

Abstract
Loss of genomic DNA methylation has been found in a variety of common human age-related diseases. Whether DNA methylation decreases over time as individuals age is unresolved. We measured DNA methylation in 1,097 blood DNA samples from 718 elderly subjects between 55–92 years of age (1–3 samples/subjects), who have been repeatedly evaluated over an 8-year time span in the Boston area Normative Aging Study. DNA methylation was measured using quantitative PCR-Pyrosequencing analysis in Alu and LINE-1 repetitive elements, heavily methylated sequences with high representation throughout the human genome. Age at the visit was negatively associated with Alu element methylation (β=−.12 %5mC/year, p=0.0005). A weaker association was observed with LINE-1 elements (β=−.06 %5mC/year, p=0.049). We observed a significant decrease in average Alu methylation over time, with a −0.2 %5mc change (p=0.012) compared to blood samples collected up to 8 years earlier. The longitudinal decline in Alu methylation was linear and highly correlated with time since the first measurement (β=−.089 %5mC/year, p<0.0001). In contrast, average LINE-1 methylation did not vary over time [p=0.51]. Our results demonstrate a progressive loss of DNA methylation in repetitive elements dispersed throughout the genome.
INTRODUCTION
DNA methylation is a mechanism of epigenetic regulation that is heritable through cell division and, in mammals, involves the addition of methyl groups to cytosine to form 5-methyl-cytosine. In-vitro and animal models have shown genomic DNA methylation loss in association with cellular senescence and organism aging (Hoal-van Helden & van Helden, 1989; Wilson & Jones, 1983; Wilson et al., 1987). In humans, lower genomic DNA methylation, including DNA hypomethylation measured in blood DNA samples (Castro et al., 2003; Guz et al., 2008; Hsiung et al., 2007; Moore et al., 2008), has been found in a variety of age-related diseases (Fraga et al., 2005), but little information is available on methylation changes during normal aging (Bjornsson et al., 2004).
Genomic DNA hypomethylation is likely to result from demethylation in transposable repetitive elements, which plays a crucial role in gene regulation and genomic stability. More than 90% of all genomic 5-methylcytosines lies within CpG islands located in transposable repetitive elements, including the Alu and LINE-1 sequences, which are those most common and well-characterized. Measurements of Alu and LINE-1 methylation have been used to estimate genomic DNA methylation (Yang et al., 2004). The presence of 5-methylcytosine limits the ability of retro-transposons to be activated and transcribed, and Alu and LINE-1 demethylation could result in increased retro-transposon activity and propagation of aberrant methylation to other genes (Asada et al., 2006).
In the present study, we determined whether DNA methylation in Alu and LINE-1 repetitive elements: i) was associated with age of the study participants; ii) decreased over time in aging individuals. DNA methylation was measured through highly quantitative PCR-Pyrosequencing (Weisenberger et al., 2005; Yang et al., 2004) in peripheral blood DNA from 718 subjects who have been repeatedly evaluated as part of the Normative Aging Study (NAS), a longitudinal study of aging in the greater Boston area.
MATERIALS AND METHODS
Subjects
Our study population consisted of 718 white males, evaluated between January 1999 and June 2007, as part of the Normative Aging Study (NAS), a longitudinal study of aging established in 1963 by the U.S. Veterans Administration (Bell et al., 1999). The NAS participants are recalled for examination every 3–5 years and at each visit all study subjects are asked to donate a 7 ml blood sample. Of all the 718 study subjects, 339 had DNA methylation measured in only one blood sample, 321 in two blood samples, and 29 in three blood samples taken at different visits, for a total of 1,097 samples. Due to some visits that were done earlier than scheduled, and to missing blood samples from some intermediate visits, we had blood samples that were taken 2–8 years apart (average 4 years). This study was approved by the Institutional Review Boards of all participating Institutions and all participants gave written informed consent to the study.
Blood Collection
7 ml of whole blood were collected by venous phlebotomy in EDTA tubes. Blood cell count was performed on fresh blood using automated methods (Coulter Cell Counter, Abbott or Bayer) within 3 hours from the time of blood drawing. Buffy coat was extracted and stored in cell lyses solution until DNA extraction. All samples were coded and frozen at –20°C.
DNA Extraction and Bisulfite Treatment of the DNA
DNA was extracted from stored frozen buffy coat of 7 mL whole blood, using the QiAmp DNA blood kits (QIAGEN, Hilden, Germany). 500 ng DNA (concentration 50 ng/µl) was treated using EZ DNA Methylation-Gold™ Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s protocol. Final elution was performed with 30 µl of M-Elution Buffer.
Repetitive Element PCR and Pyrosequencing
Analysis of repetitive element DNA methylation was performed using previously published methods (Bollati et al., 2007; Yang et al., 2004), with minor modifications. PCR primers were designed towards a consensus Alu or LINE-1 sequence and allowed the amplification of a representative pool of repetitive elements to serve as a surrogate for global DNA methylation changes. The Alu element PCR was used for Pyrosequencing-based methylation analysis. For Alu repetitive elements, a 50 µl PCR was carried out in 25 µl of GoTaq Green Master mix (Promega, Madison, WI, USA), 1 pmol of the biotinylated forward primer (biotin-TTTTTATTAAAAATATAAAAATT), 1 pmol of the reverse primer (CCCAAACTAAAATACAATAA), 50 ng of bisulfite-treated genomic DNA and water. PCR cycling conditions were 96°C for 90 s, 43°C for 60 s and 72°C for 120 s for 40 cycles. For LINE-1 repetitive element, a 50 µl PCR was carried out in 25 µl of GoTaq Green Master mix (Promega, Madison, WI, USA), 1 pmol of the forward primer (TTTTGAGTTAGGTGTGGGATATA), 1 pmol of the biotinylated reverse primer (biotin-AAAATCAAAAAATTCCCTTTC), 50 ng of bisulfite-treated genomic DNA and water. PCR cycling conditions were 95°C for 30 s, 50°C for 30 s and 72°C for 30 s for 40 cycles.
The biotin-labelled primers were used to purify the final PCR product using Sepharose beads. The PCR product was bound to Streptavidin Sepharose HP (Amersham Biosciences, Uppsala, Sweden) and the Sepharose beads containing the immobilized PCR product were purified, washed, denatured using a 0.2 M NaOH solution, and washed again using the Pyrosequencing Vacuum Prep Tool (Pyrosequencing, Inc., Westborough, MA), as recommended by the manufacturer. Then, 0.3 µM pyrosequencing primer (AATAACTAAAATTACAAAC for Alu and AGTTAGGTGTGGGATATAGT for LINE-1) was annealed to the purified single-stranded PCR product and pyrosequencing was performed using the PSQ HS 96 Pyrosequencing System (Pyrosequencing, Inc.). The degree of methylation was expressed for both Alu and LINE-1 as percentage of methylated cytosines divided by the sum of methylated and unmethylated cytosines (%5mC). We used built-in controls to verify bisulfite conversion efficiency. Every sample was tested three times for each marker to confirm reproducibility and increase precision of our results. The average of the three replicates was used in statistical analyses.
Statistical Analysis
We first evaluated the association between age at visit and DNA methylation in Alu and LINE-1 repetitive elements. Because our study included repeated measures of DNA methylation for many participants, we fitted a mixed effects model (PROC MIXED in SAS V9.0) assuming:
| [1] |
where Yit is the level of DNA methylation in either LINE-1 or Alu in subject i at time t, bo is the overall intercept, and ui is the separate random intercept for subject i. In the above X1it—Xpit are the covariates, which were included only in multivariable models. These models capture the correlation among measurements within the same subject. In multivariable models, we adjusted for the determinants of either LINE-1 or Alu methylation that were identified in exploratory univariate analyses, i.e., body mass index, systolic and diastolic blood pressure, fasting blood glucose, percent lymphocytes and neutrophils in blood count, statin use, and season of the visit, as previously reported. (Baccarelli et al., 2008, in press)
In the above model, the coefficient of age reflects both the longitudinal change in each subject over time, as well as the cross-sectional difference between subjects, based on their age. If DNA methylation is associated with survival, the latter coefficient is likely to underestimate the true rate of change with time. Differences in DNA methylation in samples collected for the same subjects over time were tested using paired t-test. Thanks to the longitudinal design of our study, we were able to estimate both associations simultaneously in a second model by fitting mixed-models which included both: i) age at the first visit (baseline); and ii) years elapsed since the first visit:
| [2] |
Where βaBaseline fits age at first visit to estimate the cross-sectional effect of age and βbYearsit fits years elapsed since the first visit to estimate the longitudinal effect of age. This model allows discrimination of the cross-sectional from the longitudinal effects of aging, at the cost of some power (since two variables are used to represent age).
For all models, we show regression coefficients (β) and their standard errors representing the change in DNA methylation for each year of age. As a sensitivity analysis, we performed a second complete set of analyses after excluding the 29 DNA methylation measurements obtained on blood samples collected during a third visit on the same study subject. Such exclusion produced only minor changes to the results (data not shown). Also we repeated all analysis by adjusting for smoking (never, former, current) and obtained results that are similar to those shown throughout the paper.
All tests were two-tailed. P<0.05 were considered statistically significant. All analyses were performed in SAS 9.1 (SAS Institute Inc., Cary, NC).
RESULTS
The characteristics of the study population are shown in Table 1. The study subjects were elderly male individuals between 55 and 92 of age (mean age 73.2 years, SD=6.6). Blood DNA methylation levels, expressed as %5mC (percentage of cytosines that are methylated), were equal to 77.2 (SD=2.2) for LINE-1 and 26.2 (SD=1.2) for Alu. DNA methylation ranged from 66.2 to 84.6 for LINE-1 elements and from 22.5 to 32.4 for Alu elements.
Table 1.
Characteristics of the study population in the Normative Aging Study [mean ± SD or n (%)]. This table includes data collected at each visit for all measurements on all subjects
| Variable | All Visits, n=1,097 | |
|---|---|---|
| Age, years (SD) | 73.2 | (6.6) |
| Body mass index, Kg/m2 (SD) | 28.2 | (4.1) |
| Smoking status, n (%) | ||
| Never smoker | 338 | (30.8) |
| Former smoker | 45 | (4.1) |
| Current smoker | 714 | (65.1) |
| Pack years of smoking,* pack × year (SD) | 30.8 | (26.7) |
| Fasting blood glucose, mg/dL (SD) | 108.5 | (26.6) |
| Diabetes, n (%) | 215 | (19.6) |
| Treatment with statins, n (%) | 434 | (39.6) |
| Systolic Blood Pressure, mmHg (SD) | 130.3 | (17.4) |
| Dyastolic Blood Pressure, mmHg (SD) | 75.5 | (9.8) |
| Blood count, | ||
| White blood cells, cells/mm3 (SD) | 6,638 | (2,955) |
| Neutrophils, % | 62.2 | (8.7) |
| Lymphocytes, % | 25.5 | (8.0) |
| Monocytes, % | 8.6 | (2.1) |
| Basophils, % | 0.6 | (0.5) |
| Eosinophils, % | 3.3 | (2.1) |
| Season of the visit, n (%) | ||
| Spring | 286 | (26.1) |
| Summer | 300 | (27.4) |
| Fall | 319 | (29.1) |
| Winter | 192 | (17.5) |
| DNA methylation, %5mC† (SD) | ||
| LINE-1 | 77.2 | (2.2) |
| Alu | 26.2 | (1.2) |
Mean and SD among ever smokers
Percentage of methylated cytosine
Association between Age at the Visit and Methylation (Cross-sectional Association)
DNA methylation in both Alu and LINE-1 elements was negatively associated with age at the visit (Figure 1). In unadjusted models, DNA methylation decreased significantly in Alu elements (β=−.023 %5mC/year, p<0.0001), while the decrease in LINE-1 elements was not statistically significant (β=−.015 %5mC/year, p=0.10).
Figure 1.
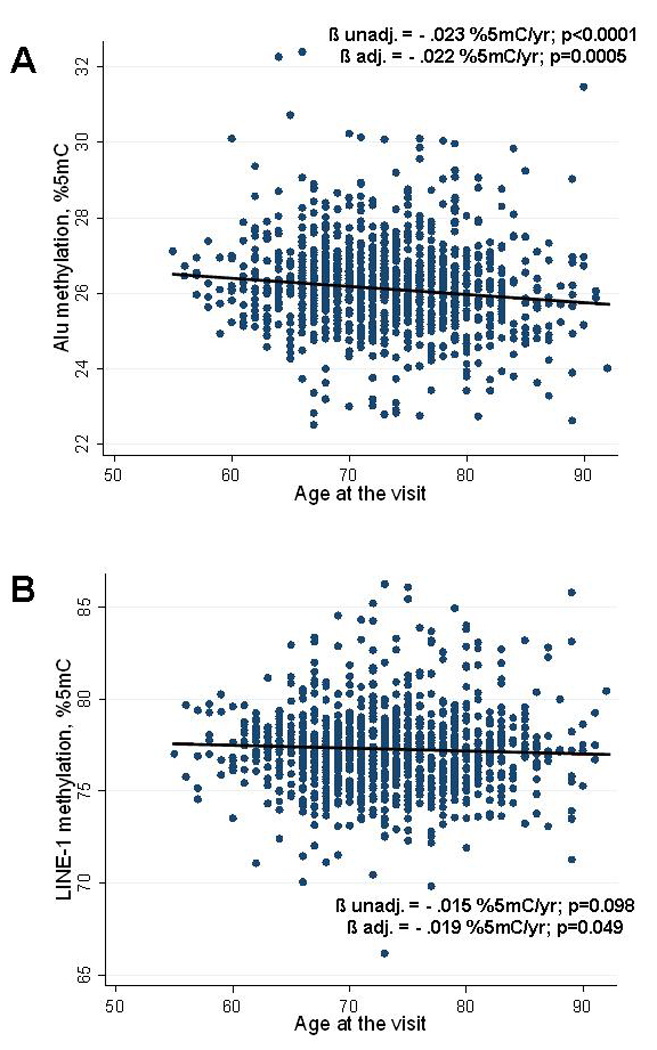
Association between Age and Methylation in Alu (A) and LINE-1 (B) repetitive elements. The graphs show the decrease of methylation (change in % 5Methyl-cytosine [%5mC]/year) in relation to the subject’s age at time of visit. Regression lines and data points represent the unadjusted values. Adjusted and unadjusted Regression coefficients (β) and p-values shown are obtained from unadjusted models and from models adjusted for body mass index, systolic and diastolic blood pressure, fasting blood glucose, percent lymphocytes and neutrophils in blood count, statin use, season of the visit.
In multivariable models adjusting for season, body mass index, systolic and diastolic blood pressure, statin use, fasting blood glucose, percent lymphocytes and neutrophils in differential blood count, DNA methylation decreased significantly both in Alu (β=−.022 %5mC/year, p=0.0005) and LINE-1 elements (β=−.019 %5mC/year, p=0.049).
Correlation between Measurements of Alu and LINE-1 Methylation Taken at Different Visits
DNA methylation was measured at least twice in 339 subjects. The time between the two first measurements varied between 2 and 8 years (mean=4.0 years, SD=1.4). The correlation between the first and the second measurement was moderate (r=.44, p<0.0001 for Alu; r=.55, p<0.0001, for LINE-1). (Figure 2)
Figure 2.
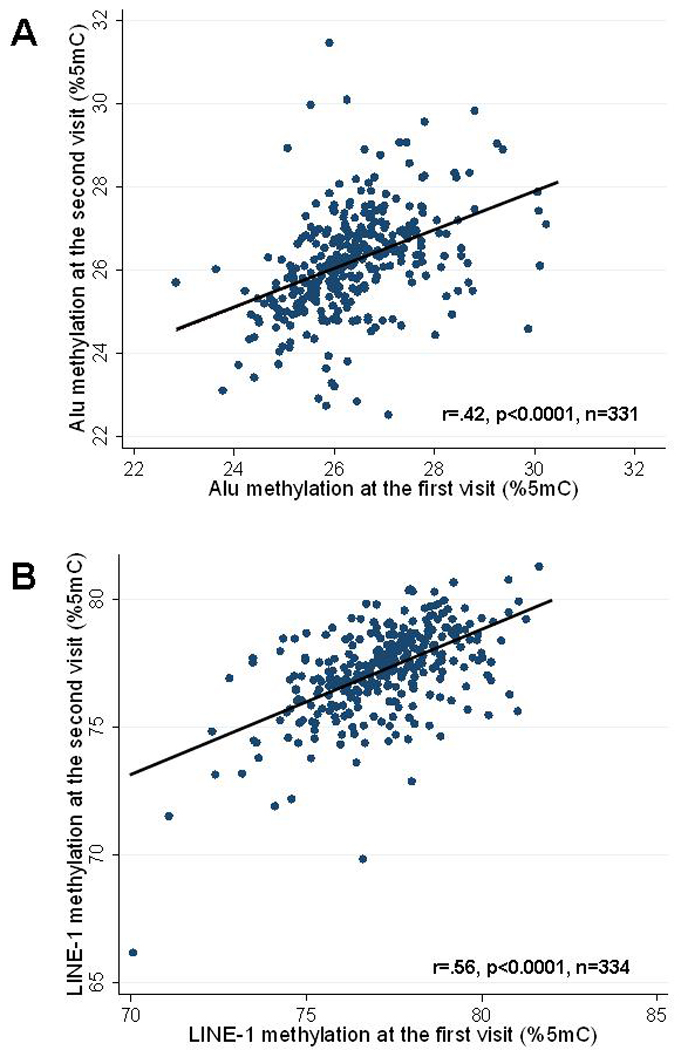
Correlation between the first and the second measurement of Alu (A) and LINE-1 (B) repetitive element methylation taken 2–8 years apart on the same subjects.
In the second set of measurements, the greatest losses in DNA methylation compared with the first set of measurements were −5.3 %5mC for Alu elements and −5.41 %5mC for LINE-1 elements, whereas the greatest gains were +5.6 %5mC for Alu elements and +4.2%5mC for LINE-1 elements. The decreases observed in Alu elements were >1 %5mC in 60 subjects (18%), >2 %5mC in 25 subjects (7%) and >3 %5mC in 8 subjects (2.4%), whereas the increases observed were >1 %5mC in 38 subjects (11%), >2 %5mC in 6 subjects (1.8%) and > 3 %5mC in 3 subjects (0.9%), hence showing a tendency towards a methylation loss in the second set of measurements.
LINE-1 elements showed a decrease in methylation as an increase, with 138 subjects (41%) showing a change of at least 1 %5mC in either directions (67 [20%] and 71 [21%] subjects showing a decrease and an increase, respectively), 64 subjects (19%) showing a change of at least 2 %5mC (34 [10%] and 30 [9%] subjects showing a decrease and an increase, respectively), and 21 subjects (6%) showing a change of 3 %5mC or more (11 [3%] and 10 [3%] subjects showing a decrease and an increase, respectively).
Changes in Alu and LINE-1 Methylation over Time (Longitudinal Association)
Among the 339 individuals who had DNA methylation measured at least twice, average Alu methylation in the second measurement (26.2 %5mc, 95% CI 26.0–26.3) was significantly decreased, compared to the first measurement (26.4 %5mc, 95% CI 26.2–26.5) taken in average 4 years earlier (p=0.012). In contrast, average LINE-1 methylation in the second sample (77.4 %5mc, 95% CI 77.2–77.5) was similar to that found in the first measurement (77.3 %5mc, 95% CI 77.1–77.5) [p=0.94].
Only twenty-nine subjects had DNA methylation measured three times. Among these subjects, Alu methylation showed a progressive decrease over time, with average levels of 26.5 (95% CI 26.1–26.9) in the first measurement, 26.3 (95% CI 25.9–26.7) in the second measurement, and 24.7 (95% CI 24.2–25.1) in the third measurement (p<0.001, test for trend across measurements). Again, average LINE-1 methylation did not show significant changes over time (79.0 %5mc, 95% CI 78.0–80.0 in the first measurement; 79.1 %5mc, 95% CI 78.7–79.5 in the second measurement; 79.2 %5mc, 95% CI 78.2–80.2 in the third measurement; p=0.82 for trend across measurements).
Discrimination of Age and Time effects on DNA Methylation
If a marker changes gradually over time as individuals age, that marker will also be associated with age at a given time in a cross-sectional analysis. However, the cross-sectional association may also be influenced by selection of the study participants by survival, as values for older subjects will represent a selected sample, including only those who survived up until that age.(Munoz & Gange, 1998) Taking advantage of the availability of measures of DNA methylation on a large sample of individuals that included a subset of measures repeated over time, we used a second set of statistical models (see statistical methods for details on modelling) that differentiated the cross-sectional effect of age (i.e, baseline effect) from the longitudinal effect (i.e, effect of years elapsed since the first visit) [Table 2]. In this model, the estimates for cross-sectional and longitudinal effects would be similar if the association between age and methylation were exclusively due to decreases occurring in the study subjects over time.
Table 2.
Discrimination of the effects of age (baseline) and time (years elapsed since the first visit) on DNA methylation of Alu and LINE-1 repetitive elements (change in %5Methyl-cytosine).
| Alu | LINE-1 | |||||
|---|---|---|---|---|---|---|
| β* | Standard Error |
p-value | β* | Standard Error |
p-value | |
| Unadjusted | ||||||
| Effect of age at baseline | −.016 | .006 | 0.007 | −.017 | .009 | 0.07 |
| Effect of years since first visits | −.082 | .015 | <0.0001 | −.000 | .024 | 0.97 |
| Adjusted† | ||||||
| Effect of age at baseline | −.016 | .006 | 0.013 | −.014 | .010 | 0.14 |
| Effect of years since first visits | −.089 | .017 | <0.0001 | −.018 | .027 | 0.51 |
Regression coefficient representing the change (% 5Methyl-cytosine/year) in ALU or LINE-1 repetitive element methylation.
Adjusted for determinants of Alu and LINE-1 methylation, including body mass index, systolic and diastolic blood pressure, fasting blood glucose, percent lymphocytes and neutrophils in blood count, statin
When our model was fit to Alu methylation, the estimate for longitudinal changes showed a stronger effect than the cross-sectional estimate (Table 2). In particular, we estimated an unadjusted −.082 %5mC decline per each year since the first examination (longitudinal effect) and a −.016 %5mC decrease per a one-year increase in baseline age in the model (cross-sectional effect). Although both effects were statistically significant (p<0.0001 for the longitudinal effect, p=0.007 for the cross-sectional effect) the longitudinal effect was approximately five-fold larger than the cross-sectional. The results were confirmed in analysis adjusted for season, body mass index, systolic and diastolic blood pressure, statins use, fasting blood glucose, percent lymphocytes and neutrophils in differential blood count (Table 2)
When fit to LINE-1 methylation, our model was not able to identify any significant association with either cross-sectional or longitudinal effects (Table 2) possibly due to insufficient statistical power.
As a sensitivity analysis, we tested for non-linear effects in a set of models in which the cross-sectional and longitudinal effects of age were fitted using non-linear terms, i.e, penalized splines with varying degrees of freedom (from 1 to 10). Our results showed that the linear models had a better fit for both the cross-sectional and longitudinal effects of age compared to any of the models with penalized splines, suggesting that the decreases in DNA methylation associated with the cross-sectional and longitudinal effects of age were linear.
DISCUSSION
Our results on an elderly population in Boston showed a gradual decrease through aging in repetitive element DNA methylation, particularly in Alu sequences. We showed that Alu element methylation was associated with age both cross-sectionally and longitudinally in repeated measures taken up to 8 years apart. In contrast, average LINE-1 methylation did not vary over time.
Genomic 5-methyl-cytosine content has been shown to decrease with age in most vertebrate tissues (Mays-Hoopes et al., 1986; Rath & Kanungo, 1989; Romanov & Vanyushin, 1981; Vanyushin et al., 1970; Vanyushin et al., 1973b; Wilson et al., 1987) and in in-vitro models (Wilson & Jones, 1983), and has been associated cross-sectionally with age in human tissues (Drinkwater et al., 1989; Fraga et al., 2005; Fuke et al., 2004; Golbus et al., 1990; Vanyushin et al., 1970; Vanyushin et al., 1973a; Vanyushin et al., 1973b; Wilson & Jones, 1983; Wilson et al., 1987). However, data from cross-sectional studies on the association of age with Alu and LINE-1 methylation (Asada et al., 2006; Bariol et al., 2003; Chalitchagorn et al., 2004; Cho et al., 2007; Iacopetta et al., 2007; Roman-Gomez et al., 2005), which have been largely used to estimate global methylation (Yang et al., 2004), are conflicting. While several works have evaluated LINE-1 and Alu element DNA methylation in relation with age in cancer tissues (Cho et al., 2007; Roman-Gomez et al., 2005; Tangkijvanich et al., 2007), just a few studies analyzed the association with age in normal tissues. Chalitchagorn et al. (Chalitchagorn et al., 2004) observed that methylation of LINE-1 measured with combined bisulfite restriction analysis PCR in normal leukocytes was independent of age. However, the number of subjects (16 women and 16 men) was likely too small to have sufficient power to detect a significant correlation between age and LINE-1 methylation. Iacopetta et al. (Iacopetta et al., 2007) showed that older age was associated with a borderline significant trend (p=.069) for lower levels of LINE-1 methylation in normal colonic mucosa, confirming a previous report in which the level of demethylation in the normal colonic mucosa, measured by analyzing the methyl-accepting capacity of DNA, correlated with advancing age of the individual (Bariol et al., 2003).
The human genome comprises approximately 1.4 million copies of Alu repetitive elements (Lander et al., 2001; Yang et al., 2004), and repetitive element methylation has been shown to correlate with total genomic methylation content (Weisenberger et al., 2005; Yang et al., 2004). Our findings suggest that genome-wide losses of methylation through aging may account, at least in part, for the increased rates of common diseases through aging (Bjornsson et al., 2004; Jiang et al., 2004; Petronis, 2001). Although future follow-up studies will be needed to determine the impact of methylation losses on disease incidence and survival, our analysis indirectly showed that lower Alu element methylation in blood DNA may predict reduced life-expectancy. Using statistical modelling of our data, we were able to separate the cross-sectional from the longitudinal decline in Alu element methylation, and found a considerably larger longitudinal decline. If the association between age and methylation were only due to decreases occurring over time as the individuals age, the estimates for cross-sectional and longitudinal effects in this model would be similar. This difference suggests that Alu methylation was associated with censoring from the cohort over time, which in the Normative Aging Study cohort was predominantly from mortality or disabling illness.
A recent article published in JAMA (Bjornsson et al., 2008) showed that, among 111 individuals between the ages of 70 and 82 years, 63% of the participants had a 0.05% or greater absolute change in the percentage of global genomic methylation compared to DNA methylation in blood samples taken approximately 10 years earlier. In that study, roughly the same number of participants showed a decrease in methylation as an increase, with a population average of DNA methylation substantially unchanged over time.
When compared with the recent results by Bjornsson and collaborators (Bjornsson et al., 2008), who found no decrease over time in DNA methylation as measured by the LUMA assay, our results indicate that age-related losses of DNA methylation are limited to specific components, albeit vastly represented, of the human genome. In our results, we also found that aging was more strongly associated with decreases in Alu methylation, while results for LINE-1 methylation were less consistent. In experimental investigations, LINE-1 methylation was shown to be regulated by mechanisms that are different from those controlling Alu elements (Ferguson-Smith & Surani, 2001; Jones & Baylin, 2002). However, the identification of the mechanisms that appear to make Alu elements more sensitive to age-related changes need to be identified. The LUMA assay measures specific methylation sequences (CCGG) that are found in gene promoters and may control gene expression. Alu element methylation has been attributed roles beyond gene regulation, including stabilization of chromosomal structures, as well as control of retrotransposon sequences and mutational event rates (Barbot et al., 2002; Mays-Hoopes et al., 1986; Ono et al., 1989; Wilson et al., 2007). Alternatively, the larger sample size of our population might have provided our study with added statistical power to detect the effects of aging on DNA methylation.
The finding of a negative association –albeit borderline significant – between LINE-1 and age in our study, with no change of LINE-1 over time, may also have resulted from greater statistical power to test the association of DNA methylation with age at the visit, as this analysis was based on more than 700 subjects with nearly 1,100 samples, whereas only approximately 350 subjects had repeated measures over time.
In our data, the decrease in DNA methylation of Alu element over time was relatively small, particularly if compared with the wide between-subject variability in DNA methylation, as well as with the changes in either direction that we observed in the same subjects over time. Our findings, along with the large variation in the degree of methylation among individuals of comparable ages that has also been observed in previous investigations (Bjornsson et al., 2008), suggest that risk factors for common diseases other than aging, such as dietary, lifestyle, environmental, and genetic factors, may contribute to the variability in DNA methylation and define individual phenotypes (Issa, 2002; Yuasa, 2002). However, at least part of the changes in DNA methylation we observed over time may have resulted from assay variability or other random variation.
In our study, we used Pyrosequencing methodology, a quantitative analysis of DNA methylation which is highly reproducible and accurate at measuring DNA methylation. In addition, we repeated DNA methylation analysis three times on each sample and used the replicate average to minimize the assay variability. Even if in the present study unfractionated peripheral blood was used as a source of DNA, all the analysis were adjusted for percent lymphocytes and neutrophils in blood count to minimize the effect on methylation caused by potential shifts in blood cell subpopulations during aging. Although we had a unique opportunity to evaluate DNA methylation changes in repeated measurements taken over time in an elderly population, the number of years covered after the first measurement was limited (<=8 years). As our results can be generalized only to an aged population that consists of older males who are almost all white, the age-related changes in women and younger populations, as well as different ethnic groups, should be addressed in future studies.
In conclusion, our results from a longitudinal study of elderly individuals demonstrated that repetitive element methylation decreases through aging, particularly in Alu element. Our data provide new insights into the molecular events related to biological aging and features in complex diseases that are not accounted for by DNA sequence-based genetics (Petronis, 2001).
ACKNOWLEDGEMENTS
This work was supported by the National Institute of Environmental Health Sciences (NIEHS) grants ES015172-01, ES00002; the Environmental Protection Agency (EPA) grants EPA R83241601 and R827353; and the CARIPLO Foundation grant 2007-5469. The VA Normative Aging Study, a component of the Massachusetts Veterans Epidemiology Research and Information Center, Boston, Massachusetts, is supported by the Cooperative Studies Program/Epidemiology Research and Information Center of the U.S. Department of Veterans Affairs.
REFERENCES
- 1.Asada K, Kotake Y, Asada R, Saunders D, Broyles RH, Towner RA, Fukui H, Floyd RA. LINE-1 Hypomethylation in a Choline-Deficiency-Induced Liver Cancer in Rats: Dependence on Feeding Period. J Biomed Biotechnol. 2006;2006:17142. doi: 10.1155/JBB/2006/17142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua A, Suh H, Zanobetti A, Sparrow D, Vokonas PS, Schwartz J. Rapid DNA Methylation Changes after Exposure to Traffic Particles AJRCCM. 2008 doi: 10.1164/rccm.200807-1097OC. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbot W, Dupressoir A, Lazar V, Heidmann T. Epigenetic regulation of an IAP retrotransposon in the aging mouse: progressive demethylation and de-silencing of the element by its repetitive induction. Nucleic Acids Res. 2002;30:2365–2373. doi: 10.1093/nar/30.11.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bariol C, Suter C, Cheong K, Ku SL, Meagher A, Hawkins N, Ward R. The relationship between hypomethylation and CpG island methylation in colorectal neoplasia. Am J Pathol. 2003;162:1361–1371. doi: 10.1016/S0002-9440(10)63932-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell AC, West AG, Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999;98:387–396. doi: 10.1016/s0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- 6.Bjornsson HT, Fallin MD, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004;20:350–358. doi: 10.1016/j.tig.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 7.Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekstrom TJ, Harris TB, Launer LJ, Eiriksdottir G, Leppert MF, Sapienza C, Gudnason V, Feinberg AP. Intra-individual change over time in DNA methylation with familial clustering. Jama. 2008;299:2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, Bertazzi PA, Yang AS. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67:876–880. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- 9.Castro R, Rivera I, Struys EA, Jansen EE, Ravasco P, Camilo ME, Blom HJ, Jakobs C, Tavares de Almeida I. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clin Chem. 2003;49:1292–1296. doi: 10.1373/49.8.1292. [DOI] [PubMed] [Google Scholar]
- 10.Chalitchagorn K, Shuangshoti S, Hourpai N, Kongruttanachok N, Tangkijvanich P, Thong-ngam D, Voravud N, Sriuranpong V, Mutirangura A. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene. 2004;23:8841–8846. doi: 10.1038/sj.onc.1208137. [DOI] [PubMed] [Google Scholar]
- 11.Cho NY, Kim BH, Choi M, Yoo EJ, Moon KC, Cho YM, Kim D, Kang GH. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J Pathol. 2007;211:269–277. doi: 10.1002/path.2106. [DOI] [PubMed] [Google Scholar]
- 12.Drinkwater RD, Blake TJ, Morley AA, Turner DR. Human lymphocytes aged in vivo have reduced levels of methylation in transcriptionally active and inactive DNA. Mutat Res. 1989;219:29–37. doi: 10.1016/0921-8734(89)90038-6. [DOI] [PubMed] [Google Scholar]
- 13.Ferguson-Smith AC, Surani MA. Imprinting and the epigenetic asymmetry between parental genomes. Science. 2001;293:1086–1089. doi: 10.1126/science.1064020. [DOI] [PubMed] [Google Scholar]
- 14.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuke C, Shimabukuro M, Petronis A, Sugimoto J, Oda T, Miura K, Miyazaki T, Ogura C, Okazaki Y, Jinno Y. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: an HPLC-based study. Ann Hum Genet. 2004;68:196–204. doi: 10.1046/j.1529-8817.2004.00081.x. [DOI] [PubMed] [Google Scholar]
- 16.Golbus J, Palella TD, Richardson BC. Quantitative changes in T cell DNA methylation occur during differentiation and ageing. Eur J Immunol. 1990;20:1869–1872. doi: 10.1002/eji.1830200836. [DOI] [PubMed] [Google Scholar]
- 17.Guz J, Foksinski M, Siomek A, Gackowski D, Rozalski R, Dziaman T, Szpila A, Olinski R. The relationship between 8-oxo-7,8-dihydro-2'-deoxyguanosine level and extent of cytosine methylation in leukocytes DNA of healthy subjects and in patients with colon adenomas and carcinomas. Mutat Res. 2008;640:170–173. doi: 10.1016/j.mrfmmm.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 18.Hoal-van Helden EG, van Helden PD. Age-related methylation changes in DNA may reflect the proliferative potential of organs. Mutat Res. 1989;219:263–266. doi: 10.1016/0921-8734(89)90027-1. [DOI] [PubMed] [Google Scholar]
- 19.Hsiung DT, Marsit CJ, Houseman EA, Eddy K, Furniss CS, McClean MD, Kelsey KT. Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2007;16:108–114. doi: 10.1158/1055-9965.EPI-06-0636. [DOI] [PubMed] [Google Scholar]
- 20.Iacopetta B, Grieu F, Phillips M, Ruszkiewicz A, Moore J, Minamoto T, Kawakami K. Methylation levels of LINE-1 repeats and CpG island loci are inversely related in normal colonic mucosa. Cancer Sci. 2007;98:1454–1460. doi: 10.1111/j.1349-7006.2007.00548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Issa JP. Epigenetic variation and human disease. J Nutr. 2002;132:2388S–2392S. doi: 10.1093/jn/132.8.2388S. [DOI] [PubMed] [Google Scholar]
- 22.Jiang YH, Bressler J, Beaudet AL. Epigenetics and human disease. Annu Rev Genomics Hum Genet. 2004;5:479–510. doi: 10.1146/annurev.genom.5.061903.180014. [DOI] [PubMed] [Google Scholar]
- 23.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 24.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 25.Mays-Hoopes L, Chao W, Butcher HC, Huang RC. Decreased methylation of the major mouse long interspersed repeated DNA during aging and in myeloma cells. Dev Genet. 1986;7:65–73. doi: 10.1002/dvg.1020070202. [DOI] [PubMed] [Google Scholar]
- 26.Moore LE, Pfeiffer RM, Poscablo C, Real FX, Kogevinas M, Silverman D, Garcia-Closas R, Chanock S, Tardon A, Serra C, Carrato A, Dosemeci M, Garcia-Closas M, Esteller M, Fraga M, Rothman N, Malats N. Genomic DNA hypomethylation as a biomarker for bladder cancer susceptibility in the Spanish Bladder Cancer Study: a case-control study. Lancet Oncol. 2008;9:359–366. doi: 10.1016/S1470-2045(08)70038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munoz A, Gange SJ. Methodological issues for biomarkers and intermediate outcomes in cohort studies. Epidemiol Rev. 1998;20:29–42. doi: 10.1093/oxfordjournals.epirev.a017970. [DOI] [PubMed] [Google Scholar]
- 28.Ono T, Takahashi N, Okada S. Age-associated changes in DNA methylation and mRNA level of the c-myc gene in spleen and liver of mice. Mutat Res. 1989;219:39–50. doi: 10.1016/0921-8734(89)90039-8. [DOI] [PubMed] [Google Scholar]
- 29.Petronis A. Human morbid genetics revisited: relevance of epigenetics. Trends Genet. 2001;17:142–146. doi: 10.1016/s0168-9525(00)02213-7. [DOI] [PubMed] [Google Scholar]
- 30.Rath PC, Kanungo MS. Methylation of repetitive DNA sequences in the brain during aging of the rat. FEBS Lett. 1989;244:193–198. doi: 10.1016/0014-5793(89)81191-3. [DOI] [PubMed] [Google Scholar]
- 31.Roman-Gomez J, Jimenez-Velasco A, Agirre X, Cervantes F, Sanchez J, Garate L, Barrios M, Castillejo JA, Navarro G, Colomer D, Prosper F, Heiniger A, Torres A. Promoter hypomethylation of the LINE-1 retrotransposable elements activates sense/antisense transcription and marks the progression of chronic myeloid leukemia. Oncogene. 2005;24:7213–7223. doi: 10.1038/sj.onc.1208866. [DOI] [PubMed] [Google Scholar]
- 32.Romanov GA, Vanyushin BF. Methylation of reiterated sequences in mammalian DNAs. Effects of the tissue type, age, malignancy and hormonal induction. Biochim Biophys Acta. 1981;653:204–218. doi: 10.1016/0005-2787(81)90156-8. [DOI] [PubMed] [Google Scholar]
- 33.Tangkijvanich P, Hourpai N, Rattanatanyong P, Wisedopas N, Mahachai V, Mutirangura A. Serum LINE-1 hypomethylation as a potential prognostic marker for hepatocellular carcinoma. Clin Chim Acta. 2007;379:127–133. doi: 10.1016/j.cca.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 34.Vanyushin BF, Tkacheva SG, Belozersky AN. Rare bases in animal DNA. Nature. 1970;225:948–949. doi: 10.1038/225948a0. [DOI] [PubMed] [Google Scholar]
- 35.Vanyushin BF, Mazin AL, Vasilyev VK, Belozersky AN. The content of 5-methylcytosine in animal DNA: the species and tissue specificity. Biochim Biophys Acta. 1973a;299:397–403. doi: 10.1016/0005-2787(73)90264-5. [DOI] [PubMed] [Google Scholar]
- 36.Vanyushin BF, Nemirovsky LE, Klimenko VV, Vasiliev VK, Belozersky AN. The 5-methylcytosine in DNA of rats. Tissue and age specificity and the changes induced by hydrocortisone and other agents. Gerontologia. 1973b;19:138–152. [PubMed] [Google Scholar]
- 37.Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, Fiala E, Ehrlich M, Laird PW. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005;33:6823–6836. doi: 10.1093/nar/gki987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775:138–162. doi: 10.1016/j.bbcan.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 39.Wilson VL, Jones PA. DNA methylation decreases in aging but not in immortal cells. Science. 1983;220:1055–1057. doi: 10.1126/science.6844925. [DOI] [PubMed] [Google Scholar]
- 40.Wilson VL, Smith RA, Ma S, Cutler RG. Genomic 5-methyldeoxycytidine decreases with age. J Biol Chem. 1987;262:9948–9951. [PubMed] [Google Scholar]
- 41.Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuasa Y. DNA methylation in cancer and ageing. Mech Ageing Dev. 2002;123:1649–1654. doi: 10.1016/s0047-6374(02)00100-8. [DOI] [PubMed] [Google Scholar]