

Abstract
We report a study of an integration-deficient lentiviral vector (IDLV) enveloped with a Sindbis virus glycoprotein mutant (SVGmu) capable of selectively binding to dendritic cells (DCs) for its potential as a vaccine carrier. The in vitro assays showed that the D64V point mutation in the catalytic domain of HIV-1 integrase efficiently inhibited the integration of the transgene upon vector transduction, while the targeting specificity of the vector to preferentially transduce and mediate durable expression in DCs was maintained. Substantial immune responses in C57BL/6 mice and complete protection against a challenge with the C57BL/6 thymoma EG.7 tumor expressing a delivered ovalbumin (OVA) antigen in mice have been achieved through the direct injection of the DC-directed IDLV encoding OVA. Thus, this DC-directed IDLV system represents a promising and efficient vector platform with remarkably improved safety for the future development of DC-based immunotherapy.
Keywords: Integration deficient lentiviral vectors, Targeting dendritic cells, Antigen-specific immune responses
1. Introduction
Although great advances have been achieved since the conception of gene therapy, the intrinsic limitations of the currently available gene delivery vehicles continues to be a major obstacle for the successful implementation of gene therapy in the clinical setting. Thus, one important ongoing effort is devoted to seeking novel gene transfer vehicles with enhanced delivery properties [1]. Viral vectors, such as the retroviral, adenoviral, and adeno-associated viral vectors, have been the focus of many delivery studies because of their high efficiency. Among these vectors, the lentiviral vector, one form of the retroviral vector, has raised a great deal of interest due to its ability of not only transducing both dividing and nondividing cells, but also maintaining long-term transgene expression through the integration of the transgene into the genome of target cells [2, 3]. On the other hand, safety issues associated with integration-induced insertional mutagenesis have seriously impaired the pace of clinical investigations of lentiviral vectors, and the challenging decision of whether or not to utilize lentiviral vectors becomes a matter of balancing therapeutic efficacy and safety considerations [3].
Inspired by the discovery of dendritic cells (DCs) as specialized antigen-presenting cells [4], DC-based immunotherapy via genetic manipulation of DCs to present specific immunogens has been extensively explored to stimulate or modulate antigen-specific immune responses [5, 6]. Lentiviral vectors have been shown to be highly efficacious in transducing both human and mouse DCs, thereby providing a powerful tool for delivery of genes into DCs [7-9]. Direct administration of lentiviral vectors has been shown to result in the transduction of DCs in vivo and therefore has been evaluated for the induction of antigen-specific immune responses in several infectious disease and tumor models [10-12]. To fully harness the great potential of DCs as the “gatekeeper” for initiating and maintaining immunity, we have recently developed a lentiviral vector system bearing a mutated glycoprotein derived from the Sindbis virus (designated as SVGmu) capable of targeting DCs through binding to the Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN) [13].
Despite the desirable advantage of lentiviral vectors to effectively deliver transgenes into DCs, vector integration has provoked safety concerns over the consequences of insertional mutagenesis [14, 15]. It has been shown that the integration of the transgene was associated with the occurrence of leukemia in children treated with retrovirus-mediated gene therapy for X-linked severe combined immunodeficiency (X-SCID) [16, 17]. In order to improve the safety profile of lentiviral vectors, considerable efforts have been made to generate integration-deficient lentiviral vectors (IDLVs) by interrupting the function of integrase or its attachment sites in the vector backbone [18, 19]. Although the integration is specifically inhibited, the resulting IDLVs can accomplish transient gene transfer to dividing cells and maintain durable transgene expression in nondividing cells [20]. To date, several reports have firmly established the utility of IDLVs for mediating gene delivery with high efficacy to a range of post-mitotic tissues [20-23] and for inducing antigen-specific immune responses upon in vivo immunization [24-27].
In this study, we investigate the immunization applications of a lentiviral vector system bearing DC-targeting specificity and a disabled propensity for integration. We generated the SVGmu-pseudotyped lentiviral vectors packaged with the defective integrase containing a D64V point mutation. This DC-directed IDLV system was shown to be effective in targeting DCs in vitro and sustaining durable transgene expression in bone marrow derived dendritic cells (BMDCs). Direct administration of the DC-directed IDLV encoding the model immunogen ovalbumin (OVA) stimulated strong and enduring antigen-specific CD8 T cell responses in vivo and afforded mice with protective antitumor immunity. It was further verified that this new vector was also efficient in inducing vaccine-specific immunity using both HIV-1 subtype B gag as an anti-HIV antigen and human gp100 as a melanoma antigen.
2. Materials and methods
2.1. Mice
C57BL/6 and Balb/c mice were purchased from Charles River Laboratories (Wilmington, MA). All mice were housed in an animal facility at the University of Southern California in accordance with institute regulations.
2.2. Plasmid construction
The packaging plasmid pMDLg/pRRE was modified to introduce the D64V point mutation in the integrase encoding region through PCR-based mutagenesis [28]. The lentiviral backbone plasmid FKOVA was generated from the FUGW construct [29] by substituting the GFP gene with the cDNA of chicken ovalbumin downstream of the human ubiquitin-C promoter. FUW is the parent construct of FUGW that lacks the GFP transgene. The cDNAs of the melanoma antigen hgp100 and HIV-1 gag were also cloned into FUW to generate the new lentiviral backbone plasmids FUW-hgp100 and FUW-gag. These lentiviral backbone plasmids (FUGW, FKOVA, FUW, FUW-hgp100 and FUW-gag) are self-inactivating (SIN) constructs with a deleted U3 region in the 3′ long terminal repeat (LTR). The packaging plasmids for making the integrating vector, pMDLg/pRRE and pRSV-REV, have been described previously [30].
2.3. Lentiviral vector production
All lentiviral vectors described in this study were pseudotyped with the mutated Sindbis virus envelope glycoprotein (SVGmu) [13]. The standard calcium phosphate precipitation procedure was utilized for transient transfection of virus-producing cells to make lentiviral vectors. HEK293T cells seeded in a 6-cm culture dish (BD Biosciences, San Jose, CA) were transiently transfected with an appropriate combination of various plasmids. The integrating lentiviral vectors were generated by the transfection of the lentiviral backbone plasmid (5 μg), packaging plasmids (pMDLg/pRRE, 2.5 μg; pRSV-REV, 2.5 μg), and the envelope plasmid SVGmu (2.5 μg). The same protocol was employed to make the integration-deficient vectors except that the mutated pMDLg/pRRE containing the D64V point mutation was used for transfection. Two days after transfection, the viral supernatant was collected and filtered through a 0.45 μm filter (Nalgene, Rochester, NY). The titers of FUGW/SVGmu(IN□) and FUGW/SVGmu(IN+) were measured by transduction of 293T.hDC-SIGN cells. The number of transduction-competant particles per ng p24 was 1.26×104 TU and 1.20×103 TU for IN+ and IN□ vectors, and was consistent across this study. The concentrated vectors for the in vivo studies were prepared by ultracentrifugation using an Optima L-90K preparative ultracentrifuge and a SW28 rotor (Beckman Coulter, Fullerton, CA) at 25,000 rpm and 4°C for 90 min. The pellets were resuspended in an appropriate volume of sterile HBSS so that the volume of concentrated vectors for immunization of each mouse was 50 μL.
2.4. Quantitative PCR analysis
293T.hDC-SIGN cells [13] were spin-transduced with either FUGW/SVGmu(IN−) or FUGW/SVGmu(IN+) and passaged everyday for two weeks. Non-transduced 293T.hDC-SIGN cells were included as a negative control. The genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). Since GFP expression from DNA espisomes was reduced rapidly to the close-to-background level through cell division within a week, vector integration was assessed by the quantitative PCR of 300 ng of genomic DNA with a primer pair specific for eGFP (FW: 5′-GGAGCGCACCATCTTCTTCA-3′; BW: 5′-AGGGTGTCGCCCTCGAA-3′; TaqMan probe: 5′-FAM-CTACAAGACCCGCGCCGAGGTG-TAMRA-3′). The assay was conducted using the TaqMan Universal PCR Master Mix and a 7300 Real Time PCR system (Applied Biosystems, Foster City, CA). The number of integrated copies per cell was calculated by utilizing the FUW plasmid to generate a standard curve and converting the 300 ng of genomic DNA to the equivalent number of 293T.hDC-SIGN cells.
2.5. Lentiviral vector transduction in vitro
Total bone marrow cells were harvested from naïve C57BL/6 mice and BMDCs were obtained by culturing bone marrow cells in the presence of GM-CSF as previously described [13]. Either bone marrow cells or BMDCs were spin-transduced twice with either the integration-deficient vector FUGW/SVGmu(IN−) or the integrating vector FUGW/SVGmu(IN+) in a 24-well plate at 2500 rpm and 27°C for 90 min. The GFP+ cells were monitored by flow cytometry analysis. At each indicated time point, BMDCs were collected and analyzed for GFP+ cells and CD11c expression; the non-transduced BMDCs were included as a control. All BMDCs were maintained by changing the conditioned medium containing GM-CSF every three days. Similarly, 293T.hDC-SIGN cells were spin-transduced and maintained to monitor GFP+ cells.
2.6. Enzyme-linked immunosorbent spot (ELISPOT) assay
Splenocytes were harvested and cocultured with either OVAp1 (SIINFEKL), OVAp2 (ISQAVHAAHAEINEAGR) or OVAp3 (KVVRFDKL) peptide (GeneScript Corp., Piscataway, NJ) overnight. On the following day, the restimulated splenocytes were counted and transferred to a MultiScreen plate (Milipore, Billerica, MA) coated with anti-mouse IL-2 antibody (BD Pharmingen, San Diego, CA). The plate was incubated at 37°C and 5% CO2 for 18~22 hours. On the third day, a biotinylated anti-mouse IL-2 antibody (BD Pharmingen) was supplied into the plate, followed by an addition of streptavidin-alkaline phosphatase (Milipore) and BCIP/NBTplus substrate (Chemicon International, Temecula, CA) to develop the spots. Spot development was stopped by thoroughly rinsing the plate with DI water. The plate was air-dried and read by a Zeiss ELISPOT reader (Carl Zeiss MicroImaging GmbH, Gottingen, Germany). The number of spot forming cells (SFC) per 106 cells was used to plot the results.
2.7. In vivo immunization of naïve mice
The fresh viral supernatant of FKOVA/SVGmu(IN−) or FKOVA/SVGmu(IN+) was concentrated in 50 μl HBSS by ultracentrifugation. The amount of viral particles present was quantified by p24 ELISA. The procedure was performed according to the protocol provided with the p24 Capture ELISA Kit from ImmunoDiagnostics (Woburn, MA). The concentrated lentiviral vectors were injected into the lower two footpads of C57BL/6 mice. The volume injected to each footpad was 25μL, which was quickly absorbed after injection and did not cause any observable problem to the mice. The immunized mice were analyzed to assess the immune responses. The integration-deficient vector encoding either the melanoma antigen hgp100 or HIV-1 gag was also generated and the results of the in vivo immunization utilizing this vector were also assessed.
2.8. Surface marker and intracellular staining
Mouse splenocytes were collected and washed in PBS. The prepared cells were stained with anti-mouse CD16/32 (BD Pharmingen, San Diego, CA) to block the Fc receptor. For tetramer staining, the cells were stained with anti-mouse CD8-PE/Cy5 (Biolengend, San Diego, CA), H-2Kb-SIINFEKL-PE tetramer (Beckman Coulter), and anti-mouse CD44-FITC (Biolengend). Anti-mouse CD62L-PE/Cy7 (Biolengend), anti-mouse CD25-PerCP (Biolengend), and anti-mouse CD69-Pacific Blue (Biolengend) were used to identify the memory T cell phenotype. For intracellular staining, the splenocytes were restimulated for 5-6 hours with OVAp1 (1 μg/ml) or OVAp3 (20 μg/ml), and with GolgiPlug (BD Pharmingen) or BFA (Aldrich Sigma, St Louis, MO) to inhibit cytokine secretion. The cell surface markers were stained with anti-mouse CD8-FITC (Biolengend) and anti-mouse CD4-PE/Cyc5 (Biolengend). The cells were then permeabilized and stained with anti-mouse IFN-γ-APC (BD Pharmingen), IL-2-PE (BD Pharmingen), and TNF-α-PE/Cy7 (BD Pharmingen). The stained cells were analyzed by flow cytometry (FACSort or LSRII, BD Biosciences).
2.9. Tumor challenge study
Two weeks after immunization with the indicated dose of either FKOVA/SVGmu(IN−), FKOVA/SVGmu(IN+), or FUW/SVGmu(IN+) as a mock control, the C57BL/6 mice were challenged with EL4 (C57BL/6, H-2b, thymoma) or EG.7 (EL4 cells stably expressing one copy of chicken OVA cDNA) tumor cells (5 × 106 cells per mouse) [13]. Tumor growth was monitored by a fine caliper and was calculated as the product of the two largest perpendicular diameters (mm2). On day 14 post-tumor transplantation, PBMCs obtained through eye-bleeding of the mice challenged with EG.7 or EL4 tumor cells were analyzed for the presence of H-2Kb-SIINFEKL-PE tetramer-positive CD8+ T cells.
3. RESULTS
3.1. Construction of IDLV directed to dendritic cells
We have previously reported an integration-competent lentiviral vector (ICLV) capable of preferentially transducing dendritic cells (DCs) [13]. The DC-targeting feature of the vector was achieved through enveloping the HIV-1-based lentiviral vector with an engineered Sindbis virus glycoprotein designated as SVGmu. Several mutations were introduced to the wild-type glycoprotein to disable its recognition to heparan sulfate structure and retain its binding to DC-SIGN, a surface protein predominantly expressed on DCs (Fig. 1A). This DC-directed ICLV could selectively deliver immunogens to DCs to efficiently stimulate antigen-specific immune responses [13]. As we are encouraged by the potential of this vaccine carrier, we are also aware of the integration characteristic of this vector, which can possibly generate insertional mutagenesis, a side effect that has been observed in a retrovirus-mediated gene therapy trial for curing the X-SCID disease [16]. To mitigate this safety concern, we decided to test the DC-directed IDLV for its ability to induce vaccine-specific immunity. A series of studies have reported a class of mutations that can specifically inhibit transgene integration while maintaining high transduction efficiency, leading to the establishment of IDLVs [18, 19]. In this study, we introduced the D64V point mutation into the catalytic domain of the integrase gene [28] (Fig. 1B) and adapted this construct into the third generation HIV-1-based lentiviral vectors to generate IDLVs.
FIGURE 1.
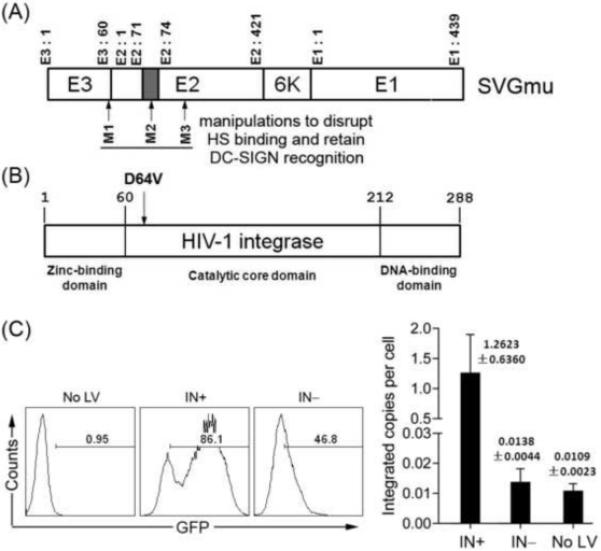
DC-directed lentiviral vectors packaged with the mutated HIV-1 integrase was defective in transgene integration. (A). The schematic representation of the engineered SVGmu glycoprotein that specifically targets the DC surface molecule DC-SIGN. The glycoprotein consists of two membrane proteins (E1 and E2) and a signal peptide (E3). A series of mutations were introduced to yield a DC-specific SVGmu (M1: deletion of amino acids 61-64 of E3; M2: insertion of an HA tag (MYPYDVPDYA) between amino acids 71 and 74 of E2; M3: mutations of 157KE158 to 157AA158). (B). The schematic structure of HIV-1 integrase harboring the point mutation D64V. (C). Quantitative PCR analysis of the integrated GFP copies in the genomic DNA from the transduced cells. HEK293T cells expressing human DC-SIGN (293T.hDC-SIGN) (1.5×105) were spin-infected with 300ng p24 of FUGW/SVGmu(IN−) or FUGW/SVGmu(IN+). Transduced cells were passaged for 2 weeks and the genomic DNA was extracted for quantitative PCR analysis with a pair of primers and a probe specific for GFP. Untransduced HEK293T cells were included as a negative control. The GFP expression (left) at one passage after transduction was analyzed by flow cytometry, and the integrated copies of GFP (right) was analyzed by quantitative PCR assay. The error bars shown in Fig. 1C represent deviation in assay duplicate points.
In order to confirm the integration deficiency afforded by the D64V point mutation, we generated the SVGmu-enveloped IDLV with FUGW as the transfer vector backbone (designated as FUGW/SVGmu(IN−)). FUGW is an HIV-1-derived lentiviral vector backbone that encodes a human ubiquitin-C promoter driving the expression of the GFP reporter gene [29]. DC-SIGN-expressing HEK293T cells (293T.hDC-SIGN) were transduced by FUGW/SVGmu(IN−) and then continuously cultured for two weeks. The integrated GFP copies were measured by a quantitative PCR assay with a pair of primers and a probe specific for GFP [31, 32]. The results showed a background-level of GFP copies in FUGW/SVGmu(IN−)-transduced 293T.hDC-SIGN, whereas significant GFP copies were detected in 293T.hDC-SIGN transduced with the integration-competent vector FUGW/SVGmu(IN+) (Fig. 1C). The reduction of integration was approximately 432 folds (calculated as [IN(+) □ No (LV)]/[IN(□) □ (No LV)]), consistent with the previous report showing that the D64V mutation can markedly reduce transgene integration by thousand-folds [22, 28].
3.2. Transduction of DCs by the SVGmu-displaying IDLV in vitro
The IDLVs have been shown to be efficacious in mediating transient transgene expression in dividing cells and relatively long-term transgene expression in nondividing cells [20]. To test the transduction efficiency of SVGmu-pseudotyped IDLVs, 293T.hDC-SIGN and bone marrow derived DCs (BMDCs) were transduced by either FUGW/SVGmu(IN−) or FUGW/SVGmu(IN+), and GFP expression was monitored over a certain period of time. In sharp contrast to the sustained GFP expression from FUGW/SVGmu(IN+), ~50% GFP+ 293T.hDC-SIGN resulted from FUGW/SVGmu(IN−) at one passage post-transduction, but its expression decreased to the background level at day 6 post-transduction (Fig. 2A). In BMDCs, both integration-competent and integration-deficient FUGW/SVGmu maintained GFP expression for up to 3 weeks, despite the fact that the FUGW/SVGmu(IN−) transduction resulted in a lower level of GFP+ BMDCs (Fig. 2B).
FIGURE 2.
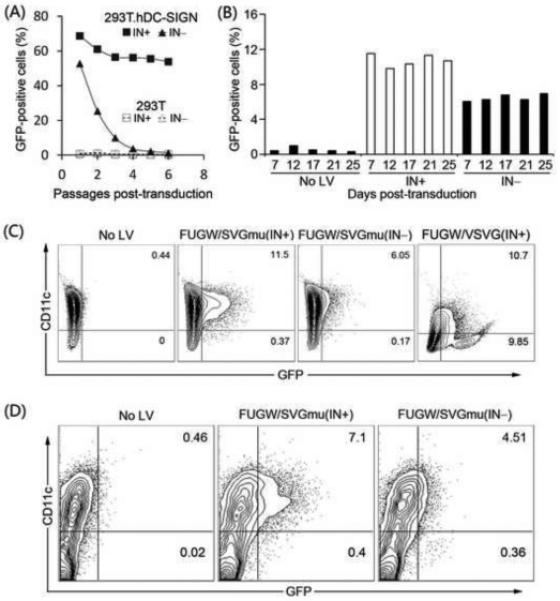
DC-directed IDLVs mediated efficient transduction and targeted DCs in vitro. (A). 293T.hDC-SIGN cells were spin-transduced with 30ng p24 of fresh viral supernatant of FUGW/SVGmu(IN−) or FUGW/SVGmu(IN+), and the GFP expression was measured over time by flow cytometry. Transduction in 293T cells served as a negative control. (B) and (C). Bone marrow cells harvested from naïve C57BL/6 mice were cultured in the presence of GM-CSF for 6 days to differentiate bone marrow derived dendritic cells (BMDCs). BMDCs were exposed to 40ng p24 of fresh viral supernatant of FUGW/SVGmu(IN−) or FUGW/SVGmu(IN+), and the variation of GFP expression over the time was recorded in (B). The co-expression of GFP and CD11c at 7 days post-transduction is shown in (C). Transduction of BMDCs with the VSVG-pseudotyped lentiviral vector (FUGW/VSVG(IN+)) was included as a non-DC-targeting control. (D). Total bone marrow cells were harvested and cultured overnight in presence of GM-CSF. On the following day, bone marrow cells were spin-transduced with 600ng p24 of concentrated FUGW/SVGmu(IN−) or FUGW/SVGmu(IN+). Two days later, the expression of GFP and CD11c was analyzed by flow cytometry.
It has been shown that ICLV pseudotyped with SVGmu can preferentially modify DCs [13]. In the present study, total bone marrow cells and BMDCs were exposed to FUGW/SVGmu(IN−), and an anti-mouse CD11c antibody was used to identify the DC population. For both of the types of cells treated with the vector, only CD11c+ cells were transduced to express GFP (Figs. 2C and 2D), indicating that the SVGmu-displaying IDLV retained a similar targeting specificity to DCs as its integration-competent vector counterpart. As a control, the VSVG-pseudotyped lentiviral vector (FUGW/VSVG(IN+)) didn’t exhibit transduction specificity when exposed to BMDCs (Fig 2C).
3.3. In vivo immunization with the SVGmu-displaying IDLV
The results above clearly demonstrated that IDLVs pseudotyped with SVGmu could target DCs in vitro and maintain persistent transgene expression in poorly proliferative BMDCs, which satisfies the prerequisite for delivering antigens to DCs in vivo for direct immunization. Therefore, we assessed the capacity of SVGmu-pseudotyped IDLVs to induce antigen-specific CD8 T cell responses in vivo using chicken ovalbimun (OVA) [33, 34] as the model antigen. Naïve C57BL/6 mice were immunized with the OVA-encoding IDLV (designated as FKOVA/SVGmu (IN−)); the vector dose was measured by the amount of p24 contained in the prepared vector. Immune responses were analyzed at 2 weeks post-injection. For mice immunized with ~1800 ng p24 of FKOVA/SVGmu(IN−), more than 17% of the CD8 T cells in the spleen were specific to the OVA epitope (OVAp1: SIINFEKL) and were able to secrete IFN-γ upon stimulation with this peptide (Figs. 3A and 3B). The magnitude of the response from immunization with FKOVA/SVGmu(IN−) increased when higher doses of the vector were used. On the contrary, an intermediate dose (~700 ng p24) of FKOVA/SVGmu(IN+) elicited the highest response (Figs. 3A and 3B). This dosage study indicated that the dose response differed between the integration-competent and the integration-deficient vectors. Nevertheless, the strongest immune responses were comparable, ~20.5% and ~19.35% OVA-specific CD8 T cells, after immunization with either FKOVA/SVGmu(IN−) or FKOVA/SVGmu(IN+), respectively. In order to exclude the possible immunization by the residual OVA protein from the vector preparation, we immunized mice with lentiviral vectors generated by the transfection protocol lacking the envelope plasmid. The resulting immune responses were similar to that from unimmunized mice (data now shown). Therefore, IDLVs bearing SVGmu are similarly effective for in vivo immunization with higher doses, as compared with their integrating counterpart.
FIGURE 3.
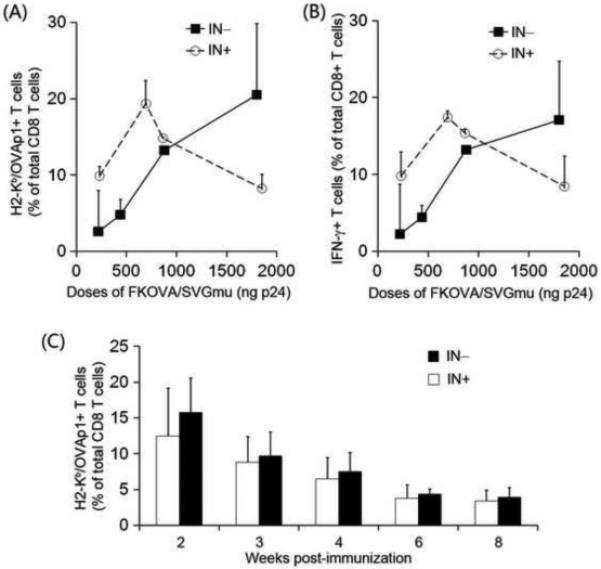
Substantial antigen-specific CD8 T cell responses induced by a single in vivo immunization with a DC-directed IDLV encoding OVA. (A) and (B). Naïve C57BL/6 mice were immunized with a single footpad injection of different doses of FKOVA/SVGmu(IN−) or FKOVA/SVGmu(IN+), indicated as the amount of p24 contained in the injected viral vectors. For each dose, two mice were immunized. Two weeks post-immunization, splenocytes isolated from treated mice were analyzed by flow cytometry. Unimmunized mice were included as a negative control, giving a less than 1% of H-2Kb-SIINFEKL-PE tetramer-positive or IFN-γ+ CD8+ T cells. (A). The frequency of OVAp1-specific CD8 T cells was measured by surface staining using the H-2Kb-SIINFEKL-PE tetramer. (B). Upon reactivation with OVAp1 for 5-6 hours, splenocytes were analyzed by intracellular staining using a PE-conjugated anti-mouse IFN-γ. (C). The kinetic study of immune responses in mice injected with 669 ng p24 of FKOVA/SVGmu(IN−) or FKOVA/SVGmu(IN+). 4 mice were included in each group. Starting at week 2 post-immunization, peripheral blood was collected by retro-orbital bleeding and harvested cells were analyzed by tetramer staining. The error bars shown in the figure represent deviation between mice within a group.
3.4. Kinetic study of the immune response upon lentiviral vector immunization
Although the transgene expression from IDLVs is mediated through the episomal forms of the vector DNA [19, 35, 36], previous studies have shown that IDLVs can achieve sustained immune responses to the encoded antigens [24-27]. Having shown that SVGmu-pseudotyped IDLVs are capable of maintaining stable transgene expression in nondividing BMDCs and inducing vigorous antigen-specific immunity in vivo, we were interested in monitoring the long-term immune response induced by FKOVA/SVGmu(IN−). Each ïve mouse was immunized with 669 ng of the vector (FKOVA/SVGmu(IN−) or (FKOVA/SVGmu(IN+)), and the resulting immune responses were monitored starting at 2 weeks post-vaccination. Consistent with the dosage study, FKOVA/SVGmu(IN□) primed a similar level of OVA-specific CD8 T cells as that primed by FKOVA/SVGmu(IN+) at 2 weeks post-immunization. The decrease patterns of the pool of OVA-specific CD8 T cells over time were similar for both vectors (Fig. 3C). Six to eight weeks after the immunizations, ~4% OVA-specific CD8 T cells were present in mice immunized with either of the two vectors, suggesting that the DC-directed IDLVs were able to maintain a similar level of long-term antigen-specific immunity as that of the integrating vector.
3.5. Functional characterization of the antigen-specific CD8 T cells
Having demonstrated the effectiveness of DC-directed IDLVs for in vivo immunizations, we further examined the characteristics of the long-lived antigen-specific CD8 T cells. Ten weeks after the immunizations, splenocytes were collected for the analysis of memory T cell phenotype and multi-cytokine secretion, which have been shown to be key indicators of the development of a high quality vaccine [37]. Flow cytometry analysis revealed that ~40% of the OVA-specific CD8 T cells were CD44+CD62L+CD25−CD69−(Figs. 4A and 4B) in both FKOVA/SVGmu(IN−) and FKOVA/SVGmu(IN+) immunized mice,which is the typical phenotype of central memory T cells [38]. As for multi-cytokine production, the frequencies of either IFN-γ+TNF-α−, IFN-γ−TNF-α+, or IFN-γ+TNF-α+ CD8 T cells were comparable between the FKOVA/SVGmu(IN−) and FKOVA/SVGmu(IN+) immunized mice upon stimulation with the H-2Kb-restricted dominant OVAp1 or subdominant OVAp3 peptide (OVAp3: KVVRFDKL) (Fig. 4C). The IL-2 signal in our intracellular multi-cytokine assay was too low to be reliably analyzed. For this reason, we utilized a more sensitive ELISpot assay to detect IL-2 production. Reactivated by the OVAp1, OVAp2 (ISQAVHAAHAEINEAGR), or OVAp3 peptides, the splenocytes exhibited similar numbers of IL-2 spot-forming cells (SFC) for mice immunized by either vector (Fig. 4D). Thus, our results indicated that, compared to the integrating vector, immunization with DC-targeted IDLVs could generate a similar quality of antigen-specific CD8 T cells as evidenced by the memory and multifunctionality analysis.
FIGURE 4.
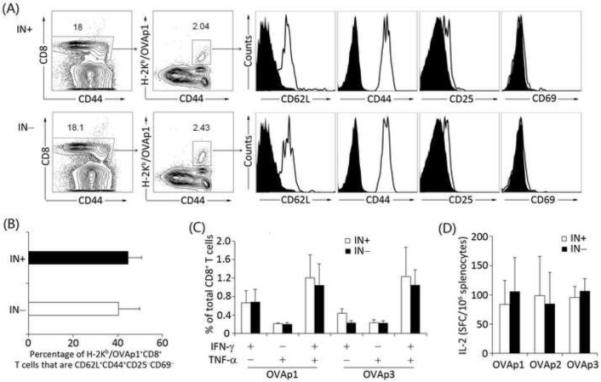
Long-lived antigen-specific CD8 T cells exhibited the central memory phenotype and were able to secrete multi-cytokines upon peptide reactivation. Naïve C57BL/6 mice were immunized with 669 ng p24 of FKOVA/SVGmu(IN−) (number of mice: n=3) or FKOVA/SVGmu(IN+) (number of mice: n=3) through a footpad injection. Ten weeks later, splenocytes were analyzed by flow cytometry analysis and an ELISPOT assay. (A). Characterization of the surface markers on OVA-specific CD8 T cells with 6-color staining. H-2Kb-SIINFEKL-PE tetramer along with anti-mouse CD8, CD25, CD69, CD44, and CD62L antibodies were used to identify OVA-specific CD8 memory T cells. (B). Quantification of the frequency of OVA-specific CD8 T cells that were displaying the central memory phenotype of CD44+CD62L+CD25−CD69−. (C). Splenocytes were cocultured with the dominant epitope, OVAp1, or the sub-dominant epitope, OVAp3, in the presence of costimulation with an anti-mouse CD28 antibody. Cytokine production was measured by multi-color intracellular staining. (D). Splenocytes were evaluated for their secretion of IL-2 by an ELISPOT assay after restimulation with OVAp1, OVAp2, or OVAp3. The error bars shown in this figure represent deviation between mice within a group.
3.6. Generation of antigen-specific antitumor immunity
We utilized the EG.7 tumor model, in which the tumor cell line EG.7 stably expresses one copy of chicken OVA cDNA [13] to further assess the antigen-specific T cell immunity generated by the SVGmu-displaying IDLV. The naïve C57BL/6 mice were immunized with either FKOVA/SVGmu(IN−) or FKOVA/SVGmu(IN+), and subsequently challenged with tumor cells two weeks post-vaccination; EL4 cells lacking the expression of the tumor antigen OVA was used as a control. By monitoring the growth of tumors, we observed a complete protection against the EG.7 challenge in the FKOVA/SVGmu(IN−) and FKOVA/SVGmu(IN+) immunized mice (Fig. 5A), which was in sharp contrast to the rapid progression of EL4 tumor cells in mice vaccinated by the same sets of vectors (Fig. 5B). In addition, flow cytometry analysis showed that the integration-competent and integration-deficient vectors elicited a similar level of OVA-specific CD8 T cells in mice challenged with either the EG.7 or the EL4 tumor (Figs. 5C and 5D), confirming that the observed antitumor immunity in EG.7-bearing mice was antigen-specific. Thus, this tumor challenge study suggested that the DC-targeted IDLVs were comparably efficacious to its integrating counterpart in inducing protective OVA-specific antitumor immunity.
FIGURE 5.
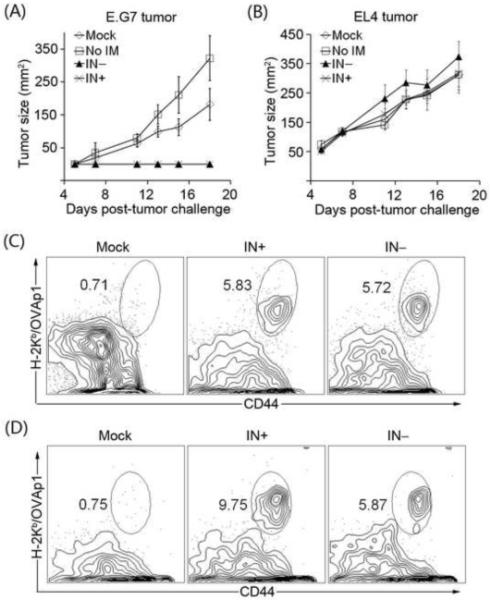
Effective antigen-specific anti-tumor immunity was generated by a single injection of the DC-directed IDLV. Naïve C57BL/6 mice were immunized with FKOVA/SVGmu(IN−), FKOVA/SVGmu(IN+), or the empty vector FUW/SVGmu(IN+) (denoted as Mock). The amount of viral particles injected into each mouse was measured as approximately 800 ng p24. Mice without immunization (represented as No IM) were included as a negative control. Two weeks later, each mouse was subcutaneously transplanted with 5×106 tumor cells of either EG.7 cells expressing OVA or EL4 cells lacking OVA expression. Tumor growth was monitored by calculating the product of the largest perpendicular length and width measured with a fine caliper. Each group was composed of four mice. (A). The growth curve of EG.7 tumors in the treated or untreated mice. (B). The growth curve of EL4 tumors in the treated or untreated mice. (C). Representative data for the frequency of OVA-specific CD8 T cells in the EG.7 tumor-challenged mice. (D). Representative data for the frequency of OVA-specific CD8 T cells in the EL4 tumor-challenged mice. The error bars shown in this figure represent deviation between mice within a group.
3.7. Lentiviral vector immunization using melanoma and HIV-1 antigens
As we have demonstrated that the DC-directed IDLV system is efficacious in delivering the model immunogen OVA to stimulate antigen-specific immunity, we further investigated whether this system could be employed to deliver some more physiologically relevant tumor and viral antigens. To this end, the sequences of the melanoma antigen human gp100 (hgp100) [39] and HIV-1 subtype B gag [40] were inserted into the lentiviral vector backbone FUW and designated as FUW-hgp100 and FUW-gag, respectively. Naïve C57BL/6 mice were immunized with the SVGmu-pseudotyped IDLV (FUW-hgp100/SVGmu(IN−)) and Balb/c mice were immunized with FUW-gag/SVGmu(IN−). Three weeks post-immunization, splenocytes were harvested from immunized animals to analyze the antigen-specific CD8 T cells responses, which revealed the presence of ~10% hgp100-specific and ~1.8% HIV-1 gag-specific CD8 T cells (Figs. 6A and 6B). As compared with the level of immune responses reported in previous studies [39, 40], the results of this immunization test indicated that this DC-directed IDLV system is a potent vaccine platform for delivering both tumor and viral antigens.
FIGURE 6.
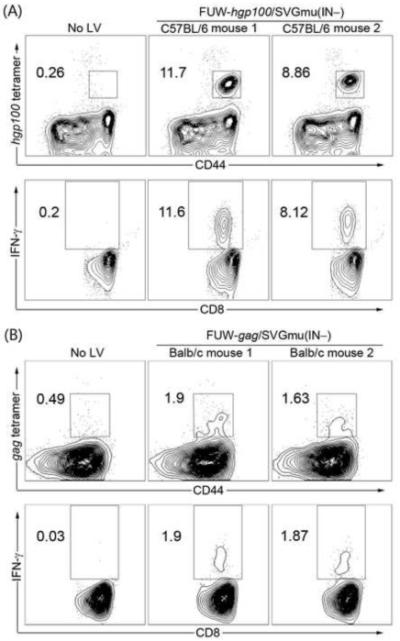
Strong antigen-specific CD8 T cells responses were elicited by DC-directed IDLVs for in vivo delivery of the melanoma antigen hgp100 or HIV-1 subtype B gag. (A). Each of the two naïve C57BL/6 mice received 1048 ng p24 of the DC-directed IDLV encoding the melanoma antigen hgp100 (denoted as FUW-hgp100/SVGmu(IN−)) through a footpad injection. A mouse receiving no immunization (No LV) was included as a negative control. At 3 weeks post-immunization, splenocytes were analyzed by tetramer staining (upper) and intracellular staining (lower). (B). Each of the two naïve Balb/c mice were administered 16200 ng p24 of the DC-directed IDLV encoding HIV-1 gag (denoted as FUW-gag/SVGmu(IN−)) through the footpad route. A mouse receiving no immunization (No LV) was included as a negative control. At 3 weeks post-immunization, splenocytes were analyzed by tetramer staining (upper) and intracellular staining (lower).
4. Discussion
We previously reported a method to disable the heparin sulfate binding ability of the Sindbis virus glycoprotein (SVG) for targeting DCs through DC-SIGN [41]. Lentiviral vectors pseudotyped with this engineered glycoprotein SVGmu have been shown to be highly effective in specifically modifying both human and mouse DCs [13]. Because of the targeting feature, this system requires many fewer infectious vector particles for in vivo immunization and has a reduced chance of transducing off-target cells, which might potentially enhance the safety and efficacy of the vaccination. However, the genotoxicity associated with insertional mutagenesis, an intrinsic property of integrating lentiviral vectors, raises serious concerns for their clinical applications. Recently, a series of studies have shown that IDLVs can retain effective transduction and relatively persistent transgene expression in less proliferative cells [18, 19]; the transgene integration of the lentiviral vector can be prevented by a class of mutations in HIV-1 integrase or the integrase attachment sites in the viral LTR. Thus, it is of great interest to evaluate the integration-deficient DC-directed lentiviral vectors for efficacious and safe targeting of DCs for in vivo immunizations.
D64V is a point mutation in the catalytic domain of HIV-1 integrase that is able to significantly reduce the occurrence of integration of vector-delivered genetic materials into target cell genomes [28]. In this present study, SVGmu-pseudotyped FUGW packaged with the defective integrase harboring the D64V mutation (FUGW/SVGmu(IN−)) was able to mediate transient GFP expression in HEK293T cells expressing hDC-SIGN, while it sustained stable GFP expression in BMDCs over three weeks, presumably due to the poorly proliferative nature of this cell type [42]. Further analysis of the total bone marrow cells and the BMDCs revealed that FUGW/SVGmu(IN−) retained the same property of targeting CD11c-positive DCs as its integrating counterpart. This is perhaps not surprising considering the fact that the displayed envelope and the encapsulated integrase perform biologically separate functions during vector transduction. The SVGmu envelope is involved in directing the vector to the DC-SIGN receptor to induce endocytosis, followed by mediating the release of the viral core from endosomes in a pH-dependent manner, whereas the D64V integration mutation prohibits vector integration of reversely transcribed proviral DNA [28].
Consistent with the preceding studies [26, 27], the DC-directed IDLVs carrying the model antigen OVA (FKOVA/SVGmu(IN−)) required a higher dose to attain comparable antigen-specific CD8 T cell responses to those of the integrating vector in vivo, probably because the level of transgene expression from IDLVs is lower than that from the integrating lentiviral vector [23]. Nevertheless, an optimal response (up to ~20% OVA-specific CD8 T cells) could be elicited by a single injection of ~1800 ng p24 of FKOVA/SVGmu(IN−). Interestingly, we found that a higher than the optimal dose of integrating vectors induced lower level of OVA-specific CD8 T cells, in sharp contrast to the highest immune response elicited by the highest dose of IDLVs. We are currently designing experiments to understand this interesting difference of the dose response between integrating and integration-deficient vectors. The kinetic study of the vaccine-specific immune response, which monitored the frequency of OVA-specific CD8 T cells in peripheral blood, demonstrated that the DC-directed IDLV was comparable to the integrating vector in maintaining durable immune responses. Furthermore, the characterization of long-lived OVA-specific CD8 T cells at 10 weeks post-immunization indicated that the integration-competent and integration-deficient vectors mounted similar levels of memory-characteristic and multi-cytokine secreting T cells. In terms of protective anti-tumor immunity, we observed that a single administration of FKOVA/SVGmu(IN−) completely prevented the growth of EG.7 tumor cells expressing the OVA antigen. Finally, the test of using DC-directed IDLVs for in vivo delivery of HIV-1 subtype B gag and the melanoma antigen hgp100 further confirmed that this system can be used as a safe and immunogenic platform for the development of novel vectored vaccines.
Although the development of IDLVs can greatly ameliorate the safety concerns over the genotoxicity triggered by vector integration, more efforts are needed to improve the limitations associated with IDLVs for vaccine applications. Due to the absence of vector integration, the transgene expression from IDLVs is mediated by the episomal vector DNA, including the linear form and circular form with one or two long terminal repeats (LTR) [18, 19, 35, 36]. This mechanism leads to a transient transgene expression in dividing cells since the episomal DNA accumulated in the nucleus after transduction can be rapidly diluted by cell division [20]. The relatively low efficiency of transcription from this episomal DNA might explain the low level of transgene expression observed in DCs. Strategies to generate replicating episomes for IDLVs could potentially circumvent this limitation [18]. A recent study reported a method to enhance transgene expression of IDLVs by deleting a large section of the U3 region that may contain the cis-acting elements negatively regulating transgene expression [23]. In addition, it is meaningful to establish assays to further characterize the potential effect of the residual integration in the insertional activation of DCs, taking into consideration the fact that, although vector integration has been significantly inhibited in IDLVs, it is unlikely to be completely eliminated [43].
In conclusion, we demonstrate that IDLVs pseudotyped with an engineered glycoprotein derived from the Sindbis virus retain the feature of targeting DCs and are able to induce a high level of antigen-specific immune responses in vivo. Compared to the integrating form of DC-directed lentiviral vectors, this vector system greatly mitigates the potential risk of insertional mutagenesis. Functional analysis indicates that both the magnitude and quality of the immune responses elicited by the DC-directed IDLVs are comparable to that of its integrating counterpart. Further studies are underway to assess this vaccine platform for DC-based immunotherapy against cancers and infectious diseases.
Acknowledgements
We thank Drs. David Baltimore and Lili Yang from California Institute of Technology for the insightful discussion on the experiment and April Tai for critical reading of the manuscript. This work was supported by a grant from the U.S. National Institutes of Health (NIH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Pfeifer A, Verma IM. Gene therapy: Promises and problems. Annu Rev Genom Hum G. 2001;2:177–211. doi: 10.1146/annurev.genom.2.1.177. [DOI] [PubMed] [Google Scholar]
- [2].Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–7. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- [3].Kootstra NA, Verma IM. Gene therapy with viral vectors. Annu Rev Pharmacol. 2003;43:413–39. doi: 10.1146/annurev.pharmtox.43.100901.140257. [DOI] [PubMed] [Google Scholar]
- [4].Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- [5].Figdor CG, de Vries IJM, Lesterhuis WJ, Melief CJM. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–80. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- [6].Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–26. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- [7].Dullaers M, Thielemans K. From pathogen to medicine: HIV-1-derived lentiviral vectors as vehicles for dendritic cell based cancer immunotherapy. J Gene Med. 2006;8:3–17. doi: 10.1002/jgm.846. [DOI] [PubMed] [Google Scholar]
- [8].Schroers R, Chen SY. Lentiviral transduction of human dendritic cells. Methods Mol Biol. 2004;246:451–9. doi: 10.1385/1-59259-650-9:451. [DOI] [PubMed] [Google Scholar]
- [9].Esslinger C, Romero P, MacDonald HR. Efficient transduction of dendritic cells and induction of a T-cell response by third-generation lentivectors. Hum Gene Ther. 2002;13:1091–100. doi: 10.1089/104303402753812494. [DOI] [PubMed] [Google Scholar]
- [10].Kim JH, Majumder N, Lin H, Watkins S, Falo LD, Jr., You Z. Induction of therapeutic antitumor immunity by in vivo administration of a lentiviral vaccine. Hum Gene Ther. 2005;16:1255–66. doi: 10.1089/hum.2005.16.1255. [DOI] [PubMed] [Google Scholar]
- [11].Dullaers M, Van Meirvenne S, Heirman C, Straetman L, Bonehill A, Aerts JL, et al. Induction of effective therapeutic antitumor immunity by direct in vivo administration of lentiviral vectors. Gene Ther. 2006;13:630–40. doi: 10.1038/sj.gt.3302697. [DOI] [PubMed] [Google Scholar]
- [12].Lemiale F, Korokhov N. Lentiviral vectors for HIV disease prevention and treatment. Vaccine. 2009;27:3443–9. doi: 10.1016/j.vaccine.2009.01.055. [DOI] [PubMed] [Google Scholar]
- [13].Yang L, Yang H, Rideout K, Cho T, Joo KI, Ziegler L, et al. Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat Biotechnol. 2008;26:326–34. doi: 10.1038/nbt1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687–96. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- [15].Bokhoven M, Stephen SL, Knight S, Gevers EF, Robinson IC, Takeuchi Y, et al. Insertional gene activation by lentiviral and gammaretroviral vectors. J Virol. 2009;83:283–94. doi: 10.1128/JVI.01865-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–9. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- [17].Nienhuis AW, Dunbar CE, Sorrentino BP. Genotoxicity of retroviral integration in hematopoietic cells. Mol Ther. 2006;13:1031–49. doi: 10.1016/j.ymthe.2006.03.001. [DOI] [PubMed] [Google Scholar]
- [18].Wanisch K, Yanez-Munoz RJ. Integration-deficient lentiviral vectors: a slow coming of age. Mol Ther. 2009;17:1316–32. doi: 10.1038/mt.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Philpott NJ, Thrasher AJ. Use of nonintegrating lentiviral vectors for gene therapy. Hum Gene Ther. 2007;18:483–9. doi: 10.1089/hum.2007.013. [DOI] [PubMed] [Google Scholar]
- [20].Philippe S, Sarkis C, Barkats M, Mammeri H, Ladroue C, Petit C, et al. Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo. Proc Natl Acad Sci USA. 2006;103:17684–9. doi: 10.1073/pnas.0606197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yanez-Munoz RJ, Balaggan KS, MacNeil A, Howe SJ, Schmidt M, Smith AJ, et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat Med. 2006;12:348–53. doi: 10.1038/nm1365. [DOI] [PubMed] [Google Scholar]
- [22].Apolonia L, Waddington SN, Fernandes C, Ward NJ, Bouma G, Blundell MP, et al. Stable gene transfer to muscle using non-integrating lentiviral vectors. Mol Ther. 2007;15:1947–54. doi: 10.1038/sj.mt.6300281. [DOI] [PubMed] [Google Scholar]
- [23].Bayer M, Kantor B, Cockrell A, Ma H, Zeithaml B, Li X, et al. A large U3 deletion causes increased in vivo expression from a nonintegrating lentiviral vector. Mol Ther. 2008;16:1968–76. doi: 10.1038/mt.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Negri DR, Michelini Z, Baroncelli S, Spada M, Vendetti S, Buffa V, et al. Successful immunization with a single injection of non-integrating lentiviral vector. Mol Ther. 2007;15:1716–23. doi: 10.1038/sj.mt.6300241. [DOI] [PubMed] [Google Scholar]
- [25].Coutant F, Frenkiel MP, Despres P, Charneau P. Protective antiviral immunity conferred by a nonintegrative lentiviral vector-based vaccine. PLoS One. 2008;3:e3973. doi: 10.1371/journal.pone.0003973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Karwacz K, Mukherjee S, Apolonia L, Blundell MP, Bouma G, Escors D, et al. Nonintegrating lentivector vaccines stimulate prolonged T-cell and antibody responses and are effective in tumor therapy. J Virol. 2009;83:3094–103. doi: 10.1128/JVI.02519-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hu B, Yang H, Dai B, Tai A, Wang P. Nonintegrating lentiviral vectors can effectively deliver ovalbumin antigen for induction of antitumor immunity. Hum Gene Ther. 2009;20:1652–64. doi: 10.1089/hum.2009.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Leavitt AD, Robles G, Alesandro N, Varmus HE. Human immunodeficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J Virol. 1996;70:721–8. doi: 10.1128/jvi.70.2.721-728.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–72. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- [30].Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–71. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Geraerts M, Willems S, Baekelandt V, Debyser Z, Gijsbers R. Comparison of lentiviral vector titration methods. BMC Biotechnol. 2006;6:34. doi: 10.1186/1472-6750-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001;7:631–4. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- [33].Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- [34].Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- [35].Vargas J, Jr., Gusella GL, Najfeld V, Klotman ME, Cara A. Novel integrase-defective lentiviral episomal vectors for gene transfer. Hum Gene Ther. 2004;15:361–72. doi: 10.1089/104303404322959515. [DOI] [PubMed] [Google Scholar]
- [36].Saenz DT, Loewen N, Peretz M, Whitwam T, Barraza R, Howell KG, et al. Unintegrated lentivirus DNA persistence and accessibility to expression in nondividing cells: analysis with class I integrase mutants. J Virol. 2004;78:2906–20. doi: 10.1128/JVI.78.6.2906-2920.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–62. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- [38].Dutton RW, Bradley LM, Swain SL. T cell memory. Annu Rev Immunol. 1998;16:201–23. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- [39].Perricone MA, Claussen KA, Smith KA, Kaplan JM, Piraino S, Shankara S, et al. Immunogene therapy for murine melanoma using recombinant adenoviral vectors expressing melanoma-associated antigens. Mol Ther. 2000;1:275–84. doi: 10.1006/mthe.2000.0029. [DOI] [PubMed] [Google Scholar]
- [40].Dai B, Yang L, Yang H, Hu B, Baltimore D, Wang P. HIV-1 Gag-specific immunity induced by a lentivector-based vaccine directed to dendritic cells. Proc Natl Acad Sci USA. 2009;106:20382–7. doi: 10.1073/pnas.0911742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhou T, Chen Y, Hao L, Zhang Y. DC-SIGN and immunoregulation. Cell Mol Immunol. 2006;3:279–83. [PubMed] [Google Scholar]
- [42].Matsuno K, Ezaki T, Kudo S, Uehara Y. A life stage of particle-laden rat dendritic cells in vivo: their terminal division, active phagocytosis, and translocation from the liver to the draining lymph. J Exp Med. 1996;183:1865–78. doi: 10.1084/jem.183.4.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gaur M, Leavitt AD. Mutations in the human immunodeficiency virus type 1 integrase D,D(35)E motif do not eliminate provirus formation. J Virol. 1998;72:4678–85. doi: 10.1128/jvi.72.6.4678-4685.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]