

Abstract
Alternative pre-mRNA splicing predominates in the nervous systems of complex organisms including humans dramatically expanding the potential size of the proteome. Cell-specific alternative pre-mRNA splicing is thought to optimize protein function for specialized cellular tasks, but direct evidence for this is limited. Transmission of noxious thermal stimuli relies on the activity of N-type CaV2.2 calcium channels in nociceptors. Using an exon replacement strategy in mice, we show that mutually exclusive splicing in the CaV2.2 gene modulates N-type channel function in nociceptors leading to a change in morphine analgesia. Exon 37a enhances μ-opioid receptor mediated inhibition of N-type calcium channels by promoting activity-independent inhibition. In the absence of e37a spinal morphine analgesia is weakened in vivo without influencing the basal response to noxious thermal stimuli. Our data suggest that highly specialized, discrete cellular responsiveness in vivo can be attributed to alternative splicing events regulated at the level of individual neurons.
Alternative pre-mRNA splicing is used extensively in the nervous systems of complex organisms including humans 1–3. Transcriptome analyses suggest that alternative pre-mRNA splicing events occur in the vast majority of multi-exon human genes 4. Furthermore, an increasing number of human diseases are linked to defects in alternative splicing 5. Neurons are thought to use alternative splicing to add and subtract discrete protein modules encoded by alternative exons to optimize protein function for specific cellular tasks. We, and others have evidence that cell-dependent inclusion of alternatively spliced exons in ion channels and receptors act like molecular switches permitting new interactions that control channel gating and receptor targeting 1, 6, 7. However, studies linking a specific splicing event in an identified population of neurons to alterations in native protein function and behavior are limited 1, 2. The challenge is perhaps greatest in the nervous system where a heterogeneous population of neurons express a vast number of multi-exon genes 2, 8. Here we study the function of one site of alternative splicing in the Cacna1b gene, which encodes the pore-forming subunit of the N-type channel.
Ion channels underlie all electrical signals in the nervous system 9. They are highly sensitive to perturbations in their structure or chemical environment. Moreover, these alterations can be monitored with precision. Studies of ion channel splice isoforms have shown that alternatively spliced exons can modify ion channel plasma membrane and sub-cellular targeting and trafficking, second messenger sensitivities, and channel gating properties 1, 6, 10.
Several groups including ours show that alternatively spliced exons in voltage-gated calcium channel genes follow tissue specific expression patterns and show that they modify channel activity and drug sensitivity 1, 7, 11–15. But we know little of the cellular consequences in neurons and nothing of the behavioral consequences of regulated alternative splicing events. We have focused on the presynaptic CaV2.2 encoding gene, Cacna1b, which is the core of the N-type calcium channel. In particular, we focus on a pair of mutually exclusive exons, 37a (e37a) and 37b (e37b), in CaV2.2 that encode 32 amino acids in the proximal region of the protein’s C-terminus. 14 of 32 amino acids differ between e37a and e37b. E37a exhibits an intriguing expression profile; CaV2.2[e37a] mRNAs are enriched in nociceptors of dorsal root ganglia (DRG) and expressed at lower levels in other regions including brain, while CaV2.2[e37b] mRNAs are ubiquitous in the nervous system 16, 17. By studying clones of CaV2.2 splice isoforms expressed in a mammalian cell line, we showed that e37a creates an inhibitory domain in the CaV2.2 protein that renders N-type calcium channels sensitive to a form of Gi/o protein-dependent inhibition that persists when cells are depolarized 7. We also found e37a promotes greater N-type current density 16, 17. In vivo silencing of CaV2.2[e37a] reduced basal thermal nociception, suggesting this isoform participates in transmission at C-fiber terminals 18. But the role of CaV2.2[e37a] channels in Gi/o protein inhibition of native N-type channels in the pain pathway remain unexplored.
With this in mind, we devised a mouse model to determine why CaV2.2[e37a] channels are enriched in nociceptors of DRG. We eliminated e37a from the mouse Cacna1b gene and replaced it with a second 37b-encoding exon. By comparing these mice to wild-type that express both e37a and e37b isoforms, we show that e37a regulates μ-opioid receptor-mediated inhibition of native N-type calcium channels in nociceptors and show that spinal level analgesia by morphine in vivo is reduced in the absence of e37a by a mechanism that does not affect basal transmission of noxious thermal stimuli.
RESULTS
Exon-substitution in the Cacna1b mouse gene
To determine how e37a impacts N-type calcium channel activity in vivo we chose an exon substitution strategy aimed at eliminating the CaV2.2[e37a] splice isoform while preserving cell-directed splicing and N-type channel expression. Figure 1 illustrates our approach. E37a and e37b are mutually exclusive, identical in size and can substitute for each other structurally and, in part, functionally. We replaced e37a in the mouse Cacna1b gene with an e37b-encoding sequence (e37b*), creating tandem 37b exons (Cacna1bb*b/b*b) and eliminating e37a. We also generated e37a-only expressing mice (Cacna1baa*/aa*) by the same strategy, replacing e37b in Cacna1b with e37a-encoding sequence (e37a*) (Fig. 1 and Supplementary Fig. 1). Correct Cacna1b targeting was verified by genomic PCR (Supplementary Fig. 1) and sequencing. Mice homozygous for each mutation, Cacna1bb*b/b*b and Cacna1baa*/aa*, were viable and fecund. Cacna1bb*b/b*b homozygotes were born at wild-type (WT) frequencies but Cacna1baa*/aa* homozygotes were born at ~50% of the expected Mendelian rate (Supplementary Table 1). Our data suggest that Cacna1bb*b/b*b homozygotes tolerate e37b* as a replacement exon for e37a, whereas Cacna1baa*/aa* homozygotes are physiologically compromised. These data point to a global disruption of N-type calcium channel function (Supplementary Table 1).
Figure 1. Exon 37-substitution in Cacna1b and resultant mRNAs in WT and mutant DRG.
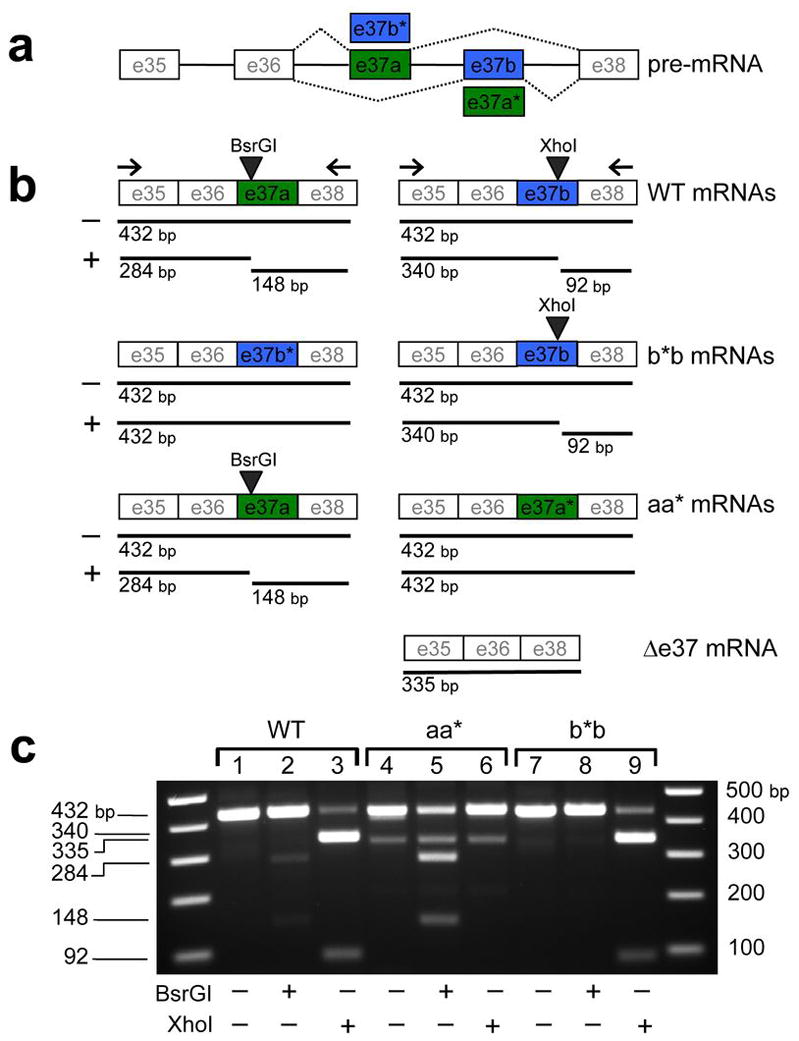
Schematic of Cacna1b gene showing mutually exclusive exons e37a (green) and e37b (blue) with constitutively expressed exons (e35, e36, and e38) (a). Exon replacement strategy illustrates predicted mRNAs (b). E37b* replaces e37a, and e37a* replaces e37b to generate Cacna1bb*b/b*b (b*b) and Cacna1baa*/aa* (aa*) mice, respectively. Sizes of predicted RT-PCR products using primers located in exons 35 and 38 from CaV2.2 mRNAs derived from DRG of WT, b*b, and aa* mice are shown. Sizes of PCR products undigested (−) and digested with BsrGI (+), and XhoI (+) are shown below each mRNA. Uncut cDNAs containing either e37a or e37b are 432 bp and cDNAs lacking either e37a or e37b (Δ37) are 335 bp. BsrGI cuts WT e37a generating 284 bp and 148 bp products and XhoI cuts WT e37b into 340 bp and 92 bp products. Mutant e37a* and mutant e37b*-containing sequences lack BsrGI and XhoI sites, respectively. Results of RT-PCR from 1 μg of total RNA isolated from mouse DRG of WT (lanes 1–3), aa* (lanes 4–6), and b*b(lanes 7–9) mice (c). PCR products untreated (lanes 1, 4, and 7) and treated with BsrGI (lanes 2, 5, and 8) or XhoI (lanes 3, 6, and 9) were separated on a 2% TAE agarose gel. 1 kb Plus DNA ladders flank lanes 1–9. A 335 bp product was amplified from DRG of aa* mice and was resistant to BsrGI and XhoI. Sequencing shows it corresponds to mRNA lacking e37 (Δ37).
To distinguish mutant (e37a* and e37b*) from WT expressed sequences (e37a and e37b) we introduced synonymous point mutations. We eliminated the BsrGI restriction site in e37a and the XhoI restriction site in e37b. A combination of RT-PCR and restriction digest analysis showed that e37b* and e37a*-containing mRNAs were expressed in DRG of mutant mice (Fig. 1b,c and Supplementary Fig. 1).
Alternative splicing patterns were comparable for Cacna1bb*b/b*b and WT mice in DRG. Although not quantitative, our PCR analyses suggest that e37b* was present in similar proportions as e37a of WT DRG (Fig. 1c; compare XhoI digest patterns, lanes 3 and 9). Similarly, e37b-derived RT-PCR products were amplified at similar proportions to e37b-derived products of WT DRG (Fig. 1c; compare lower bands in lanes 3 and 9). These results combine with our previous studies in rat to suggest that e37a-containing mRNAs are enriched in DRG relative to other parts of the nervous system but CaV2.2[e37b] mRNAs still represent the major CaV2.2 isoform in RNA of whole DRG 16, 17.
In contrast, expression patterns of CaV2.2[e37a] and CaV2.2[e37a*] mRNAs in DRG of Cacna1baa*/aa* mice are different from WT (Fig. 1c; compare BsrGI digest patterns, lanes 2 and 5). First, from Cacna1baa*/aa* DRG we amplified a PCR-product of 335 bp, resistant to both XhoI and BsrGI cleavage (Fig. 1c; lanes 4–6). DNA sequence analysis showed this cDNA product was amplified from CaV2.2 mRNAs that lacked 37 (Fig. 1b;Δ 37 mRNA). We know from our previous analyses that CaV2.2 mRNAs lacking both e37a and e37b create a shift in the reading frame, an early stop codon, and nonfunctional CaV2.2 protein 19. Second, the relative abundance of e37a- and e37a*-containing CaV2.2 mRNAs in DRG of Cacna1baa*/aa* mice is different from that of CaV2.2[e37a] and CaV2.2[e37b] mRNAs in WT mice (Fig. 1c; compare lanes 2 and 5). Below we show that N-type currents and CaV2.2 protein levels in DRG of Cacna1baa*/aa* mice are also significantly reduced relative to WT.
Ca2+ currents in DRG of b*b and WT mice are the same
We previously reported that CaV2.2[e37a] mRNAs are enriched in DRG and specifically in small diameter, nociceptors defined by their capsaicin sensitivity 16. We also showed in a mammalian expression system that e37a promotes voltage-independent inhibition of N-type currents through μ-opioid receptor activation 7. To test the role of e37a in mediating inhibition of native N-type current in capsaicin-responsive small diameter neurons of DRG we first distinguished these neurons from two other classes of neurons (large and T-rich). We did this by assessing capsaicin-sensitivity, cell capacitance (size), and calcium channel properties (Supplementary Fig. 2).
Small neurons had small membrane capacitances tightly distributed around an average value of 13 pF. These were readily separable from large neurons (37 pF) (Supplementary Fig. 2). We found the vast majority of small neurons were capsaicin-responsive (17/21; Supplementary Fig. 2d), whereas large neurons rarely responded to capsaicin (1/19; Supplementary Fig. 2d). A third group of neurons expressed large T-type calcium currents that characteristically activate at low voltages and deactivate with slow kinetics (Fig. 2b; T-rich; 20). These were intermediate in size and did not respond to capsaicin, but by virtue of their large T-type currents were easily identified and separated from small and large neurons (Supplementary Fig. 2).
Figure 2. Whole cell calcium current densities in DRG neurons of Cacna1bb*b/b*b mice are indistinguishable from WT.

Average current-voltage relationships in three types of DRG neurons from WT, Cacna1bb*b/b*b (b*b), and Cacna1baa*/aa* (aa*) mice (a–c). Currents were evoked by test pulses to − 30 mV (left) and 0 mV (right) from a holding potential of − 80 mV. A series of currents were elicited by depolarizations applied every 10 seconds to test potentials between − 70 mV and + 80 mV from a holding potential of − 80 mV. Current densities are plotted (pA/pF) for small cells (a, 13pF), large cells (b, 35pF), and T-rich cells (c, 24pF). Calcium current densities of small (a) and large (b) neurons from Cacna1baa*/aa* mice are significantly smaller compared to WT mice. Individual current voltage plots were fit with one (a, c) and two (b) Boltzmann functions using average parameters calculated from fitting individual curves (Supplementary Table 2). Scale bars correspond to 500 pA and 5 ms. Data points are mean ±s.e.m. n values shown are number of cells, data sets contain recordings from at least 9 mice. (d) Western blot of DRG lysates derived from WT, aa*, and b*b mice using a polyclonal antibody directed to CaV2.2 and a monoclonal antibody directed to Glyceraldehyde 3-Phosphate Dehydrogenase (GAPDH). CaV2.2 protein levels in DRG of aa* mice were reduced compared to WT. CaV2.2 protein runs at 260 kDa, a dash indicates the location of a 250 kDa size marker. Complete, uncropped -l blots are shown in Supplementary Fig. 5.
Whole cell calcium currents in all three groups of DRG neurons from Cacna1bb*b/b*b mice were indistinguishable from WT (Cacna1bab/ab) (Fig. 2a–c). DRG neurons express at least four different classes of calcium channels, N-type, P/Q-type, L-type, and T-type; N-type represents the largest component 16, 20, 21. We isolated pure N-type from non-N-type currents using 2 μM ω-conotoxin (ω-Ctx) GVIA and showed that N-type currents in DRG neurons of Cacna1bb*b/b*b mice were also indistinguishable from WT (Fig. 3). These data support our RT-PCR analyses that show e37b* can substitute for e37a and support WT splicing patterns in DRG neurons.
Figure 3. N-type and non-N-type current densities in sensory neurons of Cacna1bb*b/b*b mice are indistinguishable from WT.

Average N-type (a–c) and non-N-type (d–f) calcium current voltage relationships in three sub-types (small, large, and T-rich) of acutely dissociated DRG neurons from wild-type (WT), Cacna1bb*b/b*b (b*b), and Cacna1baa*/aa* (aa*) mice (a–f). Representative currents shown above current voltage plots evoked by test pulses to 0 mV from a holding potential of − 80 mV, recorded from each cell type, and each mouse line. Currents were elicited as described in Fig. 2 and plotted as pA/pF to normalize for cell size. N-type currents and non-N-type currents correspond to ω-conotoxin GVIA-sensitive and insensitive components of the whole cell current, respectively. N-type currents were isolated in each cell by subtracting non-N-type (insensitive to 2 μM ω-Ctx GVIA) from whole cell currents. N-type current densities in small (a) and large (b) neurons are significantly smaller in Cacna1baa*/aa* compared to WT mice. Individual current voltage plots were fit with one (a–e) and two (f) Boltzmann functions using average parameters calculated from fitting individual curves (Supplementary Table 2). Scale bars represent 500 pA and 5 ms. Data points are mean ±s.e.m. n values shown are number of cells but all data sets contain recordings from at least 9 mice.
N-type Ca2+ currents in aa* mice are smaller than in WT
In contrast to Cacna1bb*b/b*b, N-type currents in small and large neurons of Cacna1baa*/aa* mice were significantly smaller compared to WT (Student’s t test, P = 0.000238; Fig. 3a). In addition, T-type currents in T-rich neurons were slightly larger but not statistically significantly compared with WT (Student’s t test, P = 0.245; Figs. 2c and 3f). CaV2.2 protein levels in DRG (Fig. 2d) and in brains of Cacna1baa*/aa* mice were reduced relative to WT. This reduction was specific to CaV2.2 protein and it did not lead to compensatory changes in levels of the closely related CaV2.1 protein in brains of Cacna1baa*/aa* mice. The reduction in CaV2.2 protein levels in DRG of Cacna1baa*/aa* mice could not be explained by differences in cell size; whole cell capacitance values within each sub-set of neurons were indistinguishable among genotypes (Supplementary Fig. 2). Furthermore, we compared calcium current densities (pA pF−1) to normalize for cell size (Fig. 2 and 3). Although smaller, N-type currents in all three groups of DRG neurons from Cacna1baa*/aa* mice activated at voltages ~ 7 mV more hyperpolarized relative to N-type currents in neurons of WT and Cacna1bb*b/b*b mice (Fig. 3a–c). Our findings are remarkably consistent with our studies of cloned e37a-containing N-type channels expressed in mammalian and amphibian cells. These similarly activated at voltages ~ 7 mV more hyperpolarized relative to e37b-containing N-type channels 7, 16, 17.
The reduction of CaV2.2 protein levels in Cacna1baa*/aa* mice likely results from a disruption in alternative splicing, consistent with e37a-lacking CaV2.2 mRNAs in DRG (Fig. 1c, lanes 4–6). We next compared G protein inhibition of N-type currents in small diameter nociceptors from our various mouse lines but with our primary focus on Cacna1bb*b/b*b and WT recordings, because CaV2.2 protein levels are reduced in Cacna1baa*/aa* mice.
DAMGO inhibits Ca2+ currents in all genotypes similarly
In our studies of cloned CaV2.2[e37a] and CaV2.2[e37b] channels in a mammalian cell line, we showed that e37a-containing N-type channels promote voltage-independent inhibition of N-type currents via G protein-coupled receptors, without affecting overall inhibition of peak current 7. We next tested if e37a regulates the responsiveness of native N-type currents to μ-opioid receptor-mediated inhibition in nociceptors that we know normally express both CaV2.2[e37a] and CaV2.2[e37b] mRNAs 16.
We used a saturating concentration of DAMGO (10 μM) to activate μ-opioid receptors and monitored the effects on N-type and non-N-type currents in nociceptors of Cacna1bb*b/b*b and WT mice (Fig. 4). Overall we showed DAMGO was a more effective inhibitor of N-type (70–75%) compared to non-N-type (15–25%) current as reported by others 21. DAMGO inhibited N-type current amplitudes in nociceptors from Cacna1bb*b/b*b and WT mice equally. Similarly, the efficacy of DAMGO on N-type currents in small neurons of Cacna1baa*/aa* mice was not significantly different from WT. If e37a does not influence the overall inhibitory actions of DAMGO, does it affect the type of inhibition? We therefore tested if e37a was important for voltage-independent inhibition of N-type currents by DAMGO.
Figure 4. DAMGO inhibits N-type currents similarly in nociceptors from all three genotypes.
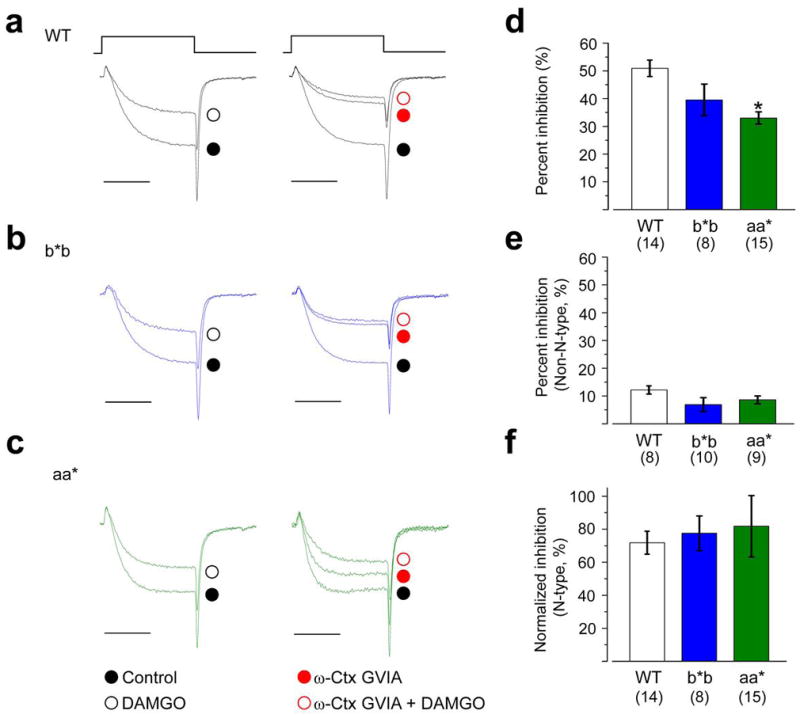
Representative currents from nociceptors isolated from WT (a), b*b (b), and aa* (c) mice show the inhibitory actions of a saturating concentration of 10 μM DAMGO on calcium currents in the absence (left; whole cell currents) and presence (right; non-N-type currents) of ω-Ctx GVIA (2 μM). For comparison, current amplitudes are scaled relative to their respective controls (a–c). Currents were elicited by test pulses to 0 mV from a holding potential of − 80 mV. Scale bar corresponds to 5 ms. Average % inhibition by DAMGO in nociceptors of WT, b*b, and aa* mice in the absence (d; whole cell current) and presence (e; non-N-type current) of 2 μM ω-Ctx GVIA. Values shown are means ±s.e.m. n values are number of cells, all data sets contain recordings from at least 9 mice. DAMGO was significantly less effective at inhibiting whole cell currents in aa* mice compared to WT (d; P = 0.0432). DAMGO’s inhibition of N-type currents in nociceptors compared among the three genotypes (f). For comparison, because N-type current density was significantly smaller in nociceptors of aa* mice, ,% inhibition by DAMGO was normalized to N-type current density as follows: {[(Icontrol−IDAMGO)whole cell − (ICtx − IDAMGO)non-N-type]/IN-type} × 100%. Average values of ICtx, IDAMGO, and IN-type were obtained from nociceptors of WT (n = 8), b*b (n = 10), aa* (n = 9) mice for these calculations.
E37a promotes voltage-independent inhibition by DAMGO
We used standard strong, brief, supra-threshold depolarizations to 80 mV just prior to the test pulse to remove voltage-dependent inhibition mediated by saturating concentrations of DAMGO (Fig. 5a–c, e–f). This allowed us to calculate the relative amounts of voltage-dependent and voltage-independent inhibition of non-N-type and N-type currents (Fig. 5d, h). DAMGO inhibited non-N-type currents by voltage-dependent and voltage-independent mechanisms, and the relative contribution of each was the same in neurons of WT, Cacna1bb*b/b*b, and Cacna1baa*/aa* mice (70% voltage-independent; Fig. 5e–h).
Figure 5. Voltage-independent inhibition by DAMGO is reduced in nociceptors that only express e37b and not e37a.

Representative currents from small neurons isolated from WT (a, e), b*b (b, f), and aa* (c, g) mice show the inhibitory actions of 10 μM DAMGO on whole cell currents in the absence (a–c; whole cell currents) and presence (e–g) of ω-Ctx GVIA (2 μM). Currents were elicited by test pulses to 0 mV from a holding potential of − 80 mV without and with a strong prepulse to +80 mV to remove voltage-dependent inhibition (a–c, e–g). For comparison, current amplitudes are scaled relative to their respective controls. Scale bar corresponds to 5 ms. Relative amount of voltage-dependent and voltage-independent inhibition of N-type (d; P = 0.0321) and non-N-type (h) currents mediated by 10 μM DAMGO in recordings from nociceptors of WT, b*b, and aa* mice. Values shown are means ±s.e.m. n values shown are number of cells, all data sets contain recordings from at least 9 mouse lines. Percent voltage-independent inhibition of N-type current was estimated as follows: {(VIwhole-cell − VINon-N-type)/total N-type inhibition} × 100%. VI corresponds to the voltage-independent component of inhibition.
However, we observed significant differences when we compared DAMGO’s effects of N-type currents in nociceptors of Cacna1bb*b/b*b mice. Even though peak N-type currents in nociceptors of all three genotypes were inhibited equally well by DAMGO (Fig. 4), its inhibitory effects were mostly voltage-dependent in neurons of Cacna1bb*b/b*b mice (75% voltage-dependent; Fig. 5b, d) compared to WT (50% voltage-dependent; Fig. 5a, d). In other words, DAMGO was significantly less able to engage voltage-independent inhibition of N-type currents in nociceptors of Cacna1bb*b/b*b mice (Fig. 5b, d). We only observed a significant shift away from voltage-independent toward voltage-dependent inhibition of N-type currents in nociceptors of Cacna1bb*b/b*b mice, whereas DAMGO’s effects on N-type currents in nociceptors of Cacna1baa*/aa* mice (Fig. 5c, d) and on non-N-type currents in Cacna1bb*b/b*b and Cacna1baa*/aa* mice (Fig. 5f,g,h) were the same as WT mice. This is generally consistent with our studies of cloned CaV2.2[e37a] and CaV2.2[e37b] channels expressed in tsA201 cells 7.
E37a influences spinal level analgesia by morphine
Our electrophysiological analyses show that e37a promotes a form of N-type channel inhibition in nociceptors that is resistant to depolarization (voltage-independent). We also compared the overall distribution of CaV2.2 protein in the dorsal spinal horn of WT, Cacna1bb*b/b*b, and Cacna1baa*/aa* mice by immunohistochemistry. The overall pattern of anti-CaV2.2 signal was similar among the three genotypes as were gross patterns of anti-CGRP signal and IB4-fluorescence. Although not quantitative, these signals suggest there are no gross disruptions of CaV2.2 distribution in afferent terminals in the superficial laminae of the dorsal spinal horn where afferents of nociceptors terminate (Fig. 6). We next used our mouse models to test the role of e37a in i) basal nociception, and ii) spinal analgesia by morphine. Small capsaicin-responsive nociceptors transmit information about noxious thermal stimuli. Intrathecal morphine, acting through μ-opioid receptors, inhibits transmission in this pathway 22, 23. At least part of morphine’s spinal level analgesia is through μ-opioid receptor-mediated inhibition of presynaptic N-type channels on nociceptor terminals in the dorsal horn of the spinal cord 24, 25. To assess this pathway we employed standard thermal stimuli to the hind paw and measured paw withdrawal latencies in all three mouse lines. This behavioral test specifically assesses spinal level circuits 26, 27. Paw withdrawal latencies measured from WT, Cacna1bb*b/b*b and Cacna1baa*/aa* mice were not significantly different (Fig. 7a). Our data suggest that amino acids unique to e37a are not necessary for normal transmission of noxious thermal stimuli.
Figure 6. Anti-CaV2.2, anti-CGRP, and FITC-IB4 signals in superficial layers of dorsal spinal horn are similar among genotypes.

Spinal cord sections from wild-type (WT), Cacna1bb*b/b*b (b*b), and Cacna1baa*/aa* (aa*) mice showing the dorsal spinal horn. The anti-CaV2.2 immunofluorescence signal is similar among the three genotypes (upper panels) as are the anti-CGRP (red signal, lower panels) and FITC-IB4 (green signal, lower panels). The lower panels show prominent CGRP and IB4 fluorescence in laminae I and II of the dorsal horn. CGRP and IB4 are often used to mark peptidergic and non-peptidergic nociceptive terminals, respectively 49, 50. A spinal cord section from WT incubated with the primary antibody pre-absorbed with the corresponding antigenic peptide is shown as control. Contrast phase image of the same section is also shown. The antigenic peptide control shows complete loss of CaV2.2 staining. The anti- CaV2.2 signal is seen throughout the dorsal spinal horn but there is an enhanced signal in the superficial laminae in the region of IB4 and CGRP signals.
Figure 7. Morphine’s spinal level analgesia is reduced in mice that lack e37a.
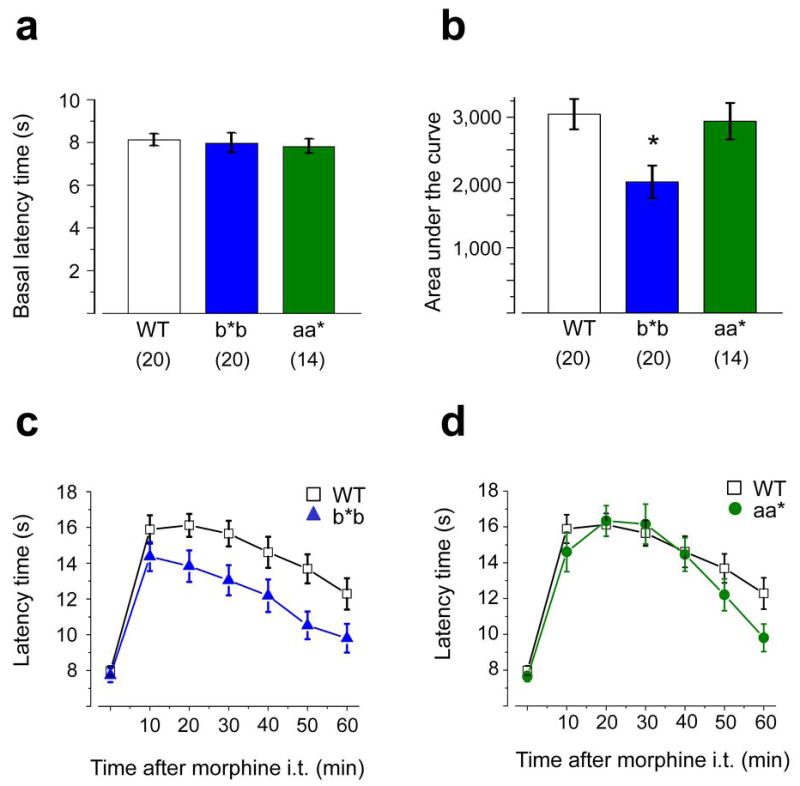
Comparison of thermal pain thresholds in WT (n = 20), b*b (n = 20), and aa* (n = 14) mice before (a) and after (b, c, d) intrathecal morphine. a, Average, basal paw withdrawal latency times (Hargreaves’s test) in response to noxious thermal stimuli measured in WT, b*b, and aa* mice were not significantly different (2 way ANOVA or Student’s t test, P > 0.5). b, Comparison intrathecal morphine analgesia expressed as the integral of the maximum possible effect (MPE) for each group as follows: (PWLmorphine − PWLbaseline) ×100/(20 − PWLbaseline) to normalize for differences in baseline responsiveness among mice. Morphine is significantly less effective against thermal stimuli in b*b mice compared to WT mice (Student’s t test P = 0.004) but has similar efficacy in aa* compared to WT mice (Student’s t test, P = 0.77). c and d, Time course of morphine analgesia following intrathecal injection (3 μg) at time 0 in WT and b*b mice (c) and in WT and aa* mice (d) measured every 10 min for 60 min. The efficacy of morphine is reduced in b*b mice relative to WT. P values at 10, 20, 30, 40, 50, and 60 min were 0.613, 0.190, 0.043, 0.023, 0.061, 0.008, and 0.044, respectively comparing average responses in b*b and WT mice (Student’s two tailed t test; c). Values shown are averages ± s.e.m.
Finally, we tested if e37a contributes to the analgesic actions of intrathecal morphine. This route of administration restricts the site of morphine action to the spinal cord and rules out contributions from supraspinal sites 28, 29. At doses higher than 3 μg, morphine induced behavior that interfered with testing. At its peak, 3 μg morphine induced analgesia to a level 70% of the maximum possible effect against the thermal stimulus similar to reports of others30. We found significant differences in the analgesic efficacy of intrathecal morphine in Cacna1bb*b/b*b compared to WT mice (Fig. 7b–d). As reported, intrathecal morphine transiently lengthens latencies of paw withdrawal in response to thermal stimuli 29. Overall morphine was 30% less effective as an analgesic at 3 μg in Cacna1bb*b/b*b compared to WT (n = 20, Student’s two tailed t test, P = 0.004; Fig. 7b). By comparison, morphine wassimilarly effective at prolonging paw withdrawal latencies in Cacna1baa*/aa* and WT mice (n = 14, P = 0.77; Fig. 7b). We compared the time courses of morphine-induced analgesia in Cacna1bb*b/b*b and Cacna1baa*/aa* mice to WT mice (Fig. 7c, d). The effect of morphine is apparent within 10 min and it peaks at 20-30 min (Fig. 7c, d). Most importantly, the analgesic effect of 3 μg morphine is reduced in Cacna1bb*b/b*b mice at all time points tested. These differences are statistically significant at 20, 30, 50, and 60 min time points (Student’s two tailed t test, P = 0.008 – 0.044; Fig. 7c). Finally, we compared the efficacy of two lower doses of intrathecal morphine in WT and Cacna1bb*b/b*b mice. While there is no significant difference in the overall efficacy of morphine at 0.1 μg and 0.3 μg, the dose-response relationships are significantly different based on their different slopes (Student’s t test, P = 0.04; Supplementary Fig. 4). Our data suggest that e37a is needed for the maximal spinal level analgesic actions of morphine at higher doses, in response to noxious thermal stimuli.
DISCUSSION
Cell-directed alternative pre-mRNA splicing is thought to represent a major step in optimizing protein function for specialized tasks, but few, if any studies have linked a single splicing event to a specific behavior in mammals 1, 2. We used an exon replacement strategy to show that it is possible to attribute a decrease in the efficacy of morphine to a single RNA processing event. We suggest that cell-specific selection of e37a during alternative pre-mRNA splicing enhances activity-independent G protein inhibition of N-type calcium channels in nociceptors. The enrichment of e37a in nociceptors may augment the analgesic actions of morphine in vivo.
Intrathecal morphine may need e37a for maximal efficacy
Our approach was helped by studying a localized splicing event in a well-described population of functionally specialized neurons and because these neurons underlie a behavior that can be measured reliably. Capsaicin-responsive small diameter nociceptors of non-myelinated C-fibers are specialized to transmit noxious thermal stimuli 22, 31. N-type CaV2.2 calcium channels at presynaptic terminals of C-fiber afferents in the spinal cord mediate transmitter release and behavioral responses to noxious stimuli 32. Morphine occludes transmission of noxious thermal stimuli in the spinal cord in large part by μ-opioid receptor-mediated inhibition of presynaptic N-type channels at nociceptor terminals 32–36. Therefore, by comparing behavioral responses in Cacna1bb*b/b*b and WT mice, we have evidence that maximal analgesic actions of intrathecal morphine may depend on e37a-containing CaV2.2 channels (Fig. 7). Collectively, our studies support the hypothesis that e37a boosts the analgesic effects of morphine via its influence on μ-opioid receptor-mediated inhibition of N-type calcium channels. E37a selectively augments voltage-independent inhibition of N-type currents without impacting the overall level of current inhibition (Figs. 4, 5 and 7). Our conclusions about the role of e37a from studies described here on native channels and receptors are reinforced by our previous studies of cloned CaV2.2 isoforms and receptors expressed in the tsA201 cell line 7.
Our analyses of Cacna1bb*b/b*b and WT mice suggest about 30% of morphine’s analgesic actions at the level of the spinal cord at higher doses, rely on e37a-dependent inhibition of N-type channels. E37a-independent actions of morphine likely also include voltage-dependent and voltage-independent inhibition of N-type calcium channels and postsynaptic GIRK potassium channel activation via μ-opioid receptors30. Notably, the GIRK-dependent component of spinal analgesia by morphine is similar in magnitude and is engaged at the same concentration of morphine as we report for e37a 30.
Paw withdrawal latencies to noxious thermal stimuli were indistinguishable among genotypes suggesting that CaV2.2[e37a] and CaV2.2[e37b] channels support transmission of nociceptive information in the spinal cord equally well. By contrast, studies using isoform specific siRNA, suggest that CaV2.2[e37a] channels have a preferred role in transmission of noxious thermal stimuli 18 in apparent contradiction to our analyses of Cacna1bb*b/b*b mice. However, these two observations might be reconcilable if CaV2.2[e37a] channels localize preferentially at transmitter release sites in nerve terminals of nociceptors of WT mice and in their absence (Cacna1bb*b/b*b), CaV2.2[e37b] channels target to these presynaptic locations and support transmission of nociception with similar efficacy. There are no gross differences in anti-CaV2.2 signals in dorsal spinal horn among genotypes (Supplementary Fig. 4), but experiments designed to quantify the density of presynaptic N-type channels would be needed to test these and other possibilities.
Cacna1baa*/aa* mice are physiologically compromised but our behavioral analyses suggest that transmission of nociceptive signals and morphine’s analgesic actions are very similar to those in WT mice. This might be unexpected because N-type currents and CaV2.2 protein levels are reduced in DRG neurons but the results areconsistent with reports that withdrawal latencies to thermal stimuli are unchanged in Cacna1b−/− null compared to WT mice 33, 37. As noted, experiments are needed to measure the relative contribution of N-type currents to transmission in the dorsal spinal horn.
Voltage-independent inhibition and morphine analgesia
Voltage-independent inhibition of N-type calcium currents has been studied for more than a decade 38–43. An appealing hypothesis, developed from these and other investigators, suggests that voltage-independent inhibition of presynaptic N-type calcium channels via G protein coupled receptors represents a mechanism for transmitter and drug-mediated inhibition of synaptic transmission independent of neuronal activity 33, 40, 41, 43, 44. By contrast, a purely voltage-dependent mechanism would inhibit N-type channels and synaptic transmission most strongly during periods of low neuronal activity, but become less effective as activity increases 40, 44, 45. Voltage-independent inhibition of N-type calcium channels through Gi/o coupled receptors and e37a, might underlie some of the analgesic actions of morphine particularly at higher doses. However, e37a may also influence other properties of N-type channels including cellular distribution and, as we have reported N-type current densities. One or all of these could influence the spinal level actions of morphine.
While morphine is a highly effective analgesic in response to noxious thermal stimuli in naïve mice and humans, it is much less effective against the thermal hyperalgesia characteristic of neuropathic pain 31. Expression levels of many genes are altered the in DRG of animals experiencing neuropathic pain. These change are implicated in the development and maintenance of chronic pain as well as in the loss of morphine efficacy 31. CaV2.2 mRNA and protein levels are not thought to change in dorsal root ganglia following peripheral nerve injury, but as we reported, CaV2.2[e37a] mRNAs are reduced 18. In light of our current study, it is possible that an injury-induced shift from e37a-containing to e37b-containing CaV2.2 channels in nociceptors, and therefore a concomitant change in inhibition of N-type calcium channels via μ-opioid receptors might contribute to decreased spinal level analgesia in response to morphine.
Cell-directed splicing of e37a
E37a is enriched in a sub-set of nociceptors in DRG16 and, given its functional importance in the pain pathway it would be extremely interesting to know the factors that control exon selection in these cells. Genome-wide analyses show involvement of several proteins in driving neuronal-specific alternative splicing 46, 47, but examples of how these factors coordinate exon selection in specific gene families within defined populations of neurons are limited. Several neuronal splicing factors can function both as repressors and enhancers, depending on binding location with respect to the target exon, and exon selection likely depends on the relative contribution of several splicing factors working in concert 2, 47, 48. The limited expression profile of e37a and the dramatic decrease in CaV2.2 protein levels in Cacna1baa*/aa* mice suggest that most neurons may contain repressor activity that prevents e37a inclusion during splicing. However, our analysis of N-type current densities among small, T-rich, and large cells of DRG from Cacna1baa*/aa* mice suggest that e37a selection depends on additional factors with differential actions in these three classes of neuron. We studied Cacna1b gene sequences at and close to e37a and e37b splice junctions but have not identified the nature of cis and trans factors that must regulate alternative splicing at this site. We hope new data from genome-wide screens of splicing factor-RNA binding events will help define cell-specific mechanisms that orchestrate alternative splicing in CaV2 pre-mRNAs in specific neurons 46–48.
The e37a/e37b splice site is conserved in all mammalian Cacna1b genes and activity unique to e37a must provide some unique evolutionary advantage. Our data suggest that at higher doses morphine engages e37a in spinal analgesia to thermal stimuli but it is likely that endogenous transmitters use this inhibitory pathway to control nociception under certain conditions. Identifying cell-specific splicing factors that control alternative splicing of e37a could offer new therapeutic approaches to increase the efficacy of drugs and neurotransmitters that work through G protein coupled receptors to inhibit N-type calcium channels. Such studies could also provide insight into cell-specific, coordinated alternative splicing of several pre-mRNAs that contribute to setting the efficacy of transmission in the pain pathway.
METHODS
Construction of genomic clones for mouse exon substitution
To create Cacna1bb*b/b*b mice, we substituted e37a with e37b*; to create Cacna1baa*/aa mice, we substituted e37b with e37a*. We derived mouse genomic DNA for all constructs from the MICER clone MHPN59f07 (Wellcome Trust Sanger Institute). Details of the cloning steps are in supplementary information. To facilitate detection of the specific exons spliced into the final mRNA products, we introduced silent mutations into restriction sites of the substituted exons; we mutated the BsrGI site in the mutant e37a to produce e37a* and mutated the XhoI site in e37b to produce e37b*.
We inserted a 2 kb loxP- NeoR-loxP cassette into each targeting construct at the AfeI site located in the intron between the normal positions of e37a and e37b. We added flanking AfeI sites to the loxP-NeoR-loxP cassette amplified from PL452 by PCR with primers SD29for and SD31rev. Finally, we re-introduced the mutagenized DNA into BamHI-EagI sites to create the final ~12 kb targeting constructs in pBSSK+.
Mouse ES cell gene targeting and mouse breeding
Animal housing and experimental procedures were in accordance with Brown institutional animal care and use committee guidelines. We targeted embryonic stem (ES) cells and generated mice as described (Nagy et al., 2003). In brief, we used 129Ola ES cells derived from a male embryo, grown on mitotically inactive SNL76/7 feeder cells. We 107 ES cells with 20 μg of a construct linearized with PvuI and G418 selected after 24 h. We selected 200 G418-resistant clones for further analysis. We used PCR to identify correctly targeted ES cell clones. We isolated E3.5 blastocysts from C57BL/6-Tyrc-Brd female mice and injected with 12-20 ES cells harvested by trypsinization from 90% confluence cultures. We then implanted injected blastocysts into day 2.5 pseudopregnant females to generate chimeras (eight to ten injected embryos per uterine horn). We mated male chimeras with C57BL/6-Tyrc-Brd females to obtain F1 progeny. We obtained germ line transmission of the mutant alleles with several targeted cell lines for each construct.
Identification of Correctly Targeted ES Cell Clones
We confirmed homologous integration by two PCR reactions on genomic DNA from each ES cell clone. Integration from the long arm was confirmed by PCR amplification of a 7 kb product using Clontech’s Advantage 2 Polymerase Mix (35 cycles, 68°C annealing, 8 min extension at 68°C) with primers SD77for (anneals 5′ to e37a, outside of the targeting construct sequence) and SD64rev (anneals to the 5′ end of the NeoR insert). Homologous integration from the short arm was confirmed by PCR amplification of a 3.5 kb product using Taq Polymerase (35 cycles, 60°C annealing, 4 min extension at 72°C; New England Biolabs) and primers SD47for (anneals to the 3′ end of the NeoR insert) and SD79rev (anneals 3′ to e37b, outside of the targeting construct sequence). We also confirmed correct targeting by sequencing through the substituted region of the Cacna1b gene of Cacna1baa*/aa and Cacna1bb*b/b*b mice.
We used BsrGI to identify the exon present in the normal e37a position. BsrGI digested PCR products amplified using primers SD62for (anneals upstream of the normal e37a position) and SD64rev (35 cycles, 60°C annealing, 2 min extension at 72°C with Taq Polymerase): e37a is digested by BsrGI (617 and 1156 bp) by contrast, e37b* is not (uncut, 1773 bp). Similarly, we used XhoI to identify the exon present in the normal e37b position. I digested PCR products amplified using primers SD47for and SD48rev (anneals downstream of the normal e37b position). E37b has two XhoI sites (generating products, 144, 216 and 636 bp) and e37a* has one XhoI site generating two different size products (144 and 852 bp).
NeoR cassette deletion
We genotyped F1 animals by genomic DNA from tails, using the four primer sets described above. We crossed > 8 positive animals identified for each of the two mutations with a Cre deletor strain [B6.FVB-Tg(EIIa-cre)C5379Lmgd/J] (Jackson Laboratory stock #003724) to delete the NeoR gene. We genotyped DNeoR mice for appropriate e37 mutations (see genotyping below). We crossed DNeoR siblings carrying appropriate mutations to produce homozygotes which were maintained by crossing sibling homozygotes. We maintain at least two separate homozygous lines for each mutation.
Genotyping
We used primers SD62for and SD64rev to genotype for DNeoR by genomic PCR. We performed this genotyping for at least two generations. Refer to Supplementary Fig. 1 for complete genotyping of Cacna1bab (WT), aa* and b*b heterozygotes and homozygotes.
Genomic DNA isolation, RNA extraction, and reverse transcription polymerase chain reaction (RT-PCR). Details are in Supplementary Information.
Western blot analysis
We pooled DRG from five mice but carried out three separate experiments on brains from three mice (2–8 months). Details of protein isolation and antibody staining are in Supplementary Information. We developed all membranes with ECL reagent (Amersham Biosciences) and used time-series exposures to ensure the signal was in the linear range. We measured signal densities (ImageQuantTM software, Amersham) from TIFF images generated from non-saturated films. After background correction we quantified CaV2.1 or CaV2.2-derived signals as a fraction of the average GAPDH signal. Complete uncropped blots are shown in Supplementary Fig. 5.
Cell isolation and electrophysiology
We harvested dorsal root ganglia from P7–P9 mice of all three genotypes. We obtain a higher yield of neurons from DRG of these young animals. However, we have also recorded N-type currents from the same age mice as those used for behavior (>2 months). Although the yield of neurons is lower, we confirmed that results from N-type currents recorded from nociceptors of older mice for all three genotypes are not significantly different from those of P7–P9 animals. Single neurons were dissociated from ganglia by dissociation in Hank’s Balanced Salt Solution (HBSS, GIBCO) medium containing 1.5 mg ml−1 collagenase (Sigma) and 2 mg ml−1 trypsin (Sigma) at 37°C for ~30 min, followed by trituration with a fire-polished pipette. We quenched protease activity by Dulbecco’s Modified Eagle Medium (DMEM, GIBCO) with 10% Fetal Bovine Serum (FBS, GIBCO). After two washes, we plated neurons on poly-D-lysine (Sigma)-coated coverslips in 10% FBS-DMEM supplemented with 1 ng ml−1Nerve Growth Factor (NGF, Sigma) and kept at 37°C with 5% CO2. We sylgard-coated and fire-polished electrodes to a resistance of 3-6 MΩ. Our external solution contained in mM: 135 TEA-Cl, 1 CaCl2, 10 HEPES, 4 MgCl2, 10−4 TTX adjusted to a pH of 7.2 with TEA-OH. Our pipette solution contained in mM: 126 CsCl, 4 MgATP, 1 EDTA(Cs)2, 10 EGTA(Cs)4, 10 HEPES adjusted to a pH of 7.2 with Cs0H. We do not include GTP in the patch pipette because we have evidence from studies of expressed channel, that low levels of GTP (0.2 – 0.3 mM) can lead to tonic inhibition of N-type currents in a voltage-independent manner in the absence of agonist. However, we obtain highly reproducible inhibition of N-type currents by brief repeat applications of DAMGO (Sigma) suggesting that components necessary for mediating inhibition do not wash out under the conditions of our experiments. We limit the cell’s exposure to DAMGO to 1 min. This also avoids μ-opioid receptor desensitization. We evoked whole-cell calcium currents by 10 ms test pulses from a holding potential of − 80 mV and tail currents monitored by a 5 ms repolarizing pulse to − 60 mV. We applied 2 μM ω-conotoxin GVIA (Alomone) via a pipette to inhibit the N-type current and voltage steps were re-applied. ω-conotoxin GVIA was removed from the recording chamber between recordings by 2% SDS and 10 mM DTT for 20 min. We used pClamp V8.1 software and the Axopatch 200A (Axon Instruments) for data acquisition, data were filtered at 2 kHz (− 3 dB) and sampled at 20 kHz. We compensated series resistance by 70% – 90% with a 7 μs lag and performed on-line leak correction with a P/– 4 protocol. All recordings were obtained at room temperatures.
Immuno-fluoresence imaging
Adult mice were deeply anesthetized with intra-peritoneal injection of 2% Pentothal, perfused transcardially with 0.9% saline solution followed by 4% paraformaldehyde. Spinal cord was removed, post-fixed over night in 4% PFA, and cryo-protected in 30% sucrose. For comparison, tissues from the three genotypes were frozen and sectioned (20 μm). Sections were dried at room temperature, hydrated in 0.01M PBS for 15 min and then blocked with 10% goat serum, 0.1% bovine serum albumin (BSA), 0.3% Triton in PBS in a humidified chamber at room temperature for 1 h. Sections were incubated with 1:200 rabbit anti-CaV2.2 or 1:800 guinea pig anti-calcitonin gene related peptide (CGRP) antibodies (Penninsula Labs) overnight at 4°C. After washing in PBS 3 times, sections were exposed to Alexa 555 conjugated goat anti-rabbit IgG (Invitrogen) for CaV2.2 at room temperature for 2 hours. To co-label CGRP and IB4, 1:100 Cy5 conjugated goat anti-guinea pig IgG (Abcam) and 1:100 FITC-conjugated IB4 (Invitrogen) were applied together for 2 hours at room temperature. After washing, sections were mounted with Vectashield (Vector labs). Regarding the specificity controls, we incubated sections with antigenic peptide pre-absorbed with primary antibody, we omitted the primary antibody and we tested CaV2.2 antibodies on tsA201 cells transfected with CaV2.1 or mock transfected, we did not observed staining under any of the listed conditions. We used polyclonal antibodies from Alomone labs to CaV2.1 and CaV2.2 and carried out controls to test for specificity. i) Because CaV2.1 and CaV2.2 are relatively homologous we used clones expressed in tsA201 cells to show that anti-CaV2.1 did not cross react with CaV2.2, and likewise that anti-CaV2.2 did not cross react with CaV2.2. In addition, we observed no signals (Western or immunofluorescence) in transfections lacking CaV2.1 and CaV2.2. We analyzed these controls by Western blotting as well as immunofluorescence. ii) All signals were lost when we omitted the primary antibody and tested with the secondary alone. iii) We showed complete loss of immunofluorescence signals in sections in which we first pre-absorbed with CaV2.2 antigenic peptide (Fig. 6). Details of the immunofluorescence signals are provided in supplementary information.
Behavioral analyses
We tested paw withdrawal threshold with Plantar Testing Instruments (IITC) 27 on 8–16 week old male mice. We applied a high-intensity beam (setting = 20%, ~45 W) to the middle of the plantar surface of a resting mouse. Paw-withdrawal latency (PWL) was measured to the nearest 0.1 s with a cutoff value of 20 s. We injected 3 μg morphine sulfate in a volume of 5 μl intrathecally under anesthesia (2% isoflurane) with a 28 gauge needle connected to a micro syringe through a PE 50 polyester catheter (15 cm) 28. We tested paw withdrawal thresholds before and 10, 20, 30, 40, 50 and 60 mins after morphine injection. We calculated and compared % maximum possible effect of morphine among three genotypes using the following: %MPE = (PWLmorphine − PWLbaseline) *100/(20 − PWLbaseline) and the area under the curve % MPE (AUC) to compare the overall effect of intrathecal morphine. Data points are mean ± s.e.m, they contain measurements from at least 10 animals for each genotype.
Statistical analysis
Data are presented as mean ± s.e.m. Comparisons of mean differences between groups were made by unpaired two-tailed Student’s t test unless otherwise stated. A probability level of P < 0.05 was considered to be statistically significant. We used the software Origin 6.1®.
Supplementary Material
Supplementary Figure 1. Genotyping mouse strains.
Supplementary Figure 2. Classification of DRG neurons from Cacna1bwt/wt, Cacna1baa*/aa*, and Cacna1bb*b/b*b mice.
Supplementary Figure 3. Western blot analysis of brain CaV2.2 and CaV2.1 channel levels in Cacna1bwt/wt, Cacna1baa*/aa*, and Cacna1bb*b/b*b mice.
Supplementary Figure 4. Effect of intrathecal morphine in WT and b*b mice
Supplementary Figure 5. Complete blots for Westerns shown in Fig. 2d and supplementary Fig. 3a
Supplementary Table 1. Survival of progeny from crosses of heterozygous Cacna1b mutant mice.
Supplementary Table 2. Boltzman-linear fit parameters
Acknowledgments
We thank J. Kysik and E. Paul in the mouse transgenic facility at Brown for helping in the initial targeting vector construction, R. Burwell for advice with animal behavior, T.D. Helton for help with image analyses, and C.G. Phillips and S.E. Allen for helpful comments on the manuscript. This work was supported by National Institutes of Health grant RO1NS055251 (D.L.) and F31NS066702 (S.M.).
Footnotes
AUTHOR CONTRIBUTIONS
All authors contributed to writing this manuscript. A.A. carried out electrophysiological studies, S.D. designed targeting constructs and generated all exon-substituted mice, J-Q.J. performed the behavioral studies and immunohistochemistry, S.M. performed Western analyses, and D.L. directed the project and oversaw all analyses.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests
References
- 1.Lipscombe D. Neuronal proteins custom designed by alternative splicing. Curr Opin Neurobiol. 2005;15:358–363. doi: 10.1016/j.conb.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- 3.Ule J, Darnell RB. RNA binding proteins and the regulation of neuronal synaptic plasticity. Curr Opin Neurobiol. 2006;16:102–110. doi: 10.1016/j.conb.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 5.Dredge BK, Polydorides AD, Darnell RB. The splice of life: alternative splicing and neurological disease. Nat Rev Neurosci. 2001;2:43–50. doi: 10.1038/35049061. [DOI] [PubMed] [Google Scholar]
- 6.Mu Y, Otsuka T, Horton AC, Scott DB, Ehlers MD. Activity-dependent mRNA splicing controls ER export and synaptic delivery of NMDA receptors. Neuron. 2003;40:581–594. doi: 10.1016/s0896-6273(03)00676-7. [DOI] [PubMed] [Google Scholar]
- 7.Raingo J, Castiglioni AJ, Lipscombe D. Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat Neurosci. 2007;10:285–292. doi: 10.1038/nn1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hille B. Ion Channels of Excitable Membranes. Sinauer Associates, Inc; Sunderland, MA: 2001. [Google Scholar]
- 10.Xie J, Black DL. A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature. 2001;410:936–939. doi: 10.1038/35073593. [DOI] [PubMed] [Google Scholar]
- 11.Mich PM, Horne WA. Alternative splicing of the Ca2+ channel beta4 subunit confers specificity for gabapentin inhibition of Cav2.1 trafficking. Mol Pharmacol. 2008;74:904–912. doi: 10.1124/mol.108.045153. [DOI] [PubMed] [Google Scholar]
- 12.Liao P, Zhang HY, Soong TW. Alternative splicing of voltage-gated calcium channels: from molecular biology to disease. Pflugers Arch. 2009;458:481–487. doi: 10.1007/s00424-009-0635-5. [DOI] [PubMed] [Google Scholar]
- 13.Zhong X, Liu JR, Kyle JW, Hanck DA, Agnew WS. A profile of alternative RNA splicing and transcript variation of CACNA1H, a human T-channel gene candidate for idiopathic generalized epilepsies. Hum Mol Genet. 2006;15:1497–1512. doi: 10.1093/hmg/ddl068. [DOI] [PubMed] [Google Scholar]
- 14.Bourinet E, et al. Splicing of alpha 1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- 15.Adams PJ, et al. Ca(V)2.1 P/Q-type calcium channel alternative splicing affects the functional impact of familial hemiplegic migraine mutations: implications for calcium channelopathies. Channels (Austin) 2009;3:110–121. doi: 10.4161/chan.3.2.7932. [DOI] [PubMed] [Google Scholar]
- 16.Bell TJ, Thaler C, Castiglioni AJ, Helton TD, Lipscombe D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron. 2004;41:127–138. doi: 10.1016/s0896-6273(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 17.Castiglioni AJ, Raingo J, Lipscombe D. Alternative splicing in the C-terminus of CaV2.2 controls expression and gating of N-type calcium channels. J Physiol. 2006;576:119–134. doi: 10.1113/jphysiol.2006.115030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altier C, et al. Differential role of N-type calcium channel splice isoforms in pain. J Neurosci. 2007;27:6363–6373. doi: 10.1523/JNEUROSCI.0307-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan JQ, Lipscombe D. Alternative splicing in the cytoplasmic II-III loop of the N-type Ca channel alpha 1B subunit: functional differences are beta subunit-specific. J Neurosci. 2000;20:4769–4775. doi: 10.1523/JNEUROSCI.20-13-04769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson MT, Joksovic PM, Perez-Reyes E, Todorovic SM. The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J Neurosci. 2005;25:8766–8775. doi: 10.1523/JNEUROSCI.2527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taddese A, Nah SY, McCleskey EW. Selective opioid inhibition of small nociceptive neurons. Science. 1995;270:1366–1369. doi: 10.1126/science.270.5240.1366. [DOI] [PubMed] [Google Scholar]
- 22.Scherrer G, et al. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell. 2009;137:1148–1159. doi: 10.1016/j.cell.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glaum SR, Miller RJ, Hammond DL. Inhibitory actions of delta 1-, delta 2-, and mu-opioid receptor agonists on excitatory transmission in lamina II neurons of adult rat spinal cord. J Neurosci. 1994;14:4965–4971. doi: 10.1523/JNEUROSCI.14-08-04965.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bao J, Li JJ, Perl ER. Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J Neurosci. 1998;18:8740–8750. doi: 10.1523/JNEUROSCI.18-21-08740.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- 26.Wilson SG, Mogil JS. Measuring pain in the (knockout) mouse: big challenges in a small mammal. Behav Brain Res. 2001;125:65–73. doi: 10.1016/s0166-4328(01)00281-9. [DOI] [PubMed] [Google Scholar]
- 27.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 28.Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- 29.Wang YX, Pettus M, Gao D, Phillips C, Scott Bowersox S. Effects of intrathecal administration of ziconotide, a selective neuronal N-type calcium channel blocker, on mechanical allodynia and heat hyperalgesia in a rat model of postoperative pain. Pain. 2000;84:151–158. doi: 10.1016/s0304-3959(99)00197-9. [DOI] [PubMed] [Google Scholar]
- 30.Marker CL, Lujan R, Loh HH, Wickman K. Spinal G-protein-gated potassium channels contribute in a dose-dependent manner to the analgesic effect of mu- and delta- but not kappa-opioids. J Neurosci. 2005;25:3551–3559. doi: 10.1523/JNEUROSCI.4899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowersox SS, et al. Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. J Pharmacol Exp Ther. 1996;279:1243–1249. [PubMed] [Google Scholar]
- 33.Altier C, Zamponi GW. Targeting Ca2+ channels to treat pain: T-type versus N-type. Trends Pharmacol Sci. 2004;25:465–470. doi: 10.1016/j.tips.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 34.Scott DA, Wright CE, Angus JA. Actions of intrathecal omega-conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the rat. Eur J Pharmacol. 2002;451:279–286. doi: 10.1016/s0014-2999(02)02247-1. [DOI] [PubMed] [Google Scholar]
- 35.Matthews EA, Dickenson AH. Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain. 2001;92:235–246. doi: 10.1016/s0304-3959(01)00255-x. [DOI] [PubMed] [Google Scholar]
- 36.Malmberg AB, Yaksh TL. Effect of continuous intrathecal infusion of omega-conopeptides, N-type calcium-channel blockers, on behavior and antinociception in the formalin and hot-plate tests in rats. Pain. 1995;60:83–90. doi: 10.1016/0304-3959(94)00094-U. [DOI] [PubMed] [Google Scholar]
- 37.Saegusa H, Matsuda Y, Tanabe T. Effects of ablation of N- and R-type Ca(2+) channels on pain transmission. Neurosci Res. 2002;43:1–7. doi: 10.1016/s0168-0102(02)00017-2. [DOI] [PubMed] [Google Scholar]
- 38.Hille B, et al. Multiple G-protein-coupled pathways inhibit N-type Ca channels of neurons. Life Sci. 1995;56:989–992. doi: 10.1016/0024-3205(95)00038-8. [DOI] [PubMed] [Google Scholar]
- 39.Tedford HW, Zamponi GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol Rev. 2006;58:837–862. doi: 10.1124/pr.58.4.11. [DOI] [PubMed] [Google Scholar]
- 40.Elmslie KS. Neurotransmitter modulation of neuronal calcium channels. J Bioenerg Biomembr. 2003;35:477–489. doi: 10.1023/b:jobb.0000008021.55853.18. [DOI] [PubMed] [Google Scholar]
- 41.Kammermeier PJ, Ruiz-Velasco V, Ikeda SR. A voltage-independent calcium current inhibitory pathway activated by muscarinic agonists in rat sympathetic neurons requires both Galpha q/11 and Gbeta gamma. J Neurosci. 2000;20:5623–5629. doi: 10.1523/JNEUROSCI.20-15-05623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shapiro MS, Hille B. Substance P and somatostatin inhibit calcium channels in rat sympathetic neurons via different G protein pathways. Neuron. 1993;10:11–20. doi: 10.1016/0896-6273(93)90237-l. [DOI] [PubMed] [Google Scholar]
- 43.Luebke JI, Dunlap K. Sensory neuron N-type calcium currents are inhibited by both voltage-dependent and -independent mechanisms. Pflugers Arch. 1994;428:499–507. doi: 10.1007/BF00374571. [DOI] [PubMed] [Google Scholar]
- 44.Ikeda SR, Dunlap K. Calcium channels diversify their signaling portfolio. Nat Neurosci. 2007;10:269–271. doi: 10.1038/nn0307-269. [DOI] [PubMed] [Google Scholar]
- 45.Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Adv Second Messenger Phosphoprotein Res. 1999;33:131–151. doi: 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- 46.Zhang C, et al. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 2008;22:2550–2563. doi: 10.1101/gad.1703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Licatalosi DD, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boutz PL, et al. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636–1652. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silverman JD, Kruger L. Selective neuronal glycoconjugate expression in sensory and autonomic ganglia: relation of lectin reactivity to peptide and enzyme markers. J Neurocytol. 1990;19:789–801. doi: 10.1007/BF01188046. [DOI] [PubMed] [Google Scholar]
- 50.Nagy JI, Hunt SP. Fluoride-resistant acid phosphatase-containing neurones in dorsal root ganglia are separate from those containing substance P or somatostatin. Neuroscience. 1982;7:89–97. doi: 10.1016/0306-4522(82)90155-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Genotyping mouse strains.
Supplementary Figure 2. Classification of DRG neurons from Cacna1bwt/wt, Cacna1baa*/aa*, and Cacna1bb*b/b*b mice.
Supplementary Figure 3. Western blot analysis of brain CaV2.2 and CaV2.1 channel levels in Cacna1bwt/wt, Cacna1baa*/aa*, and Cacna1bb*b/b*b mice.
Supplementary Figure 4. Effect of intrathecal morphine in WT and b*b mice
Supplementary Figure 5. Complete blots for Westerns shown in Fig. 2d and supplementary Fig. 3a
Supplementary Table 1. Survival of progeny from crosses of heterozygous Cacna1b mutant mice.
Supplementary Table 2. Boltzman-linear fit parameters