 1994;269:5735–5741. [PubMed] [Google Scholar]
- 11.Hui E., Bai J., Chapman E.R. Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys. J. 2006;91:1767–1777. doi: 10.1529/biophysj.105.080325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiavo G., Gu Q.-M., Rothman J.E. Calcium-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc. Natl. Acad. Sci. USA. 1996;93:13327–13332. doi: 10.1073/pnas.93.23.13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai J., Tucker W.C., Chapman E.R. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- 14.Shin O.H., Xu J., Südhof T.C. Differential but convergent functions of Ca2+ binding to synaptotagmin-1 C2 domains mediate neurotransmitter release. Proc. Natl. Acad. Sci. USA. 2009;106:16469–16474. doi: 10.1073/pnas.0908798106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li C., Ullrich B., Südhof T.C. Ca2+-dependent and -independent activities of neural and non-neural synaptotagmins. Nature. 1995;375:594–599. doi: 10.1038/375594a0. [DOI] [PubMed] [Google Scholar]
- 16.Chapman E.R., Hanson P.I., Jahn R. Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J. Biol. Chem. 1995;270:23667–23671. doi: 10.1074/jbc.270.40.23667. [DOI] [PubMed] [Google Scholar]
- 17.Schiavo G., Stenbeck G., Söllner T.H. Binding of the synaptic vesicle v-SNARE, synaptotagmin, to the plasma membrane t-SNARE, SNAP-25, can explain docked vesicles at neurotoxin-treated synapses. Proc. Natl. Acad. Sci. USA. 1997;94:997–1001. doi: 10.1073/pnas.94.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rickman C., Jiménez J.L., Davletov B. Conserved prefusion protein assembly in regulated exocytosis. Mol. Biol. Cell. 2006;17:283–294. doi: 10.1091/mbc.E05-07-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X., Kim-Miller M.J., Martin T.F. Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron. 2002;34:599–611. doi: 10.1016/s0896-6273(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 20.Tucker W.C., Edwardson J.M., Chapman E.R. Identification of synaptotagmin effectors via acute inhibition of secretion from cracked PC12 cells. J. Cell Biol. 2003;162:199–209. doi: 10.1083/jcb.200302060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tucker W.C., Weber T., Chapman E.R. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science. 2004;304:435–438. doi: 10.1126/science.1097196. [DOI] [PubMed] [Google Scholar]
- 22.Bhalla A., Chicka M.C., Chapman E.R. Ca2+-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat. Struct. Mol. Biol. 2006;13:323–330. doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- 23.Damer C.K., Creutz C.E. Synergistic membrane interactions of the two C2 domains of synaptotagmin. J. Biol. Chem. 1994;269:31115–31123. [PubMed] [Google Scholar]
- 24.Chapman E.R., Davis A.F. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J. Biol. Chem. 1998;273:13995–14001. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- 25.Davis A.F., Bai J., Chapman E.R. Kinetics of synaptotagmin responses to Ca2+ and assembly with the core SNARE complex onto membranes. Neuron. 1999;24:363–376. doi: 10.1016/s0896-6273(00)80850-8. [DOI] [PubMed] [Google Scholar]
- 26.Herrick D.Z., Sterbling S., Cafiso D.S. Position of synaptotagmin I at the membrane interface: cooperative interactions of tandem C2 domains. Biochemistry. 2006;45:9668–9674. doi: 10.1021/bi060874j. [DOI] [PubMed] [Google Scholar]
- 27.Shahin V., Datta D., Edwardson J.M. Synaptotagmin perturbs the structure of phospholipid bilayers. Biochemistry. 2008;47:2143–2152. doi: 10.1021/bi701879g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Araç D., Chen X., Rizo J. Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- 29.Mackler J.M., Reist N.E. Mutations in the second C2 domain of synaptotagmin disrupt synaptic transmission at Drosophila neuromuscular junctions. J. Comp. Neurol. 2001;436:4–16. [PubMed] [Google Scholar]
- 30.Loewen C.A., Lee S.M., Reist N.E. C2B polylysine motif of synaptotagmin facilitates a Ca2+-independent stage of synaptic vesicle priming in vivo. Mol. Biol. Cell. 2006;17:5211–5226. doi: 10.1091/mbc.E06-07-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kertz J.A., Almeida P.F.F., Hinderliter A. The cooperative response of synaptotagmin I C2A. A hypothesis for a Ca2+-driven molecular hammer. Biophys. J. 2007;92:1409–1418. doi: 10.1529/biophysj.106.087197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y., He Y., Sui S.F. Visualization of synaptotagmin I oligomers assembled onto lipid monolayers. Proc. Natl. Acad. Sci. USA. 2003;100:2082–2087. doi: 10.1073/pnas.0435872100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshimura S.H., Takahashi H., Takeyasu K. Development of glutathione-coupled cantilever for the single-molecule force measurement by scanning force microscopy. FEBS Lett. 2006;580:3961–3965. doi: 10.1016/j.febslet.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 34.Kienberger F., Pastushenko V.P., Hinterdorfer P. Static and dynamic properties of single poly(ethylene glycol) molecules investigated by force spectroscopy. Single Mol. 2000;1:123–128. [Google Scholar]
- 35.Otsuka S., Iwasaka S., Yoshimura S.H. Individual binding pockets of importin-β for FG-nucleoporins have different binding properties and different sensitivities to RanGTP. Proc. Natl. Acad. Sci. USA. 2008;105:16101–16106. doi: 10.1073/pnas.0802647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawrence J.C., Saslowsky D.E., Henderson R.M. Real-time analysis of the effects of cholesterol on lipid raft behavior using atomic force microscopy. Biophys. J. 2003;84:1827–1832. doi: 10.1016/s0006-3495(03)74990-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sprong H., van der Sluijs P., van Meer G. How proteins move lipids and lipids move proteins. Nat. Rev. Mol. Cell Biol. 2001;2:504–513. doi: 10.1038/35080071. [DOI] [PubMed] [Google Scholar]
- 38.Tamm L.K., McConnell H.M. Supported phospholipid bilayers. Biophys. J. 1985;47:105–113. doi: 10.1016/S0006-3495(85)83882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kienberger F., Kada G., Hinterdorfer P. Single molecule studies of antibody-antigen interaction strength versus intra-molecular antigen stability. J. Mol. Biol. 2005;347:597–606. doi: 10.1016/j.jmb.2005.01.042. [DOI] [PubMed] [Google Scholar]
- 40.Stevens C.F., Sullivan J.M. The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron. 2003;39:299–308. doi: 10.1016/s0896-6273(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 41.Robinson I.M., Ranjan R., Schwarz T.L. Synaptotagmins I and IV promote transmitter release independently of Ca2+ binding in the C2A domain. Nature. 2002;418:336–340. doi: 10.1038/nature00915. [DOI] [PubMed] [Google Scholar]
- 42.Fernández-Chacón R., Königstorfer A., Südhof T.C. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- 43.Rhee J.-S., Li L.Y., Rosenmund C. Augmenting neurotransmitter release by enhancing the apparent Ca2+ affinity of synaptotagmin 1. Proc. Natl. Acad. Sci. USA. 2005;102:18664–18669. doi: 10.1073/pnas.0509153102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mackler J.M., Drummond J.A., Reist N.E. The C2B Ca2+-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature. 2002;418:340–344. doi: 10.1038/nature00846. [DOI] [PubMed] [Google Scholar]
- 45.Nishiki T., Augustine G.J. Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J. Neurosci. 2004;24:8542–8550. doi: 10.1523/JNEUROSCI.2545-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stein A., Radhakrishnan A., Jahn R. Synaptotagmin activates membrane fusion through a Ca2+-dependent trans interaction with phospholipids. Nat. Struct. Mol. Biol. 2007;14:904–911. doi: 10.1038/nsmb1305. [DOI] [PubMed] [Google Scholar]
- 47.Gaffaney J.D., Dunning F.M., Chapman E.R. Synaptotagmin C2B domain regulates Ca2+-triggered fusion in vitro: critical residues revealed by scanning alanine mutagenesis. J. Biol. Chem. 2008;283:31763–31775. doi: 10.1074/jbc.M803355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frazier A.A., Roller C.R., Cafiso D.S. Membrane-bound orientation and position of the synaptotagmin I C2A domain by site-directed spin labeling. Biochemistry. 2003;42:96–105. doi: 10.1021/bi0268145. [DOI] [PubMed] [Google Scholar]
- 49.Rufener E., Frazier A.A., Cafiso D.S. Membrane-bound orientation and position of the synaptotagmin C2B domain determined by site-directed spin labeling. Biochemistry. 2005;44:18–28. doi: 10.1021/bi048370d. [DOI] [PubMed] [Google Scholar]
- 50.Kuo W., Herrick D.Z., Cafiso D.S. The calcium-dependent and calcium-independent membrane binding of synaptotagmin 1: two modes of C2B binding. J. Mol. Biol. 2009;387:284–294. doi: 10.1016/j.jmb.2009.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li L.Y., Shin O.-H., Rosenmund C. Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J. Biol. Chem. 2006;281:15845–15852. doi: 10.1074/jbc.M600888200. [DOI] [PubMed] [Google Scholar]
- 52.Desai R.C., Vyas B., Chapman E.R. The C2B domain of synaptotagmin is a Ca2+-sensing module essential for exocytosis. J. Cell Biol. 2000;150:1125–1136. doi: 10.1083/jcb.150.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]