

Abstract
Tissue-resident mast cells (MCs) are important in allergic diseases. In a mouse model of allergic airways inflammation, an increase in peribronchiolar MCs was associated with increased concentrations of the chemokine CCL2 in lung lavage. MC progenitors (MCps) arising in bone marrow (BM) are recruited to tissues by transendothelial migration, and we found that CCL2 is chemotactic for MCps in freshly isolated BM in vitro. Immature, but not mature, BM-derived MCs migrated in response to CCL2 when cultured in IL-3+stem cell factor (SCF) but not when cultured in IL-3 alone. However, the cells under both culture conditions expressed mRNA for CCR2, the receptor for CCL2, and bound the radiolabeled chemokine with similar affinities, highlighting SCF as a key mediator in coupling CCR2 to downstream events, culminating in chemotaxis. Immature BM-derived MCs from IL-3 +SCF cultures, when administered i.v., accumulated at skin sites injected with CCL2 in vivo. MCp recruitment to the allergen-sensitized/challenged lung was significantly reduced in CCR2−/− and CCL2−/− mouse strains. However, reconstitution studies of sublethally irradiated and BM-reconstituted mice indicated that BM cells and stromal elements could provide CCL2, whereas the CCR2 function resided with stromal elements rather than BM cells. These experiments revealed a new function of SCF in chemokine receptor coupling, but they suggest a complex role of the CCL2/CCR2 axis in recruiting MCps during pulmonary inflammation.
Mast cells (MCs) are long-lived tissue-resident cells that have important functions in several pathophysiological systems, including allergic reactions, responses to helminth and bacterial infection, atherosclerosis, angiogenesis, and tissue repair (1). In common with other leukocytes, MCs are derived from hematopoietic stem cells in the bone marrow (BM) (2–5). However, MCs do not mature in the BM and are released into the circulation as committed MC progenitors (MCps) (5, 6). Circulating progenitors accumulate in different tissues of the body and mature under the influence of locally produced factors that determine the final phenotype of the MCs (7, 8).
The importance of MCs in host defense to helminths and in allergic reactions is emphasized by the their increased numbers in the affected tissues (9). Patients with allergic rhinitis have increased MC numbers in the nasal mucosa (10–12), and patients with asthma have increased MC numbers in airway smooth muscle, mucus glands, and epithelium (13, 14). An increase in circulating MCps is observed in asthmatic patients compared with normal subjects (15). Furthermore, in models of Ag-induced Th2-mediated pulmonary inflammation in mice, there is a marked increase in airway MCs, although the lungs of normal laboratory-bred mice have few MCs (16, 17) or their progenitors (18, 19).
The mechanisms underlying the population of tissues with MCs are not well understood, but they involve recruitment of MCps by adhesion to the microvascular endothelium and migration and further expansion in the tissue during maturation by cytokine-dependent proliferation. Mice deficient in β7 integrins on MCps or wild-type (WT) mice subjected to mAb blockade of α4 and β7 or the counterligands, VCAM-1 and mucosal addressin cell adhesion molecule-1, have impaired homing of MCps to the small intestine and have no detectable mature MCs at this site (18–20). Naive mice deficient in the chemokine receptor CXCR2 also have reduced numbers of intestinal MCps, suggesting that chemokines, such as CXCL1 (KC) and CXCL2 (MIP-2), are involved in trafficking, whereas there was no reduction in the numbers of intestinal MCps in CCR2−/−, CCR3−/−, or CCR5−/− mice (18). In sensitized and Ag-challenged mice, MCp recruitment to the lung is dependent on the expression of α4-integrins on the MCp and the expression of VCAM-1 and CXCR2 on the lung endothelium (21, 22). The diminished numbers of recruited lung MCps in CXCR2-deficient mice were associated with diminished numbers of mature intraepithelial MCs 1 wk later (22).
We showed that mouse BM-derived MCps respond chemotactically to leukotriene B4 (LTB4), acting through the high-affinity BLT1R, but this receptor is lost during cell maturation (23). Further, we showed that immature and mature mouse BM-derived MCs (BMMCs) respond chemotactically to PGE2 (24). In these studies, the BMMCs were cultured with IL-3 alone. Stem cell factor (SCF) was used as an active reference chemoattractant.
In this study, we found that an increase in peribronchial MCs in allergen-sensitized and aerosol-challenged mice was associated with an increase in bronchoalveolar lavage (BAL) CCL2. Further, CCL2 was highly chemotactic for MCps in freshly isolated BMs in vitro. BMMCs cultured with IL-3+SCF also responded chemotactically to CCL2 in vitro, an effect that was lost during further maturation. Interestingly, cells cultured in IL-3 alone did not respond chemotactically to CCL2, but they had similar levels of CCR2 mRNA to cells cultured in IL-3+SCF. Further, cells cultured in IL-3 alone or in IL-3+SCF bound radiolabeled CCL2 similarly, demonstrating that SCF is important in coupling the receptor to transduction pathways mediating chemotaxis. In vivo, we showed that the CCR2/CCL2 axis is central to Ag-induced recruitment of MCps to the lung, with the participation of stromal and BM elements. However, experiments with sublethal irradiation and BM reconstitution in vivo indicated that the donated BMs could provide CCL2, whereas CCR2 on a radioresistant cell or cells in the stroma is important for MCp trafficking in the allergic airways model used.
Materials and Methods
Reagents
All tissue culture reagents were purchased from Invitrogen (Paisley, U.K.). Tyrode’s buffer, DNP IgE, methacholine, and OVA were from Sigma-Aldrich (Poole, U.K.). Anti-mouse CD117 and anti-mouse Gr-1 were from BD Biosciences (Oxford, U.K.). CellTracker Green 5-chloromethylfluorescein diacetate (CMFDA) was from Invitrogen. Microbeads were from Miltenyi Biotec (Bisley, U.K.). TaqMan Universal PCR Master Mix, CCR2, GAPDH, and 18S-specific primers were from Applied Biosystems (Foster City, CA).
Mice
Female BALB/c mice were purchased from Harlan (Loughborough, U.K.) or from Taconic (Germantown, NY). CCR2-deficient mice on a BALB/c background were originally obtained from William Kuziel (Protein Design Laboratories, Fremont, CA at backcross number 6 to BALB/c) and were backcrossed to number 8 at the Dana Farber Cancer Institute for these experiments. CCL2-deficient mice (25) on a BALB/c background (backcross number 10) were maintained in the Dana Farber Cancer Institute animal facility. All animals were 6–16 wk old when used. U.K. Home Office guidelines for animal welfare, based on the Animals (Scientific Procedures) Act of 1986, were strictly observed. The CCR2−/− and CCL2−/− mice experiments were approved by the Institutional Animal Care and Use Committee at the Dana Farber Cancer Institute, and the studies were carried out in accordance with the guidelines for animal care of the National Institutes of Health and the Public Health Service.
Sensitization and challenge of mice
Mice were sensitized and challenged according to the protocol described by Williams and Galli (26) (Supplemental Fig. 1A). Briefly, female BALB/c mice were sensitized by seven i.p. injections of OVA in PBS (200 μl containing 10 μg OVA) every other day. Forty days after the first sensitization, anesthetized mice were challenged intranasally three times every 3 d (200 μg OVA in 20 μl PBS or PBS alone for the control group). Airway hyperresponsiveness (AHR) was assessed on day 44 by whole-body plethysmography in response to inhaled methacholine (Sigma-Aldrich) at concentrations of 3–100 mg/ml. This was confirmed on day 47 by direct measurements of lung resistance and compliance in anesthetized and tracheostomized mice in response to inhaled methacholine at 3–100 mg/ml. Lung function-measurement equipment was from EMMS (Bordon, U.K.). Following lung resistance and compliance measurement, mice were killed, and tissues were harvested for processing.
For the assessment of lung MC progenitors, mice received i.p injections of 10 μg OVA (Sigma-Aldrich, Grand Island, NY) adsorbed to 1 mg alum (Pierce, Rockford, IL) in 200 μl sterile HBSS on days 0 and 7. Mice were challenged with 1% aerosolized OVA in HBSS for 30 min per day using a PARI nebulizer (Midlothian, VA) on days 17–19 and were euthanized for the determination of MCp on day 20 (Supplemental Fig. 1B).
Lung histopathology
Lungs were fixed in 10% normal buffered formalin. Four-micrometer paraffin-embedded sections were stained with H&E to assess inflammation and murine MC protease (mMCP)-1 to quantify MCs. Briefly, after blocking for 15 min in a high-salt buffer (0.5 M NaCl), paraffin-embedded lung sections were stained with 2 μg/ml rat anti-mouse mMCP-1 (a kind gift from Dr. Jeremy Brown, University of Edinburgh, Edinburgh, U.K.) or isotype control for 1 h. After washing, slides were incubated with biotinylated goat anti-rat IgG secondary Ab (Jackson ImmunoResearch, Newmarket, U.K.) prior to incubation with avidin-biotin complex-HRP complex, according to the manufacturer’s instructions (Vector Laboratories, Peterborough, U.K.). Staining was detected using diaminobenzidine substrate (Vector Laboratories), and sections were counterstained with Mayer’s hematoxylin. Lung sections were examined using a Nikon Eclipse 80i light microscope with a Nikon Plan Apo 20×/0.75 objective (Nikon, Kingston-Upon-Thames, U.K.). The total number of MCs was divided by the area of lung sections to determine the number of MCs/cm2. Peribronchial MCs were MCs found within 0.1 mm of airways. The total number of peribronchial MCs was divided by the total distance of airways per section to give the number of MCs/cm of airway. The total distance of airways and the total area of each section were measured using Q imaging software (Media Cybernetics, Marlow, U.K.). Images were taken on a Leica DM2500 light microscope using a Leica DFC 300 FX camera and analyzed using Leica Application Suite software (version 2.8.1) (Leica, Milton Keynes, U.K.).
Bronchoalveolar lavage
The airways of the mice were lavaged three times with 0.4 ml PBS via a tracheal cannula. BAL fluid was centrifuged at 1200 rpm for 5 min at 4°C, and the supernatant was stored at −80°C until future use for cytokine and chemokine analysis.
Cytokine and chemokine analysis
Cytokine and chemokine levels in bronchoalveolar lavage (BAL) fluid were determined by immunoassay using an electro-chemiluminescence multiplex system Sector Imager 3000 from Meso Scale Discovery (Gaithersburg, MD), according to the manufacturer’s instructions. The mouse Th1/Th2 multiplex kit was used, which contains assays for the following cytokines/chemokines: IFN-γ; IL-1β, -2, -4, -5, -10, and -12; CXCL1; and TNF-α. In addition, CCL2 was measured in a singleplex assay (Meso Scale Discovery).
Mononuclear cell preparation and MCp assessment
Mice were euthanized by CO2 asphyxiation, and lungs and spleen were harvested. Lung and spleen were placed separately in 20 ml RPMI 1640 complete medium (RPMI 1640 containing 100 U/ml penicillin, 100 μg/ml streptomycin, 10 μg/ml gentamicin, 2 mM l-glutamine, 0.1 mM nonessential amino acids, and 10% heat-inactivated FCS; Sigma-Aldrich, Grand Island, NY) and were processed essentially as previously described (19, 22). Briefly, the lungs, perfused with 10 ml HBSS administered via the right ventricle, were removed, finely chopped with scalpels, and digested with collagenase type 4 (Worthington, Lakewood, NJ). Three sequential enzymatic digestions of remaining lung tissue were carried out for ~20 min each at 37°C. The liberated lung cells were spun on 44/67% Percoll (Sigma-Aldrich) gradients (400 × g; 20 min), and the mononuclear cells (MNCs) were collected from the interface and washed in RPMI 1640 complete medium. The number of viable cells was determined by trypan blue dye exclusion on a hemocytometer. The cells were serially diluted 2-fold in RPMI 1640 complete medium and grown in standard 96-well flat-bottom microtiter plates (Corning, Corning, NY) with gamma-irradiated (30 Gy) splenic feeder cells plus cytokines (mouse IL-3 at 20 ng/ml and mouse SCF at 100 ng/ml). Lung MNCs were plated beginning at 20,000 cells/well, with 24 wells plated for each cell concentration. The cultures were placed in humidified 37°C incubators with 5% CO2 for 12–14 d, and wells containing MC colonies were counted with an inverted microscope. The MC colonies were easily distinguished as large colonies of nonadherent, small- to medium-sized cells (19, 27). The MCp concentration is expressed as the number of MCps/106 MNCs isolated from the lung. The number of MCps/lung was derived by multiplying the concentration of MCps by the MNC yield.
Reconstitution of recruitment by adoptive transfer of BM
For the reconstitution of MCp recruitment in sensitized, Ag-challenged mice, the mice were immunized on days 0 and 7, sublethally irradiated on day 14 with 500 rad, and reconstituted with 15 × 106 BM cells 2–4 h later. The BM cells were flushed from the femur and tibia of donor mice with RPMI 1640 complete medium, spun down, counted, and kept on ice at a concentration of 75 × 106 cells/ml until transfer into the tail vein of the recipient mice. The reconstituted mice were challenged with 1% aerosolized OVA on days 21–23, and the determination of MCp was performed on day 24.
BMMC culture
Mouse BMMCs were cultured for up to 10 wk at 37°C in 5% CO2 from BALB/c femoral BM cells at 5 × 105/ml in RPMI 1640 with 10% FBS, 50 μM 2-ME, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 5 ng/ml murine IL-3, with or without 50 ng/ml SCF (PeproTech, London, U.K.).
Mouse BMMC chemotaxis
Thirty microliters of agonist or buffer were added to the lower wells of a 96-well chemotaxis plate (5 μm pore size; NeuroProbe, Gaithersburg, MD). Twenty microliters of BMMCs were added to the top of 96-well chemotaxis plates, on top of the membrane, at 2 × 106 cells/ml. After a 3-h incubation at 37°C and 5% CO2, migrated cells were stained with c-kit and Gr-1 for flow cytometry, as described previously (23). Assays were performed in duplicate, and the results are expressed as absolute number of cells migrating.
Chemotaxis of freshly isolated mouse BM cells
Chemotaxis of freshly isolated BM cells was measured, as described previously (23). Thirty microliters of agonist or buffer were added to the lower wells of a 96-well chemotaxis plate, and 20 μl BM cells, at 107 cells/ml, were added to the upper wells of chemotaxis plates immediately after isolation from femurs. After 3 h of incubation, migrated cells were removed and cultured for 14 d in DMEM with 10% FCS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 2.5 μg/ml fungizone, 1 mM sodium pyruvate, 1 ng/ml TGF-β1, 1 ng/ml murine IL-3, 5 ng/ml murine IL-9, and 50 ng/ml murine SCF (28). Assays were performed in quadruplicate; after lysis, cells were enumerated by specific ELISA for mMCP-1 and -2 (29).
Real-time PCR analysis
Total RNA was extracted from purified BMMCs, and cDNA was synthesized as previously described (23). PCR was performed in triplicate using the sequence-detection system (models ABI Prism 7500 and 7700; Applied Biosystems), with TaqMan Universal PCR Master Mix and FROUNT, CCR2, GAPDH, and 18S-specific primers. Samples were standardized to 18S, and CCR2 and FROUNT mRNA were quantified using the comparative threshold for detection method (http//:appliedbiosystems.com).
Radioligand binding
[125I]CCL2 was purchased from PerkinElmer (Wellesley, MA). Ligand binding was performed by adding increasing concentrations of unlabeled CCL2 with [125I]CCL2 to the BMMCs and using centrifugation through oil to separate bound chemokine from free chemokine, as previously described (30). Data are presented as a percentage of maximum binding.
In vivo MC tracking
BMMCs cultured for 2 wk in IL-3+SCF were purified using anti–c-kit microbeads and labeled with 25 μM CellTracker Green CMFDA for 40 min at 37°C. The isolated cells (2 × 106 cells/mouse) were injected i.v., 5 min prior to intradermal (i.d.) injections of mediator (50 μl in Tyrode’s buffer) into the dorsal skin of the mouse. Mice were killed 1 h after i.d. injection, and the skin was removed and fixed in 4% paraformaldehyde for 24 h and processed for fluorescent microscopy, as previously described (23). Eight-millimeter-diameter biopsies (Biopsy Punch, Schuco International, London, U.K.) of the injection site were taken and counterstained with 10 μg/ml Alexa 568-conjugated GSL-1 isolectin B4, which is a marker for mouse endothelial cells and neutrophils (P. Clark and A. Al-Kashi, unpublished observations). The biopsies were whole mounted in a hardset mounting medium (VECTASHIELD; Vector Laboratories) and counted for accumulated fluorescent cells, using a Leica DM IRBE microscope with standard epifluorescence and a 20× PL-FLUOROSTAR objective magnification, and by confocal microscopy, using a Leica TCS NT system; images were acquired using Leica lite software (all from Leica).
Statistics
Groups of three or more were analyzed using a Kruskal-Wallis test, with pairs of groups compared using the Dunn multiple-comparison test. The Mann-Whitney U test was used for groups of two. The Bonferroni test was used to compare groups at multiple concentrations. Graph generation and statistical analyses were performed with GraphPad Prism, version 4.00 (GraphPad, San Diego, CA). The statistical significance for the lung MCp assays was determined using a two-tailed Student t test. A p value < 0.05 was considered significant.
Results
Increased numbers of MCs in sensitized and challenged mouse lung are associated with elevated CCL2 levels
An in vivo model of allergic airways disease developed by other investigators to show a role for MCs in airway hyperreactivity and chronic inflammation was used to investigate changes in the numbers of mature or maturing MCs in OVA-sensitized and -challenged lung (26).
Relative to PBS-treated controls, the OVA-sensitized and -challenged mice showed increased AHR, as assessed by enhanced pause (Supplemental Fig. 2A); statistically significant increases in airway resistance at methacholine doses of 10, 30, and 100 mg/ml (p < 0.05, p <0.001, and p <0.01, respectively; Supplemental Fig. 2B); and a trend toward decreased dynamic compliance (Supplemental Fig. 2C). OVA-challenged mice showed increased infiltration of inflammatory cells around the bronchovascular bundles (Supplemental Fig. 2E) compared with PBS-challenged controls (Supplemental Fig. 2D) and statistically significant increases in the serum levels of IgE (10.9 ± 2 μg/ml [OVA] versus 0.35 ± 0.2 μg/ml [PBS], p = 0.0025; Supplemental Fig. 2F). The Th2 cytokines IL-4 and -5 in OVA-sensitized and -challenged mice (747.2 ± 172.8 pg/ml, p = 0.0303; 386.7 ± 89.73 pg/ml, p = 0.0101 respectively; Supplemental Fig. 2G, 2H) were elevated compared with PBS-challenged controls (119.5 ± 35.9 pg/ml and 55.2 ± 27.2 pg/ml, respectively). However, there was no difference in the levels of Th1 cytokines (data not shown). These data demonstrated that the OVA sensitization and challenge protocol used induced many features of Ag-induced Th2 cytokine-mediated allergic airway inflammation.
In the same set of experiments, as determined by immuno-detection of the mucosal MC (MMC)-specific protease, mMCP-1, there was a significant (10.4-fold) increase in the total number of MCs per lung section (p = 0.018; Fig. 1A). The majority of these cells were located in and around the airways, and their number was significantly increased (8.6-fold) with OVA challenge compared with PBS-challenged controls (Fig. 1B; p = 0.018). Fig. 1C shows MMCs stained for mMCP-1 in a bronchiole of an OVA-sensitized and -challenged mouse. Consistent with the mMCP-1 staining for MMCs, an 11.4-fold increase in bronchiolar MC staining was also observed with chloroacetate esterase reactivity in the lungs of OVA-sensitized and -challenged mice compared with PBS-challenged controls (2.5 ± 0.8/cm [n = 7] and 0.22 ± 0.22/cm [n = 5], respectively; p = 0.018).
FIGURE 1.

OVA sensitization and challenge induces increased MC accumulation in the lung that is associated with increased CCL2 in BAL fluid. Total MCs/cm2 (A) and peribronchial MCs/cm of airway (B) were determined in lung sections from OVA-sensitized and -challenged mice compared with PBS controls by immunohistochemistry using the MC-specific marker mMCP-1 and counterstained with hematoxylin. C, Representative photomicrograph of mMCP-1+ MCs located intraepithelially around a bronchiole; arrows indicate mMCP-1+ MCs (original magnification ×20). Scale bar, 50 μm. BAL fluid was analyzed for concentrations of the chemokines CCL2 (D) and CXCL1 (E) by immunoassay. Data are mean ± SEM (n = 5–7). *p < 0.05, Mann-Whitney U test.
To identify chemoattractants mediating the increase in peribronchial MCs, BAL fluid was analyzed for cytokines and the chemokines CCL2 and CXCL1. CCL2 was significantly increased (7.1-fold; 792 ± 162 pg/ml) in BAL fluid from OVA-sensitized and -challenged mice compared with controls (110.5 ± 46.9 pg/ml; p = 0.01; Fig. 1D). In contrast, there was no significant difference in the concentration of CXCL1 in the BAL fluid of OVA- and PBS-challenged mice (1132.6 ± 173.9 pg/ml and 883.2 ± 57.2 pg/ml, respectively, p = 0.27; Fig 1E).
Freshly isolated BM progenitors migrate to CCL2
To investigate the potential involvement of CCL2 in the recruitment of MCps to the lung, the dose-dependent ability of CCL2 to induce the migration of MCps present in freshly isolated BMs was determined. Migrated cells were recovered and cultured in the presence of TGF-β1, IL-3 and -9, and SCF for 2 wk to establish their lineage. These conditions promote the rapid maturation and proliferation of BM-derived MC progenitors (BM-MCps) into BMMCs containing mMCP-1 and -2 (31). CCL2 induced the migration of BM-MCps with a typical bell-shaped dose/response curve. The migration was evident from microscopic inspection of the culture plate and was quantified by measuring the concentration of mMCP-1 and -2 in lysed cells by ELISA. Significant migration was achieved in response to 1 nM CCL2 (mMCP-1: 633 ± 275 ng/ml; p = 0.017 and mMCP-2: 707 ∓ 198 ng/ml; p = 0.047) and 0.1 nM CCL2 (mMCP-1: 622 ± 294; p = 0.016) compared with buffer control (Fig. 2). There was no chemotactic response of the BM-MCps to other CC chemokines, including CCL3, CCL5, CCL11, and CCL24, or to the CXC chemokines CXCL1, CXCL2, CXCL9, CXCL10, and CXCL11. SCF (0.1–10 nM) did not induce significant migration of BM-MCps, as previously reported (23) (data not shown).
FIGURE 2.

Freshly isolated BM MCps migrate to CCL2. Freshly isolated BM cells were tested for their ability to migrate to CCL2 (0.1–100 nM) in a chemotaxis assay. After a 3-h incubation, migrated cells were cultured for 14 d in the presence of TGF-β1, SCF, and IL-3 and -9 and then were frozen, lysed, and assayed for levels of mMCP-1 and -2 by ELISA. CCL2-induced migration of cells that developed into mMCP-1– and -2–expressing cells (MCs). Results were compared with migration to buffer alone. Data are mean ± SEM (n = 5) for different BM cell preparations. Statistical significance was determined with the Kruskal-Wallis test with the Dunn posttest to compare migration to concentrations of CCL2 with buffer alone. *p < 0.05 for mMCP-1; +p < 0.05 for mMCP-2, compared with buffer alone.
Immature BMMCs cultured with IL-3+SCF, but not IL-3 alone, migrate in response to CCL2
To further assess CCL2 as a chemoattractant, we used BMMCs cultured for different intervals with IL-3, with or without SCF. Immature BMMCs cultured >2 wk in IL-3 alone were unable to migrate to CCL2, as previously reported (23) (Fig. 3A, Table I). In contrast, BMMCs cultured for 2 wk in the presence of IL-3+SCF were very responsive (Fig. 3A). Significant migration of BMMCs cultured for 2 wk in IL-3+SCF was achieved at CCL2 concentrations of 1 nM (p = 0.048) and 10 nM (p = 0.013) compared with BMMCs cultured for 2 wk in IL-3 alone. Under both culture conditions, 10-wk-old mature BMMCs did not migrate to CCL2. However, 2- and 10-wk-old BMMCs migrated equally well toward SCF, indicating that there was no global defect in the overall migratory capacity of the cells as they matured (Table I). The addition of 50 ng/ml SCF to IL-3–cultured BMMCs for 1 or 24 h prior to assay did not induce the ability of these cells to migrate to CCL2 (Fig. 3B).
FIGURE 3.

CCL2 is chemotactic for immature BMMCs cultured for 2 wk. A, CCL2 (0.1–100 nM) was assayed for chemoattractant activity against BMMCs cultured for 2 or 10 wk with 5 ng/ml IL-3 or 5 ng/ml IL-3 + 50 ng/ml SCF. B, CCL2 (0.1–100 nM) was assayed for chemoattractant activity against BMMCs cultured for 2 wk with 5 ng/ml IL-3 alone and after incubation with an additional 50 ng/ml SCF for 1 or 24 h. Results were compared with BMMCs cultured for 2 wk with 5 ng/ml IL-3 + 50 ng/ml SCF. C, CCL2 was added to the top or bottom of a chemotaxis plate, and BMMCs cultured for 2 wk with IL-3 + SCF were added to the top. After a 3-h incubation, migrated cells were removed, and c-kit+ BMMCs were counted. No cells migrated to assay buffer alone. Data are mean ± SEM for n = 3 (B, C) and n = 6–8 (A). Statistical significance was determined using two-way ANOVA with the Bonferroni posttest. *p < 0.05; **p < 0.01; ***p < 0.001, compared with IL-3 cultures.
Table I.
BMMC migration to different chemotactic mediators
| Cell Age and Culture Conditions |
|||||
|---|---|---|---|---|---|
| Freshly Isolated BM |
2 Wk Immature |
10 Wk Mature |
|||
| Chemoattractant | IL-3 | IL-3+SCF | IL-3 | IL-3+SCF | |
| CC chemokine | |||||
| CCL2 (MCP-1/JE) | + | − | + | − | − |
| Lipid mediator | |||||
| LTB4 | + | + | + | − | − |
| PGE2 | − | + | + | + | + |
| Growth factor | |||||
| SCF | − | + | + | + | + |
−, no migration
+, migration.
The CCL2-induced migration of BMMCs cultured for 2 wk was a chemotactic response requiring a gradient, because cells only migrated when CCL2 was placed in the bottom well. No chemokinetic response representing increased random movement was observed when CCL2 was included at the same concentration in the upper chamber with the cells (Fig. 3C; p = 0.0002).
CCR2 expression in BMMCs cultured in IL-3, with and without SCF
To determine whether the differences in chemotaxis to CCL2 between BMMCs cultured for 2 wk in IL-3 and IL-3+SCF were related to CCR2R expression, mRNA levels were compared under both culture conditions. BMMCs cultured for 2 wk with IL-3 and IL-3+SCF expressed CCR2 mRNA that decreased significantly to low levels after 6 and 10 wk of culture (Fig. 4A, 4B), as assessed by real-time PCR. In BMMCs cultured for 2 wk with IL-3, the relative level of CCR2 mRNA decreased by 7.7-fold over the next 8 wk (Fig. 4A; p = 0.023). Similarly, the relative level of CCR2 mRNA in BMMCs cultured with IL-3+SCF for 2 wk decreased by 17.4-fold over the next 8 wk in culture (Fig. 4B; p = 0.036).
FIGURE 4.

CCR2 is expressed on BMMCs. BMMCs cultured in 5 ng/ml IL-3 (A) or 5 ng/ml IL-3 + 50 ng/ml SCF (B) expressed CCR2 mRNA, which decreased as BMMCs matured (assessed by real-time PCR). Data are mean ± SEM for n = 4–7, using RNA isolated from separate cell cultures. Statistical significance was determined using the Kruskal-Wallis test; *p < 0.05. C, BMMCs cultured for 2 wk in IL-3 or IL-3+SCF bind [125I]CCL2 with similar affinities. Data are mean ± SEM for three independent BMMC preparations. D, Relative mRNA expression of FROUNT on BMMCs cultured for 2 wk in IL-3 or IL-3+SCF. Data are mean ± SEM for n = 9–10, using RNA isolated from separate cell cultures. *p < 0.05, Mann-Whitney U test.
To assess any differences in CCR2 cell surface expression or affinity for CCL2 between BMMCs cultured in IL-3 and IL-3+SCF, competition binding was conducted with [125I]CCL2 and unlabeled CCL2. Both BMMC cultures were able to bind CCL2 (Fig. 4C), and there was no difference in the logEC50 between BMMCs cultured in IL-3 (−9.2 M) and those cultured in IL-3+SCF (−9.1 M), indicating that CCR2 can bind CCL2 with equal affinity in both cell-culture conditions.
Cells from both types of 2-wk cultures expressed comparable levels of the MC markers c-kit, CD34, T1/ST2, CD13, and FcεR1 and appeared to be similar in size and granularity, as indicated by forward and side scatter (data not shown). These 2-wk cultures had lower granularity (less side scatter) compared with their respective 10-wk cultures, indicating an immature phenotype (data not shown). Two-week BMMCs from both cultures stained positively for Alcian blue, with little staining for safranin (Supplemental Fig. 3A, 3B). By 10 wk of culture, IL-3–cultured BMMCs only stained positive for Alcian blue, whereas BMMCs cultured in IL-3+SCF stained positively for Alcian blue and safranin (Supplemental Fig. 3C, 3D).
In monocytes and mesenchymal stem cells, CCR2-mediated chemotaxis is dependent on engagement of the receptor with a cytoplasmic protein FROUNT (32, 33). Real-time PCR was used to determine whether differences in CCL2-mediated chemotaxis of BMMCs cultured for 2 wk in IL-3 or IL-3+SCF were related to FROUNT expression. BMMCs cultured in IL-3+SCF expressed significantly more FROUNT mRNA compared with those cultured in IL-3 (40.39 ± 7.958 versus 16.16 ± 2.384, respectively, p = 0.0172l; Fig 4D).
In vivo recruitment of BMMCs in response to i.d. injection of CCL2
To examine whether i.d. injection of CCL2 is able to recruit circulating BMMCs (23, 24), 2-wk-old c-kit+ BMMCs grown in the presence of IL-3+SCF were labeled with CMFDA. Immediately after i.v. administration of 2 × 106 labeled BMMCs, each mouse received an i.d. injection of CCL2 or vehicle (Tyrode’s buffer) into the dorsal skin. Relative to sites injected with Tyrode’s buffer, CCL2 induced a dose-dependent recruitment of circulating BMMCs to the i.d. injection site, with significant accumulation in response to 15 pmol CCL2 (5.8-fold increase; p = 0.001) and 150 pmol CCL2 (5.4 fold increase; p = 0.0039) (Fig. 5A). Maximal BMMC accumulation was achieved in response to 15 pmol CCL2. A representative confocal photomicrograph of a whole-mounted skin biopsy of the CCL2 i.d. injection site shows CMFDA+ BMMCs (green fluorescence) close to blood microvessels in the skin (Fig. 5B).
FIGURE 5.

Immature BMMCs localize to CCL2 administered i.d. A, Increased numbers of 2-wk CMFDA-labeled, c-kit+ BMMCs were noted in skin injected with 0.15, 15, or 150 pmol CCL2 compared with vehicle control (Tyrode’s buffer). Data are mean ± SEM for n = 8–12 mice. Statistical significance was determined with the Kruskal-Wallis test with the Dunn posttest. **p < 0.01; ***p < 0.001, compared with injection of Tyrode’s buffer i.d. B, Representative confocal photomicrograph of whole-mount mouse skin from the site injected with 150 pmol CCL2 showing extravascular CMFDA+ BMMCs (arrows). CMFDA-labeled c-kit+ BMMCs appear green in skin samples counterstained with GSL-1 isolectin B4, staining endothelium and neutrophils red (original magnification ×40). Scale bar, 80 μM.
The CCR2/CCL2 axis is involved in Ag-induced MCp recruitment to the lung of sensitized and challenged mice
To determine whether the absence of CCL2/CCR2 influences the Ag-induced recruitment of MCps to lung, we used a previously published protocol that assesses recruitment of MCps to the lung several days prior to their maturation into MMC (21). Similar low baseline numbers of MCps/106 lung MNCs were found in sensitized, unchallenged, WT CCL2−/− and CCR2−/− mice (Fig. 6A, 6C). Following an aerosolized-OVA challenge, CCL2−/− and CCR2−/− mice had significantly reduced numbers of lung MCps/106 MNCs compared with WT mice challenged in parallel (CCL2−/−, 59% reduction, p = 0.009, Fig. 6A; CCR2−/−, 55% reduction, p = 0.003, Fig. 6D). This decrease was specific to MCps, because the increases in total lung MNC numbers after OVA challenge were similar between the WT mice challenged in parallel and the CCL2−/− or CCR2−/− mice (Fig. 6B, 6D).
FIGURE 6.

MCp recruitment to the lung is significantly reduced in sensitized and Ag-challenged CCL2- and CCR2-deficient mice. Average concentration of MCps in individual mouse lungs (MCps/106 MNCs) (A) and number of lung MNCs recovered per mouse (B) from OVA-sensitized WT and CCL2−/− mice with and without aerosolized OVA challenges. Values are the mean ± SEM from three separate experiments. Average concentration of MCps in individual mouse lungs (MCps/106 MNCs) (C) and number of lung MNCs recovered per mouse (D) from OVA-sensitized WT and CCR2−/− mice with and without aerosolized OVA challenges. Values are the mean ± SEM from four separate experiments. *p < 0.05, two-tailed Student t test.
CCR2 expression is required on a lung stromal component for recruitment of MCps to the lung of sensitized, sublethally irradiated, BM-reconstituted and Ag-challenged mice
To evaluate whether the decreased recruitment of lung MCps in the CCR2−/− mice was due to the absence of CCR2 expression on the MCps, sensitized, sublethally irradiated WT mice were reconstituted with WT or CCR2−/− BMs and then challenged 1 wk later. Sensitized, sublethally irradiated, Ag-challenged WT mice that were not reconstituted with BM prior to challenge had very few MCps/106 MNCs and lung MNCs per mouse compared with the nonirradiated, sensitized, Ag-challenged WT mice (Fig. 7A, 7B), indicating the sensitivity of the response to sublethal irradiation in the absence of BM reconstitution. Reconstitution of sensitized, sublethally irradiated, Ag-challenged WT mice with WT or CCR2−/− BM restored the recruitment of lung MCps to the level of sensitized and challenged nonirradiated mice (Fig. 7A, 7B). The number of lung MNCs also was fully restored with WT or CCR2−/− BM. Furthermore, analysis of five of the lung MCp colonies generated in vitro following reconstitution of the WT mice with CCR2−/− BM revealed that all were of the donor CCR2−/− genotype.
FIGURE 7.

CCR2 expression by stroma, but not lung, MCps is critical for OVA-induced recruitment of lung MCps. A and B, CCR2−/− BM reconstitutes MCp recruitment to the lung of sensitized, sublethally irradiated, Ag-challenged WT mice. A, Average concentration of MCps in individual mouse lungs (MCps/106 MNCs) from sensitized, Ag-challenged WT mice (WT ch); sensitized, sublethally irradiated, Ag-challenged WT mice without BM reconstitution (WT irr ch; sensitized, sublethally irradiated, Ag-challenged, WT mice reconstituted with WT BM (WT-BM ch); and sensitized, sublethally irradiated, Ag-challenged WT mice reconstituted with CCR2−/− BM (CCR2BM ch). Values are the mean ± SE from three to four mice in two separate experiments. B, Number of lung MNCs recovered per mouse in the same mice as in A. C and D, Average concentration of MCps in individual mouse lungs (MCps/106 MNCs) from sensitized, sublethally irradiated, Ag-challenged WT mice without BM reconstitution (WT irr ch); sensitized, sublethally irradiated, Ag-challenged, WT mice reconstituted with WT BM (WT + WTBM ch); and sensitized, sublethally irradiated, Ag-challenged CCR2−/− mice reconstituted with WT BM (CCR2−/− + WTBM ch). D, Number of lung MNCs recovered per mouse in the same mice as in C. Values are the mean ± SE from 9–12 mice analyzed individually in four separate experiments.
To determine whether the absence of CCR2 on a radiation-resistant lung parenchymal cell could account for the reduced lung MCp recruitment in sensitized, Ag-challenged CCR2−/− mice, we compared the capacity of WT BM to reconstitute sensitized, sublethally irradiated, Ag-challenged WT and CCR2−/− mice for MCp recruitment. Compared with similarly treated WT mice, CCR2−/− mice reconstituted with the WT BM cells had a significant reduction (41%) in the concentration of MCps/106 MNCs (p = 0.04; Fig. 7C), whereas the number of lung MNCs per mouse was similar (Fig. 7D). These results indicate that CCR2 expression on BM cells is not critical to MCp recruitment to lung and that CCR2 needs to be expressed on a non–BM-derived radiation-resistant cell in the lung parenchyma.
CCL2 expression by stromal and BM components is needed for optimal recruitment of MCps to the lung of sensitized, sublethally irradiated, BM-reconstituted, Ag-challenged mice
To assess whether the decreased recruitment of MCps to inflamed lung in the CCL2−/− mice is due to the loss of CCL2 expression in the MCps, sensitized, sublethally irradiated WT mice were reconstituted with WT or CCL2−/− BM and then challenged 1 wk later. Sensitized, sublethally irradiated, Ag-challenged WT mice that were not reconstituted with BM before challenge had very few lung MCps or MNCs (Fig. 8A, 8B). Reconstitution of sensitized, sublethally irradiated, Ag-challenged WT mice with WT BM restored the recruitment of lung MCps, whereas sensitized, sublethally irradiated, Ag-challenged WT mice that were reconstituted with CCL2−/− BM showed significantly reduced numbers of MCps/106 MNCs (63% reduction, p = 0.0001; Fig. 8A). The number of lung MNCs per mouse were similar (Fig. 8B), indicating a selective effect on the BM-derived MCps when CCL2 is absent in the BM.
FIGURE 8.
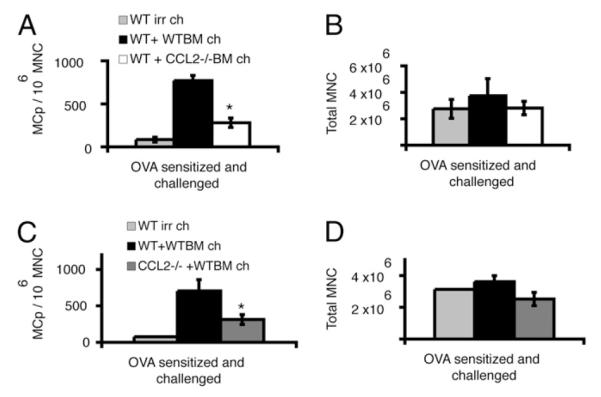
Stroma- and BM-derived cells are required to express CCL2 for lung MCp recruitment. WT BM does not reconstitute MCp recruitment to the lung of sensitized, sublethally irradiated, Ag-challenged CCRL−/− mice. A, Average concentration of MCps in individual mouse lungs (MCps/106 MNCs) from sensitized, sublethally irradiated, Ag-challenged WT mice without BM reconstitution (WT irr ch); sensitized, sublethally irradiated, Ag-challenged WT mice reconstituted with WT BM (WT+WTBM ch); and sensitized, sublethally irradiated, Ag-challenged WT mice reconstituted with CCL2−/− BM (WT+CCL2−/− BM ch). Values are the mean ± SE from six mice in two separate experiments. B, Number of lung MNCs recovered per mouse in the same mice as in A. C and D, Average concentration of MCps in individual mouse lungs (MCps/106 MNCs) from sensitized, sublethally irradiated, Ag-challenged WT mice without BM reconstitution (WT irr ch); sensitized, sublethally irradiated, Ag-challenged WT mice reconstituted with WT BM (WT + WTBM ch); and sensitized, sublethally irradiated, Ag-challenged CCL2−/− mice reconstituted with WT BM (CCL2−/− + WTBM ch). Values are the mean ± SE from six or seven mice analyzed individually from two separate experiments. D, Number of lung MNCs recovered per mouse in the same mice as in C. *p <0.05, two-tailed Student t test.
Because CCR2 stromal expression was needed for lung MCp recruitment, we also investigated whether the functional expression of CCL2 by radiation-resistant lung parenchymal cells could contribute to MCp recruitment. Compared with sensitized, sublethally irradiated, Ag-challenged WT mice reconstituted with WT BM, similarly treated CCL2−/− mice reconstituted with the WT BM cells had a significant reduction in the number of MCps/106 MNCs (61% reduction, p = 0.03; Fig. 8C). The number of lung MNCs per mouse was similar in the two groups (Fig. 8D). These results indicate that CCL2 expression by the BM-derived cells and by non–BM-derived radiation-resistant cells in the lung parenchyma are needed for MCp recruitment to the inflamed lung.
Discussion
Our focus on the role of the CCL2/CCR2 axis in Ag-induced pulmonary inflammation arose from two in vivo studies that differed in their design. In a conventional model of MC-dependent pulmonary inflammation mediated by Th2 cytokines, as defined by elevated levels of IL-4 and -5 in the BAL fluid, cellular infiltration of the bronchovascular bundles in the lung, and elevation of serum IgE, there was significant expansion of the MC numbers. The concomitantly increased concentration of CCL2, but not CXCL1, in the BAL fluid raised the possibility that CCL2 might contribute to the 10-fold increment in MMCs by recruiting their progenitors. A separate protocol developed to assess for lung MCps within the MNC population before their maturation to mucosal or connective tissue MCs was used to determine whether CCR2 or CCL2 was involved in this response. The increment in pulmonary MCps is assessed by limiting dilution and clonal expansion with IL-3/SCF and has been validated by FACS sorting for the MCps, with selective expansion only to MCs, despite a full array of hematopoietic cytokines (22). Mice with targeted disruption of CCR2 or CCL2 had a significant reduction in the Ag-induced recruitment of MCps. These results supported the involvement of CCL2 in the recruitment of MCps to lung, possibly by their directed migration. To address this possibility, we proceeded with an in vitro analysis of the chemotactic effect of CCL2 on BM-MCps and determined the effects of their further maturation to immature or more mature BMMCs on this response. We also conducted an in vivo analysis of whether the CCR2 expression by MCps was directly involved in their Ag-induced pulmonary recruitment.
Chemotactic assays of fresh suspensions of BM cells showed that MCps within the BM population responded chemotactically to CCL2, and the responding cells differentiated into BMMCs when cultured with SCF, IL-3 and -9, and TGF-β. The responsiveness to CCL2 was lost when BM cells were cultured for 2 wk with IL-3. However, BMMCs cultured for 2 wk with IL-3+SCF were highly responsive to CCL2 in chemotaxis assays in vitro and were able to accumulate in skin sites injected with CCL2 in vivo when administered i.v. Addition of SCF for 1 or 24 h did not restore responsiveness to CCL2 in BMMCs cultured with IL-3. Interestingly, the changes in chemotactic responses to CCL2 mirror those to SCF; MCps in fresh BM do not respond to SCF in chemotaxis assays, but this response is acquired when the cells are cultured in IL-3 (23). BMMCs cultured under both conditions expressed similar levels of CCR2 mRNA and bound radiolabeled CCL2 with similar affinity, indicating similar cell-surface expression of the receptor. CCR2 mRNA decreased to low levels at 6 and 10 wk in culture, and chemotactic responsiveness was lost in those cells cultured with IL-3+SCF.
The SCF-induced coupling of the chemokine receptor to transduction pathways mediating chemotaxis suggests the upregulation of a key component of the signaling pathway or, alternatively, the downregulation of an inhibitory component. A cytoplasmic adaptor molecule, FROUNT, was shown to be essential for CCR2-mediated migration in CHO cell lines (32) and mesenchymal stem cells (33). Upon CCL2 binding, FROUNT translocated and bound to CCL2–CCR2 complexes at the leading edge of the migrating cell, allowing receptor clustering and a link to the PI3K-Rac-lamellipodium protrusion cascade (32, 33). We observed that BMMCs cultured in IL-3+SCF expressed significantly more FROUNT mRNA than did cells cultured with IL-3 alone, suggesting that FROUNT may be a component in the receptor coupling that is stimulated by SCF.
Previous studies using mice deficient in SCF or its receptor demonstrated that these animals are essentially devoid of mature tissue MCs, although BMMCs can be cultured from the BM (34). Our results demonstrated a hitherto unrecognized activity of SCF in that it is able to induce coupling of a chemokine receptor to transduction pathways mediating chemotaxis. Whether this activity relates to the lack of tissue MCs observed in mice deficient in SCF or its receptor remains to be established.
We assessed the importance of CCR2 and CCL2 expression in MCp recruitment in an in vivo model of pulmonary inflammation at a time point that precedes any increment in MMCs to avoid any effect of their presence or proliferation on the estimate of MCps. Mice deficient in CCL2 had a significant reduction in lung MCp concentration/106 MNCs compared with WT BALB/c mice, indicating a role for CCL2 in Ag-induced MCp recruitment in vivo. Mice deficient in CCR2 also had significant reductions in Ag-induced recruitment of lung MCps/106 MNCs compared with sensitized and challenged WT mice, indicating that the CCL2/CCR2 pairing is central to the Ag-induced recruitment of MCps.
Unexpectedly, reconstitution of sublethally irradiated WT mice with BM showed a reduction in the concentration of MCps/106 MNCs when the donors were CCL2 deficient compared with reconstitution with WT BM cells. This finding implies that the donor BM supplies the CCL2 in this model. More surprising was the finding that reconstitution of sublethally irradiated WT mice with BM from CCR2-deficient mice was intact, indicating that CCR2-expressing hematopoietic cells (including lung MCps) were not essential for the recruitment of donor MCps to the lung. Instead, the latter findings indicated that radio-resistant lung cells expressing CCR2 were needed for MCp recruitment. The additional finding that sublethally irradiated CCR2−/− and CCL2−/− mice reconstituted with normal BM had a significant reduction in MCps compared with irradiated WT recipients reconstituted with normal BM again revealed a role for radio-resistant host cells expressing CCR2 and also indicated a second required source for CCL2. Although these latter studies with attempted reconstitution of sublethally irradiated mutants might have other complex interpretations, the classical reconstitution of irradiated WT mice with WT versus mutant BM establishes that the donor BM is a source of CCL2 and that the CCR2 critical to MCp recruitment is provided by radioresistant host cells.
CCL2 is produced by a wide variety of cells, including MC (35–38), in response to inflammatory stimuli and, therefore, could provide a mechanism for rapidly recruiting MCps to the affected tissues. Elevated lung mRNA for CCL2 was detected in an OVA-sensitization/challenge model (39). In addition, CCL2 neutralization reduced AHR in a Schistosoma egg Ag-sensitization model (40), as well as in a cockroach Ag-sensitization and -challenge model in which a significant reduction in the MC mediator, histamine, was observed in the BAL fluid (41). Marked increases in the numbers of airway MCs were observed in a mouse model of Ag-induced inflammation, with enhanced airway responses to methacholine and chronic inflammation (17). Increased CCL2 levels also were shown in the BAL fluid of allergic asthmatic patients (42), and CCL2 was shown to be released into asthmatic airways following endobronchial allergen challenge (43). Several radioresistant cell types, such as endothelial cells (44, 45), epithelial cells (46), and fibroblasts (47), express CCR2 and may account for the CCR2-dependent recruitment of MCps to lung.
We now must ask how we can reconcile the obvious importance of the CCR2/CCL2 pairing to Ag-induced recruitment of MCps to the lung, apparent from the impaired MCp response in each null strain, with a failure to find a functional requirement for CCR2 in BM cells in adoptive transfer after sublethal irradiation of a WT recipient. Additionally, how can we reconcile these findings with the in vitro and in vivo evidence that CCL2 can recruit MCps from BM and direct movement of immature 2-wk-cultured BMMCs? We considered the possibility that the CCR2/CCL2 axis might regulate the efflux of MCps from BM or spleen but could not demonstrate retention of BM-MCps in CCR2−/− mice (data not shown). CCR2 on MCps may be involved in trafficking under basal conditions, but other receptors might be able to substitute in the context of allergic reactions in which a plethora of mediators are generated. Relevant mediators, such as LTB4 (23), may be generated in the challenged lung by a CCL2/CCR2-dependent mechanism. In addition, CCR2 may have a role, in this or other disease models, in repositioning maturing cells within the tissue subsequent to recruitment.
We previously showed that CXCR2-deficient mice are significantly impaired in the Ag-induced recruitment of MCps (22). However, the ability of BM from CXCR2−/− mice to reconstitute sensitized, sublethally irradiated WT mice for Ag-induced recruitment of MCp to the lung was intact and similar to that obtained with BM from normal donors. The radioresistant stromal target of CXCR2 was VCAM-1 expression, which mediates the α4 integrin-dependent adhesion essential for transendothelial migration of MCps. In that study, we could not define the CXCR2 ligand or its source. In the current study, we again found that the radioresistant chemokine receptor-bearing cell is in the tissue and can add that the ligand is in the BAL fluid, but we do not know the functional consequences that lead to MCp recruitment.
This study revealed a novel and potentially important activity of SCF, in that it is able to couple surface CCR2 to transduction pathways mediating chemotaxis of immature MCs. CCR2 seems to already be coupled to those pathways in MCps in the BM, but it is lost on culture in the absence of SCF. For CCL2 to induce chemotaxis, it needs to couple to a coordinated sequence of events mediating changes in the intracellular cytoskeleton and expression and/or activation of surface integrin molecules controlling attachment to extracellular surfaces. We have not established at what level SCF acts. However, an action at the level of integrins is possible, because a previous study showed that the magnitude of MC chemotaxis induced by CCL2 is dependent on the nature of the surface of the filter used (48). In these studies, coating with vitronectin or laminin considerably enhanced the chemotaxis of cells cultured with IL-3. We did not coat filters in our studies but will investigate whether SCF alters the pattern of integrin expression. Because CCL2 is generated in the sensitized and allergen-challenged lung, is it tempting to speculate that this chemokine is involved in the recruitment of MCps from the blood and possibly their release from the BM. Experiments with CCR2 and CCL2 gene-deleted animals support this hypothesis. However, experiments using sublethal irradiation and BM transfer suggest that CCR2 expression on donor MCps is not essential for their recruitment to the lung. These results using BM transfer, which echo those from a previous study of CXCR2, suggest that receptor expression on a radioresistant cell in the lung is important for donor MCp recruitment. Taken as a whole, the study suggests an unexpected complexity in mechanisms underlying MCp trafficking in the in vivo setting.
Supplementary Material
Acknowledgments
We thank Gill Martin (Leukocyte Biology, Imperial College London) for culture of BMMCs, Lorraine Lawrence for histopathology, and Profs. Clare Lloyd and Kate Choy for assistance with the lung-function measurements. We also thank Dr. J.K. Brown, University of Edinburgh, for supplying anti–mMCP-1 mAb and the mMCP-1 and -2 ELISA reagents; Jenny Willis and Dr. Zarin Brown, Novartis Institutes for Biomedical Research, for assistance with cytokine and chemokine immunoassay; and Dr. William Kuziel, Protein Design Laboratories, for CCR2-deficient mice.
This work was supported by a Ph.D. studentship provided by Novartis Institutes for Biomedical Research, the Asthma UK Centre, the Swedish Society for Medical Research, and the National Institutes of Health (Grant NIH R01-AI083516).
Abbreviations used in this paper
- AHR
airway hyperresponsiveness
- BAL
bronchoalveolar lavage
- BM
bone marrow
- BM-MCp
bone marrow-derived mast cell progenitor
- BMMC
bone marrow-derived mast cell
- CMFDA
5-chloromethylfluorescein diacetate
- i.d.
intradermal
- LTB4
leukotriene B4
- MC
mast cell
- MCp
mast cell progenitor
- MMC
mucosal mast cell
- mMCP
murine mast cell protease
- MNC
mononuclear cell
- SCF
stem cell factor
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures The authors have no financial conflicts of interest.
References
- 1.Marshall JS. Mast-cell responses to pathogens. Nat. Rev. Immunol. 2004;4:787–799. doi: 10.1038/nri1460. [DOI] [PubMed] [Google Scholar]
- 2.Kitamura Y, Go S, Hatanaka K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood. 1978;52:447–452. [PubMed] [Google Scholar]
- 3.Jamur MC, Grodzki AC, Berenstein EH, Hamawy MM, Siraganian RP, Oliver C. Identification and characterization of undifferentiated mast cells in mouse bone marrow. Blood. 2005;105:4282–4289. doi: 10.1182/blood-2004-02-0756. [DOI] [PubMed] [Google Scholar]
- 4.Chen CC, Grimbaldeston MA, Tsai M, Weissman IL, Galli SJ. Identification of mast cell progenitors in adult mice. Proc. Natl. Acad. Sci. USA. 2005;102:11408–11413. doi: 10.1073/pnas.0504197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arinobu Y, Iwasaki H, Gurish MF, Mizuno S, Shigematsu H, Ozawa H, Tenen DG, Austen KF, Akashi K. Developmental checkpoints of the basophil/mast cell lineages in adult murine hematopoiesis. Proc. Natl. Acad. Sci. USA. 2005;102:18105–18110. doi: 10.1073/pnas.0509148102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodewald HR, Dessing M, Dvorak AM, Galli SJ. Identification of a committed precursor for the mast cell lineage. Science. 1996;271:818–822. doi: 10.1126/science.271.5250.818. [DOI] [PubMed] [Google Scholar]
- 7.Gurish MF, Pear WS, Stevens RL, Scott ML, Sokol K, Ghildyal N, Webster MJ, Hu X, Austen KF, Baltimore D, et al. Tissue-regulated differentiation and maturation of a v-abl-immortalized mast cell-committed progenitor. Immunity. 1995;3:175–186. doi: 10.1016/1074-7613(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 8.Friend DS, Ghildyal N, Austen KF, Gurish MF, Matsumoto R, Stevens RL. Mast cells that reside at different locations in the jejunum of mice infected with Trichinella spiralis exhibit sequential changes in their granule ultrastructure and chymase phenotype. J. Cell Biol. 1996;135:279–290. doi: 10.1083/jcb.135.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Befus AD, Bienenstock J. Immunologically mediated intestinal mastocytosis in Nippostrongylus brasiliensis-infected rats. Immunology. 1979;38:95–101. [PMC free article] [PubMed] [Google Scholar]
- 10.Pipkorn U, Karlsson G, Enerbäck L. Phenotypic expression of proteoglycan in mast cells of the human nasal mucosa. Histochem. J. 1988;20:519–525. doi: 10.1007/BF01002650. [DOI] [PubMed] [Google Scholar]
- 11.Fokkens WJ, Godthelp T, Holm AF, Blom H, Mulder PG, Vroom TM, Rijntjes E. Dynamics of mast cells in the nasal mucosa of patients with allergic rhinitis and non-allergic controls: a biopsy study. Clin. Exp. Allergy. 1992;22:701–710. doi: 10.1111/j.1365-2222.1992.tb00194.x. [DOI] [PubMed] [Google Scholar]
- 12.Nouri-Aria KT, Pilette C, Jacobson MR, Watanabe H, Durham SR. IL-9 and c-Kit+ mast cells in allergic rhinitis during seasonal allergen exposure: effect of immunotherapy. J. Allergy Clin. Immunol. 2005;116:73–79. doi: 10.1016/j.jaci.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 13.Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast-cell infiltration of airway smooth muscle in asthma. N. Engl. J. Med. 2002;346:1699–1705. doi: 10.1056/NEJMoa012705. [DOI] [PubMed] [Google Scholar]
- 14.Carroll NG, Mutavdzic S, James AL. Increased mast cells and neutrophils in submucosal mucous glands and mucus plugging in patients with asthma. Thorax. 2002;57:677–682. doi: 10.1136/thorax.57.8.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mwamtemi HH, Koike K, Kinoshita T, Ito S, Ishida S, Nakazawa Y, Kurokawa Y, Shinozaki K, Sakashita K, Takeuchi K, et al. An increase in circulating mast cell colony-forming cells in asthma. J. Immunol. 2001;166:4672–4677. doi: 10.4049/jimmunol.166.7.4672. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda RK, Miller M, Nayar J, Walker L, Cho JY, McElwain K, McElwain S, Raz E, Broide DH. Accumulation of peribronchial mast cells in a mouse model of ovalbumin allergen induced chronic airway inflammation: modulation by immunostimulatory DNA sequences. J. Immunol. 2003;171:4860–4867. doi: 10.4049/jimmunol.171.9.4860. [DOI] [PubMed] [Google Scholar]
- 17.Yu M, Tsai M, Tam SY, Jones C, Zehnder J, Galli SJ. Mast cells can promote the development of multiple features of chronic asthma in mice. J. Clin. Invest. 2006;116:1633–1641. doi: 10.1172/JCI25702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abonia JP, Austen KF, Rollins BJ, Joshi SK, Flavell RA, Kuziel WA, Koni PA, Gurish MF. Constitutive homing of mast cell progenitors to the intestine depends on autologous expression of the chemokine receptor CXCR2. Blood. 2005;105:4308–4313. doi: 10.1182/blood-2004-09-3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurish MF, Tao H, Abonia JP, Arya A, Friend DS, Parker CM, Austen KF. Intestinal mast cell progenitors require CD49dbeta7 (alpha4beta7 integrin) for tissue-specific homing. J. Exp. Med. 2001;194:1243–1252. doi: 10.1084/jem.194.9.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Artis D, Humphreys NE, Potten CS, Wagner N, Müller W, McDermott JR, Grencis RK, Else KJ. Beta7 integrin-deficient mice: delayed leukocyte recruitment and attenuated protective immunity in the small intestine during enteric helminth infection. Eur. J. Immunol. 2000;30:1656–1664. doi: 10.1002/1521-4141(200006)30:6<1656::AID-IMMU1656>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 21.Abonia JP, Hallgren J, Jones T, Shi T, Xu Y, Koni P, Flavell RA, Boyce JA, Austen KF, Gurish MF. Alpha-4 integrins and VCAM-1, but not MAdCAM-1, are essential for recruitment of mast cell progenitors to the inflamed lung. Blood. 2006;108:1588–1594. doi: 10.1182/blood-2005-12-012781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hallgren J, Jones TG, Abonia JP, Xing W, Humbles A, Austen KF, Gurish MF. Pulmonary CXCR2 regulates VCAM-1 and antigen-induced recruitment of mast cell progenitors. Proc. Natl. Acad. Sci. USA. 2007;104:20478–20483. doi: 10.1073/pnas.0709651104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weller CL, Collington SJ, Brown JK, Miller HR, Al-Kashi A, Clark P, Jose PJ, Hartnell A, Williams TJ. Leukotriene B4, an activation product of mast cells, is a chemoattractant for their progenitors. J. Exp. Med. 2005;201:1961–1971. doi: 10.1084/jem.20042407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weller CL, Collington SJ, Hartnell A, Conroy DM, Kaise T, Barker JE, Wilson MS, Taylor GW, Jose PJ, Williams TJ. Chemotactic action of prostaglandin E2 on mouse mast cells acting via the PGE2 receptor 3. Proc. Natl. Acad. Sci. USA. 2007;104:11712–11717. doi: 10.1073/pnas.0701700104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams CM, Galli SJ. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J. Exp. Med. 2000;192:455–462. doi: 10.1084/jem.192.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crapper RM, Schrader JW. Frequency of mast cell precursors in normal tissues determined by an in vitro assay: antigen induces parallel increases in the frequency of P cell precursors and mast cells. J. Immunol. 1983;131:923–928. [PubMed] [Google Scholar]
- 28.Brown JK, Knight PA, Wright SH, Thornton EM, Miller HR. Constitutive secretion of the granule chymase mouse mast cell protease-1 and the chemokine, CCL2, by mucosal mast cell homologues. Clin. Exp. Allergy. 2003;33:132–146. doi: 10.1046/j.1365-2222.2003.01571.x. [DOI] [PubMed] [Google Scholar]
- 29.Pemberton AD, Brown JK, Wright SH, Knight PA, McPhee ML, McEuen AR, Forse PA, Miller HR. Purification and characterization of mouse mast cell proteinase-2 and the differential expression and release of mouse mast cell proteinase-1 and -2 in vivo. Clin. Exp. Allergy. 2003;33:1005–1012. doi: 10.1046/j.1365-2222.2003.01720.x. [DOI] [PubMed] [Google Scholar]
- 30.Martinelli R, Sabroe I, LaRosa G, Williams TJ, Pease JE. The CC chemokine eotaxin (CCL11) is a partial agonist of CC chemokine receptor 2b. J. Biol. Chem. 2001;276:42957–42964. doi: 10.1074/jbc.M103933200. [DOI] [PubMed] [Google Scholar]
- 31.Miller HR, Wright SH, Knight PA, Thornton EM. A novel function for transforming growth factor-beta1: upregulation of the expression and the IgE-independent extracellular release of a mucosal mast cell granule-specific beta-chymase, mouse mast cell protease-1. Blood. 1999;93:3473–3486. [PubMed] [Google Scholar]
- 32.Terashima Y, Onai N, Murai M, Enomoto M, Poonpiriya V, Hamada T, Motomura K, Suwa M, Ezaki T, Haga T, et al. Pivotal function for cytoplasmic protein FROUNT in CCR2-mediated monocyte chemotaxis. Nat. Immunol. 2005;6:827–835. doi: 10.1038/ni1222. [DOI] [PubMed] [Google Scholar]
- 33.Belema-Bedada F, Uchida S, Martire A, Kostin S, Braun T. Efficient homing of multipotent adult mesenchymal stem cells depends on FROUNT-mediated clustering of CCR2. Cell Stem Cell. 2008;2:566–575. doi: 10.1016/j.stem.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Eklund KK, Ghildyal N, Austen KF, Friend DS, Schiller V, Stevens RL. Mouse bone marrow-derived mast cells (mBMMC) obtained in vitro from mice that are mast cell-deficient in vivo express the same panel of granule proteases as mBMMC and serosal mast cells from their normal littermates. J. Exp. Med. 1994;180:67–73. doi: 10.1084/jem.180.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oliveira SH, Lukacs NW. Stem cell factor and igE-stimulated murine mast cells produce chemokines (CCL2, CCL17, CCL22) and express chemokine receptors. Inflamm. Res. 2001;50:168–174. doi: 10.1007/s000110050741. [DOI] [PubMed] [Google Scholar]
- 37.Baghestanian M, Hofbauer R, Kiener HP, Bankl HC, Wimazal F, Willheim M, Scheiner O, Füreder W, Müller MR, Bevec D, et al. The c-kit ligand stem cell factor and anti-IgE promote expression of monocyte chemoattractant protein-1 in human lung mast cells. Blood. 1997;90:4438–4449. [PubMed] [Google Scholar]
- 38.Lin DA, Boyce JA. IL-4 regulates MEK expression required for lysophosphatidic acid-mediated chemokine generation by human mast cells. J. Immunol. 2005;175:5430–5438. doi: 10.4049/jimmunol.175.8.5430. [DOI] [PubMed] [Google Scholar]
- 39.Gonzalo JA, Lloyd CM, Wen D, Albar JP, Wells TN, Proudfoot A, Martinez-A C, Dorf M, Bjerke T, Coyle AJ, Gutierrez-Ramos JC. The coordinated action of CC chemokines in the lung orchestrates allergic inflammation and airway hyperresponsiveness. J. Exp. Med. 1998;188:157–167. doi: 10.1084/jem.188.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lukacs NW, Strieter RM, Warmington K, Lincoln P, Chensue SW, Kunkel SL. Differential recruitment of leukocyte populations and alteration of airway hyperreactivity by C-C family chemokines in allergic airway inflammation. J. Immunol. 1997;158:4398–4404. [PubMed] [Google Scholar]
- 41.Campbell EM, Charo IF, Kunkel SL, Strieter RM, Boring L, Gosling J, Lukacs NW. Monocyte chemoattractant protein-1 mediates cockroach allergen-induced bronchial hyperreactivity in normal but not CCR2-/- mice: the role of mast cells. J. Immunol. 1999;163:2160–2167. [PubMed] [Google Scholar]
- 42.Alam R, York J, Boyars M, Stafford S, Grant JA, Lee J, Forsythe P, Sim T, Ida N. Increased MCP-1, RANTES, and MIP-1alpha in bronchoalveolar lavage fluid of allergic asthmatic patients. Am. J. Respir. Crit. Care Med. 1996;153:1398–1404. doi: 10.1164/ajrccm.153.4.8616572. [DOI] [PubMed] [Google Scholar]
- 43.Holgate ST, Bodey KS, Janezic A, Frew AJ, Kaplan AP, Teran LM. Release of RANTES, MIP-1 alpha, and MCP-1 into asthmatic airways following endobronchial allergen challenge. Am. J. Respir. Crit. Care Med. 1997;156:1377–1383. doi: 10.1164/ajrccm.156.5.9610064. [DOI] [PubMed] [Google Scholar]
- 44.Sanchez O, Marcos E, Perros F, Fadel E, Tu L, Humbert M, Dartevelle P, Simonneau G, Adnot S, Eddahibi S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2007;176:1041–1047. doi: 10.1164/rccm.200610-1559OC. [DOI] [PubMed] [Google Scholar]
- 45.Dzenko KA, Song L, Ge S, Kuziel WA, Pachter JS. CCR2 expression by brain microvascular endothelial cells is critical for macrophage transendothelial migration in response to CCL2. Microvasc. Res. 2005;70:53–64. doi: 10.1016/j.mvr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Christensen PJ, Du M, Moore B, Morris S, Toews GB, Paine R., III Expression and functional implications of CCR2 expression on murine alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286:L68–L72. doi: 10.1152/ajplung.00079.2003. [DOI] [PubMed] [Google Scholar]
- 47.Hogaboam CM, Bone-Larson CL, Lipinski S, Lukacs NW, Chensue SW, Strieter RM, Kunkel SL. Differential monocyte chemoattractant protein-1 and chemokine receptor 2 expression by murine lung fibroblasts derived from Th1- and Th2-type pulmonary granuloma models. J. Immunol. 1999;163:2193–2201. [PubMed] [Google Scholar]
- 48.Taub D, Dastych J, Inamura N, Upton J, Kelvin D, Metcalfe D, Oppenheim J. Bone marrow-derived murine mast cells migrate, but do not degranulate, in response to chemokines. J. Immunol. 1995;154:2393–2402. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.