

Abstract
The development of a large animal model of fulminant hepatic failure produced with acetaminophen that should be useful in the development and evaluation of potential medical therapies for the important clinical problem of fulminant hepatic failure is described. Acetaminophen in dimethyl sulfoxide (600 mg/ml) given as three subcutaneous injections, with the first dose (750 mg/kg body wt) being given at noon, the second dose (200 mg/kg body wt) being given 9 h later, and the third dose (200 mg/kg body wt) being given 24 h after the initial dose consistently produces fulminant hepatic failure in dogs. The dimethyl sulfoxide vehicle, injected intramuscularly, does not influence either animal survival or hepatic function in control-treated dogs. No deaths occur within the first 36 h. By 72 h after initial drug administration, the mortality is 90%. Histopathological and biochemical investigations demonstrate a high degree of hepatocellular necrosis in nonsurviving animals without appreciable damage to the kidneys, lungs, or heart. The drug schedule and preparation outlined avoids the administration of large volumes of vehicle and results in prolonged high levels of acetaminophen in the blood sufficient to induce severe hepatic injury. Ranitidine (120 mg/kg body wt i.m.) given 30 min before each acetaminophen dose significantly reduces the mortality and hepatic necrosis produced using this model. This model satisfies all criteria established by Miller et al. for the production of a suitable large animal model of fulminant acute hepatic failure.
A major deterrent to the development of new therapies for the important clinical problem of fulminant hepatic failure is the lack of a safe, reliable, and inexpensive large animal model to use in such studies (1–3). Currently, the two most frequently used large animal models of fulminant hepatic failure are produced either as a result of ischemic hepatic injury or acetaminophen-induced hepatotoxicity. Ischemic hepatic necrosis necessitates surgical intervention, requires considerable technical expertise, involves great expense, and is associated with a high degree of model-to-model variability (4–10). The second model capitalizes on the well-known hepatotoxicity of acetaminophen (11–15) and has been studied widely. Unfortunately, it has never been standardized or been shown to be reproducible, producing inconsistent toxicity from animal to animal and between experiments (16–18).
Potentially, there are three important reasons for this lack of reproducible results with the acetaminophen model. First, no attention has been given to the determination of acetaminophen blood levels in the animals being studied despite the fact that it is known that high blood levels of the drug are required to produce hepatic necrosis. Second, the duration of elevated levels of the drug necessary to produce consistent panlobular necrosis has not been determined. Third, no current model of drug-induced hepatic necrosis exists using 10-ml volumes of acetaminophen solution, which can be easily injected subcutaneously. The model reported considers each of these three issues.
Materials and Methods
Chemicals
Acetaminophen and dimethyl sulfoxide (DMSO) were purchased from Sigma Chemical Co., St. Louis, Mo. Pentothal was purchased from Abbott Laboratories, North Chicago, Ill. Ranitidine was obtained from Glaxo, Inc., Research Triangle Park, N.C. Xylocaine was purchased from Astra Pharmaceutical Product, Inc., Worchester, Mass. Formaldehyde was obtained from Fisher Scientific, Pittsburgh, Pa.
Animals
One hundred twenty-seven male beagle-mix dogs were purchased from Russel B. Hutton Farms, St. Thomas, Pa. The dogs were housed in a large animal care facility and maintained at a constant temperature of 20° ± 1°C with a 6 am to 6 pm light cycle. All dogs were quarantined and allowed to acclimatize in the animal facility for a minimum of 1 wk before being used. All dogs were given a standard dry dog food and water ad libitum. Their body weights ranged from 9 to 14 kg.
Study Design
The animals were randomly divided into four groups.
Group 1 (n = 31) (intravenous administration of acetaminophen with and without sodium pentothal induction)
Thirty-one dogs received an intravenous infusion of 1% acetaminophen in physiologic saline over a period of 45 min. In 7 dogs (1a), a dose of 700 mg/kg body wt was infused. In the remaining 24 dogs (1b), a dose of 400 mg/kg body wt was infused. Sodium pentothal (25 mg/kg body wt i.m.) as an inducer of microsomal enzymes was administered daily to the animals in group 1b for 4 days before and on the day of acetaminophen administration.
Group 2 (n = 24) (intramuscular administration of acetaminophen dissolved in dimethyl sulfoxide)
Twenty-one dogs were given an intramuscular injection of acetaminophen (900 mg/kg body wt dissolved in DMSO at a concentration of 600 mg/ml). All of the dogs received sodium pentothal (25 mg/kg body wt i.v.) as an anesthetic on the day of acetaminophen administration. Three dogs were used as controls for this group and were treated with the same volume of DMSO given the treated animals but without any acetaminophen.
Group 3 (n = 20) (subcutaneous administration of acetaminophen dissolved in dimethyl sulfoxide)
Twenty dogs were treated with a subcutaneous injection of acetaminophen at a dose of 1600 rng/kg body wt, which was administered as a solution of acetaminophen in DMSO (600 mg/ml). Novocaine was used as a local anesthetic, being injected in the areas surrounding the acetaminophen injections.
Group 4 (n = 52) (multiple subcutaneous injections of acetaminophen)
Fifty-two dogs received a total of three time-spaced subcutaneous injections of acetaminophen in DMSO at a concentration of 600 mg/ml. The first injection of acetaminophen (750 mg/kg body wt) was given at noon; the second injection (200 mg/kg body wt) was given 9 h later; the third dose (200 mg/kg body wt) was given 24 h after the initial dose. This treatment protocol was chosen to produce a concentration of drug in serum of ≥140 μg/ml for a period of at least 20 h. Previous studies have shown that it is important to attain and maintain this level for a period of 24 h to produce consistent hepatic necrosis (19). Further, this dosing schedule was selected because it is not complicated by the production of methemoglobinemia. Twelve of the 52 dogs in this group were also given ranitidine (120 mg/kg body wt i.m.) 30–60 min before each dose of acetaminophen.
Biochemical Determinations
Blood levels of acetaminophen
Five animals in each group were used to determine blood acetaminophen levels (20). A blood sample of 7 ml was taken from each animal at 0, 24, 48, 60, and 72 h after initial administration of acetaminophen.
Biochemical parameters
Ten dogs in groups 1, 2, and 3, and all of the dogs in group 4, had serum glutamic-pyruvic transaminase, ammonia, urea nitrogen, albumin, bilirubin, and cholesterol levels determined before and at various times after acetaminophen administration. Standard laboratory methodology was used for each measure. A coagulation profile comprising nine parameters (fibrinogen, factors II, V, VII, VIII, IX, X, XI, and XII) was obtained at each such assessment. Fibrinogen levels were expressed as milligrams per deciliter and the various factors as units per milliliter (21–24). Plasma amino acid profiles were determined using deproteinized plasma obtained with 4% sulfosalicylic acid treatment of the plasma. The resultant supernatant was applied to an amino acid analyzer (Beckman Instruments, Somerset, N.J.) and the levels of free branched chain and aromatic amino acids (including tryptophan) in the plasma were determined.
Hematology
In addition to all of the above measures, the hematocrit and the presence of hemoglobinuria and methemoglobinemia were assessed in all of the dogs in group 4. The methemoglobinemia was quantified in a hematology laboratory using a standard methodology (25).
Histology
All nonsurviving dogs underwent necropsies that included a full histologic evaluation of the liver, kidneys, lungs, and heart. Tissues were fixed in 10% neutral buffered formalin, sectioned at 6 μm and stained with hematoxylin and eosin.
The assessment of the degree of hepatic necrosis was initially performed using a semiquantitative scale. The scale was based on two observations and performed by a single staff pathologist who was blinded as to the treatment group: (I) the extent of necrosis within individual lobules [(a) mild = less than one-third of the lobule, (b) moderate = one-third to two-thirds of the lobule, and (c) greater than two-thirds of the lobule]. This initial assessment was then combined within (II) an estimation of the number of lobules affected [(a) less than one-third, (b) one-third to two-thirds, and (c) greater than two-thirds of the lobules examined being affected].
Statistical Analysis
Statistical analyses were performed using a one-way analysis of variance program in the SPEC/PC statistical software (SPSS, Inc., Chicago, Ill.) package on an IBM-AT microcomputer.
Results
Animal Survival
The survival data for the animals in groups 1–4 are summarized in Figure 1. All of the animals in group 1a survived, whereas 80% of the dogs in group 1b died within 48 h of drug administration. The transaminase levels in the group 1b dogs increased only moderately (<180 U/L) (Figure 1), suggesting that the mortality experienced in this group may not have been due to drug-induced liver injury per se, a conclusion that was confirmed by the histopathological data.
Figure 1.
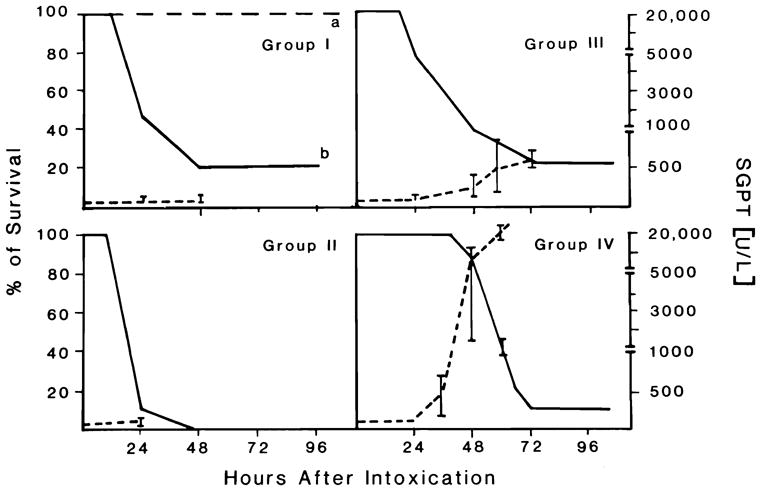
Survival rate and serum glutamic-pyruvic transaminase levels plotted across time in groups 1–4. Each panel shows the survival rate in groups 1b, 2–4 (solid line) and in group 1a (long dash line) expressed as a percentage, and the serum glutamic-pyruvic transaminase (alanine aminotransaminase) levels (short dash line) expressed as units per liter.
All of the dogs in group 2 died within 48 h of drug administration, with most of the mortality occurring in the first 24 h. Only modestly increased transaminase levels were found in these animals. In contrast to the treated animals, no change in transaminase levels or alteration of liver and kidney histology occurred in the 3 DMSO-treated control animals. Moreover, none of the controls died.
The mortality rate was 25% within 24 h, 60% at 48 h, and 80% at 72 h for group 3. The transaminase levels in this group increased markedly up to 600 U/L, suggesting substantial hepatocellular injury in these animals.
No deaths occurred in the first 24 h in group 4. Moreover, only a 10% mortality was seen in the next 24 h. From 48 through 72 h, a progressive increase in mortality was observed and achieved a level of 90% at 72 h. This group of animals demonstrated a very impressive increase in their serum transaminase levels. Peak serum glutamic-pyruvic transaminase levels (20–25 × 103 U/L) occurred between 48 and 60 h after initial dosing. It is of interest to note that the peak in the transaminase levels often paralleled the mortality.
The effect of ranitidine treatment on the survival rate of group 4 dogs is shown in Figure 2. Ranitidine had a significant protective effect. At 72 h, the survival rate of animals treated with a combination of ranitidine and acetaminophen was 80% as compared to only 10% in those given acetaminophen alone.
Figure 2.

Survival rate in group 4 dogs with and without ranitidine treatment. The treated animals received a dose of 120 mg/kg body wt i.m. 30–60 min before each acetaminophen injection.
Histopathological Results
Histologic examinations of the livers obtained at the time of autopsy or at death revealed striking differences between groups. The liver tissue obtained from dogs in groups 1, 2, and 3 demonstrated only occasional areas of centrilobular necrosis involving less than one-fifth of the lobules and less than one-fifth of the radius of the hepatic lobule, with the central vein being considered as being in the center of each lobule. Prominent centrilobular sinusoidal congestion was seen also. A mild micro-vesicular vacuolization of hepatocytes was present in several of the animals. In contrast, the liver tissue obtained from animals in group 4 demonstrated a severe zonal (centrilobular) necrosis, involving all of the lobules, with reticulin collapse and occasional central-central bridging necrosis (see Figures 3A and 3B). The degree of necrosis within individual lobules varied from involvement of one-third of the lobule in the least severely affected ones to almost total involvement in the more affected lobules. The hepatocellular necrosis seen in these animals was associated with prominent centrilobular congestion, hemorrhage, and a mild influx of neutrophils. The necrotic cells in the livers of the group 4 animals demonstrated cytoaggregation and eosinophilia as well as nuclear pyknosis, karyorrhexsis, and karyolysis. The few remaining viable periportal hepatocytes in these animals demonstrated cellular swelling and microvacuolization. No histologic abnormalities were found in the kidneys, lungs, or hearts of these animals.
Figure 3.
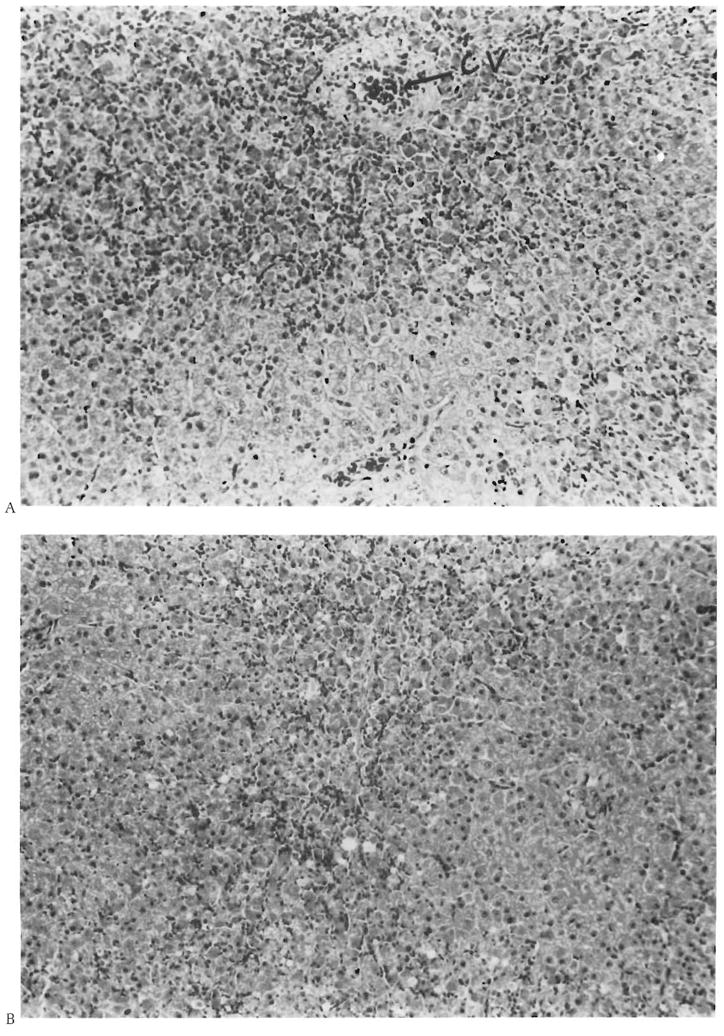
A. Histopathological appearance of the liver of group 4 animals. Note severe centrilobular necrosis and hemorrhage (CV = central vein) with preservation of some of the periportal hepatocytes (portal tract at bottom center) (H&E, × 200). B. The damage has resulted in central-central bridging necrosis (top center to bottom center) with preservation of periportal areas (left center and bottom right) (H&E, × 200).
Biochemical Results
Plasma acetaminophen levels
Blood acetaminophen levels in the dogs from groups 1–4 are depicted in Figure 4. In the animals injected intravenously, intramuscularly, or subcutaneously (groups 1, 2, and 3) with only a single dose of acetaminophen, a rapid increase in acetaminophen levels was seen for the first 2–3 h. This was followed by a rapid decline in acetaminophen levels over the next 12–16 h. In the animals in group 4 that received repeated subcutaneous injections of acetaminophen, the blood level increased for 12 h, at which point they began to slowly decline over the next 12 h such that an acetaminophen level >140 μg/ml was maintained for a period of 4–20 h.
Figure 4.

Blood acetaminophen levels in the animals in groups 1–4. Each panel shows the acetaminophen blood level expressed as micrograms per milliliter.
Biochemical parameters
The biochemical parameters monitored in the animals from groups 1, 2, and 3 (data not shown) were inconsistent with a significant degree of hepatic necrosis. Table 1 lists the various biochemical determinations obtained in the animals in group 4. Nonsurviving animals demonstrated a significant increase in transaminase levels and a significant decrease in cholesterol, albumin, and urea nitrogen levels, all of which occurred within 48 h of acetaminophen administration. Plasma ammonia levels increased rapidly. The maximum transaminase level in nonsurviving animals was 21,253 ± 3746 U/ml. The maximum transaminase level achieved in surviving animals was 1398 ± 503 U/ml. Moreover, the levels of cholesterol, albumin, urea, and ammonia reached to almost the same levels in surviving animals as that achieved in the nonsurviving animals (Table 1), but recovery to the normal level was observed during the last 48 h. Table 1 also lists the data for the plasma branched chain amino acids, aromatic amino acids, and ratio determinations of the two obtained in the animals in group 4. An increase in the plasma aromatic amino acids was observed in all group 4 animals and, as expected, a more pronounced increase was observed in the nonsurviving dogs. Bilirubin levels increased slowly in both cases without reaching a significant statistical value. Table 2 reports the results of the various coagulation tests obtained in the surviving and nonsurviving animals in group 4. A profound depression of all factors measured was observed with time in the nonsurviving animals, which is more pronounced than in surviving animals.
Table 1.
Biochemical Features of Group 4 Dogs
| 0 h | 24 h | 48 h | 60 h | 72h | 96 h | 120 h | |
|---|---|---|---|---|---|---|---|
| Nonsurviving | |||||||
| SGPT (U/L) | 41 ± 4 | 75 ± 5 | 8206 ± 3000a,b | 21,253 ± 3746a,b | |||
| Bilirubin (mg/100 ml) | 1.1 ± 0.2 | 0.9 ± 0.5 | 1.2 ± 0.4 | 2.0 ± 0.8 | |||
| Cholesterol (mg/100 ml) | 108 ±14 | 91 ± 9 | 66 ± 22a | 54 ± 18c | |||
| Albumin (g/100ml) | 3 ± 0.2 | 2 ± 0.4 | 1.9 ± 0.4a | 1.8 ± 0.3a | |||
| Ammonia (μmol/L) | 82 ± 23 | 304 ± 161a | 525 ± 133a,b | ||||
| Urea nitrogen (mg/100 ml) | 26.6 ± 2.8 | 12 ± 2.1a | 6.4 ± 1.9a,b | ||||
| AAA (μmol/100 ml) | 16 ± 2 | 69.4 ± 3.1a,b | |||||
| BCAA (μmol/100 ml) | 50.8 ± 3.5 | 61.4 ± 2.7a | |||||
| BCAA/AAA | 3.14 ± 0.23 | 0.88 ± 0.08a | |||||
| Surviving | |||||||
| SGPT (U/L) | 51 ± 16 | 71 ± 16 | 642 ± 163a | 932 ± 432a | 1398 ± 603a | 907±717a | |
| Bilirubin (mg/100 ml) | 0.8 ± 0.2 | 0.9 ± 0.7 | 1.3 ± 0.5 | 1.2 ± 0.5 | 2.0 ± 1.0a | 1.8 ± 0.7a | 1.3 ± 0.8 |
| Cholesterol (mg/100 ml) | 90 ± 1 | 83 ± 7 | 72 ± 20 | 92 ± 28 | 85 ± 43 | 89 ± 10 | 95 ± 3 |
| Albumin (g/100 ml) | 3 ± 0.2 | 1.63 ± 0.1a | 1.41 ± 0.3a | 1.63 ± 0.6a | 1.64 ± 0.1a | 1.8 ± 0.2a | 2.2 ± 0.1a |
| Ammonia (μmol/L) | 89 ± 14 | 148 ± 21a | 207 ± 19a | 284 ± 35a | 270 ± 12a | 241 ± 15a | |
| Urea nitrogen (mg/100 ml) | 25.1 ± 7 | 12.2 ± 1.5a | 10.1 ± 1.5a | 5.89 ± 3.6a | 7.81 ± 2.3a | 14.11 ± 3.5a | 20.9 ± 8.5 |
| AAA (μmol/100 ml) | 18.3 ± 1.2 | 45 ± 1a | 20 ± 3 | ||||
| BCAA (μmol/100 ml) | 46.3 ± 1.7 | 64.7 ± 0.7a | 50 ± 1 | ||||
| BCAA/AAA | 2.55 ± 0.21 | 0.99 ± 0.09a | 2.5 ± 0.12 | ||||
AAA, aromatic amino acids; BCAA, branched chain amino acids; SGPT, serum glutamic-pyruvic transaminase. The values are expressed as mean ± SD.
p < 0.05 when the values are compared with the basal levels.
p < 0.05 when the values of nonsurviving dogs are compared with the values of the surviving dogs.
Table 2.
Coagulation Profile in Dogs of Group 4 (Surviving and Nonsurviving at Different Times After Intoxication)
| 0 h | 24 h | 48 h | 60 h | 72 h | 96 h | |
|---|---|---|---|---|---|---|
| Nonsurviving | ||||||
| Fibrinogen (mg/100 ml) | 284 ± 65 | 169 ± 70a | 50 ± 38a | 23 ± 32a,b | ||
| II (U/ml) | 1.6 ± 1 | 0.6 ± 0.4 | 0.42±0.1a | 0.25 ± 0.01a,b | ||
| V (U/ml) | 6.2 ± 2.7 | 1.8 ± 0.4a | 0.52 ± 0.5a | 1.01 ± 0.9a | ||
| VII (U/ml) | 4.58 ± 2 | 0.81 ± 0,5a | 0.29 ± 0.06a | 0.20 ± 0.04a | ||
| VIII (U/ml) | 4.15 ± 0.75 | 4.18 ± 1.2 | 2 ± 0.2a,b | 2.74 ± 0.6a,b | ||
| IX (U/ml) | 1.79 ± 0.76 | 1.02 ± 0.41 | 0.55 ± 0.la,b | 0.41 ± 0.13a,b | ||
| X (U/ml) | 3.09 ± 1.2 | 0.84 ± 0.68a | 0.26 ± 0.08a | 0.20 ± 0.02a | ||
| XI (U/ml) | 3.2 ± 0.44 | 1.99 ± 0.12a | 1.58 ± 0.04a | 1.50 ± 0.15a | ||
| XII (U/ml) | 1.1 ± 0.18 | 0.75 ± 0.2a | 0.61 ± 1.5 | 0.62 ± 0.11a | ||
| Surviving | ||||||
| Fibrinogen (mg/100 ml) | 266 ± 74 | 253 ± 45 | 100 ± 6a | 198 ± 45 | 145 ± 77 | 335 ± 194 |
| II (U/ml) | 1.3 ± 0.5 | 0.60 ± 0.1a | 0.5 ± 0.3 | 0.5 ± 0.13a | 0.8 ± 0.21 | 1.1 ± 0.32 |
| V (U/ml) | 7.25 ± 1 | 1.60 ± 1.13a | 2.2 ± 0.7a | 1.5 ± 1a | 1.1 ± 0.8a | 4.9 ± 2.1a |
| VII (U/ml) | 2.70 ± 1 | 0.85 ± 0.3a | 0.66 ± 0.35a | 0.55 ± 0.33a | 0.4 ± 0.1a | 3.5 ± 2.4 |
| VIII (U/ml) | 4.8 ± 0.9 | 4.9 ± 0.7 | 5.07 ± 2.2 | 4.82 ± 1.7 | 2.4 ± 0.2a | 8.6 ± 2.3a |
| IX (U/ml) | 1.5 ± 0.2 | 1.2 ± 0.2 | 1 ± 0.3 | 0.91 ± 0.37 | 0.7 ± 0.16a | 2.14 ± 0.6 |
| X (U/ml) | 2.2 ± 0.5 | 0.7 ± 0.12a | 0.5 ± 0.29a | 0.46 ± 0.36a | 0.18 ± 0.02a | 2 ± 1.8 |
| XI (U/ml) | 3.2 ± 0.3 | 2.1 ± 0.4a | 1.8 ± 0.34a | 1.82 ± 0.5a | 1.38 ± 0.02a | 2.4 ± 0.4a |
| XII (U/ml) | 1.9 ± 0.3 | 0.9 ± 0.04a | 0.8 ± 0.2a | 0.7 ± 0.18a | 0.6 ± 0.2a | 1.1 ± 0.3a |
The values are expressed as mean ± SD.
p < 0.05 when the values are compared with the basal levels.
p < 0.05 when the values of nonsurviving dogs are compared with the values of the surviving dogs.
Hematology
A slight reduction in the hematocrit level was found in ~60% of the dogs in group 4. In 40% of the dogs in group 4, it decreased to levels as low as 35% as a result of intestinal hemorrhage occurring as part of the syndrome of fulminant hepatic failure. Hemoglobinuria and methemoglobinemia were not present in any of the dogs (data not shown). Similar findings have been reported previously in dogs subjected to acetaminophen-induced hepatic injury (18).
Clinical Features
All of the animals in group 4 demonstrated a clinical picture consistent with fulminant hepatic failure that paralleled the biochemical and histologic data. Two different stages of the clinical course of these animals could be identified. The first stage was characterized by gastrointestinal signs and symptoms such as anorexia, vomiting, and diarrhea. The second stage was characterized by the appearance of neurologic symptoms that rapidly progressed to coma.
Discussion
Fulminant hepatic failure in humans carries a very high mortality rate and usually represents the end stage of one of several different types of hepatic injury: viral infection, drug or chemical injury, or vascular insult. The onset and clinical course of fulminant hepatic failure is unpredictable, thus it has been difficult, if not impossible, to organize multicenter studies to evaluate potential therapeutic modalities that might be applied to this clinical problem using a standard protocol. The three studies that have been attempted in such cases in humans have yielded conflicting results (26–28).
A large animal model of fulminant hepatic failure suitable for the development and evaluation of potential therapeutic modalities for this condition has yet to be described. During the last 10 yr, acetaminophen toxicity (16–18) and surgically produced ischemic necrosis (4–10) have been the most widely utilized models of acute hepatic failure. Galactosamine intoxication, a model developed principally in rats (29–32), has not been studied extensively in large animals because of the considerable expense of the drug. A rabbit model of fulminant hepatic failure has been reported by Blitzer et al. (33) but has not been widely accepted principally because the rabbit is a very difficult animal with which to work. The surgical models in current use require considerable technical expertise and expensive operating room resources. Moreover, because of the expense and constraints on operating room time, it is difficult to acquire enough animals to obtain results that can be handled statistically. Worse yet, the results with such models are often inconsistent and are not reproducible from animal to animal.
Acetaminophen is an inexpensive and readily available drug. It has been used previously as a hepatotoxic agent for the production of acute hepatic failure in both large and small animals but with quite variable results. Nonetheless, the biochemical consequences and mechanisms of acetaminophen-induced liver injury have been investigated extensively. Liver damage has been shown to be related to the biotransformation of the drug into an active metabolite that binds to hepatocyte macromolecules, leading to cellular injury and death (12–15). Binding occurs only after cellular stores of glutathione have become depleted by >70% (13, 15). The hepatic toxicity of acetaminophen can be modified by either inducers or inhibitors of drug-metabolizing enzymes as well as by procedures that modify the level of protective electrophilic sulfhydryl compounds within the liver (12–15, 34).
The reported results of various experimental models (11–18) have been compared with the results reported by Prescott et al. (19) of a series of 30 patients with paracetamol overdose. In humans, the plasma acetaminophen level obtained 12 h after ingestion has been shown to be the best predictor of hepatic injury due to the drug (19).
None of the previous studies that have evaluated acetaminophen toxicity in large animals (11–19) have considered this observation. In the present report, a procedure has been developed that maintains plasma concentrations of acetaminophen for up to 20 h at a high enough concentration to create a reproducible form of severe hepatic injury that is not complicated by the appearance of methemoglobinemia. As such this would appear to be the best large animal model of fulminant hepatic failure reported to date.
It was impossible to achieve consistent hepatotoxicity with only a single administration of acetaminophen, whether given intravenously or intragastrically (16). A single dose of acetaminophen when given either with or without prior induction of microsomal enzymes results in significant animal mortality which is not due to hepatic injury but is due to factors such as methemoglobinemia and cardiorespiratory failure (35–38). In contrast, the use of a multidose drug administration protocol, with DMSO as a solvent to allow a slow steady release of the drug from the injection site, produces consistent severe hepatocellular injury. Importantly, the DMSO used as a vehicle does not influence either animal survival or hepatic function when used alone in the control animals. Thus, the multiple injection protocol appears to be the best and easiest currently available method to achieve sufficiently high blood levels of acetaminophen required to induce both consistent and severe hepatic injury. This is demonstrated clearly in Figures 1 and 4, in which both the survival rates and blood levels of acetaminophen achieved in the different groups of animals studied are shown. Group 4 animals had a high mortality rate beginning at 48 h, which peaked at 72 h. Importantly, mortality coincided with the peak transaminase levels.
All of the animals in group 1 that were not given sodium pentothal survived. In contrast, 80% of the animals treated with acetaminophen in combination with the inducer died. Neither the biochemical nor the histopathological data from these animals were consistent with fulminant hepatic necrosis. Similar results were obtained using a single dose of acetaminophen dissolved in DMSO, as was the case for groups 2 and 3. In both groups, acetaminophen levels increased quickly during the first 4 h after drug administration but then rapidly decreased reaching nonhepatotoxic levels by 8 h after administration. As was the case in group 1, all of the animals in groups 2 and 3 that died failed to demonstrate any biochemical or pathologic evidence of fulminant hepatic failure. It is therefore most likely that these animals died as a result of a combination of methemoglobinemia, kidney failure, or acute cardiovascular failure, each of which has been shown to occur in earlier studies (32–35). In contrast, the data obtained from the animals in group 4 demonstrate that multiple injections of acetaminophen maintain high blood levels of the drug, which leads to consistent severe hepatic injury. The transaminase levels in the group 4 animals rise to levels 10–500 times basal values. In addition, levels of cholesterol, albumin, and various coagulation factors in blood decrease, a finding consistent with severe hepatic injury. Moreover, a sharp increase in ammonia levels that mirrored the transaminase levels was observed. The pattern of plasma amino acid levels rapidly changed to that found in acute hepatic failure. Most importantly, the histopathological findings demonstrated massive hepatic necrosis. An elevation of serum bilirubin levels did occur (Table 1), but was not statistically significant and was lower than that reported by others (18). Nonetheless, the complete absence of hemoglobinuria and histologic evidence of renal, lung, and heart injury clearly document the fact that the animals in group 4 died as a result of acute hepatic necrosis.
An interesting observation shown in Figure 2 is that ranitidine was almost completely protective. This observation is consistent with reports demonstrating a protective effect of ranitidine therapy in rats subjected to hepatotoxic doses of acetaminophen (39). The protective effect of ranitidine is reported to occur only at very high dosages of ranitidine (39). At such concentrations, ranitidine binds to cytochrome P450 and prevents the oxidation of acetaminophen to its hepatotoxic metabolite (39–41).
The data presented with this model of fulminant hepatic failure support the following conclusions: (a) the amount of acetaminophen used in these experiments does not produce clinically important injury in organs other than the liver; (b) the DMSO used as a vehicle did not affect the results; (c) acetaminophen can be used to induce a reproducible form of severe hepatic injury in dogs; and (d) ranitidine, presumably by inhibiting the oxidation of acetaminophen, prevents the development of fulminant hepatic failure despite very high levels of acetaminophen in dogs.
In conclusion, our results describe for the first time a model of acute hepatic failure in a large animal, which can easily be reproduced in any laboratory, and which should be very useful in the study of mechanisms and therapeutic approaches to the important clinical problem of fulminant hepatic failure. This model satisfies all of the criteria outlined by Miller et al. (18) for a suitable animal model of acute hepatic failure, i.e., the damage is specific to the liver, it is effective in an animal, which allows eventual human application, the mortality exhibited following the administration of the hepatotoxic drug occurs in a defined time range, the hepatic failure is highly reproducible, and most importantly, it is safe for the investigators involved.
Acknowledgments
This study was supported by awards from the Veterans Administration and Project grants AM-29961, AM-3000l, and AM-31577, from the National Institutes of Health, Bethesda, Maryland, and by grant 887/12/9144 from the Consiglio Nazionale delle Ricerche, Italy.
References
- 1.Okuda K. Fulminant hepatic failure: a review. In: Picazo J, editor. Glucagon in gastroenterology and hepatology. Pharmacological, clinical and therapeutic implications. Boston, Mass: MTP Press; 1982. [Google Scholar]
- 2.Trey CG, Davidson CS. The management of fulminant hepatic failure. In: Popper H, Shaffner F, editors. Progress in liver disease. Vol. 3. New York: Grune & Stratton; 1970. p. 382. [PubMed] [Google Scholar]
- 3.Standardization of nomenclature, diagnostic criteria and diagnostic methodology. Proceedings No. 22. [DHEW Publications No. (NIH) 77–725] Washington, D.C: Fogarty International Center; 1977. Diseases of the liver and biliary tract. [Google Scholar]
- 4.DeGrott GH, Reuvers CG, Schalm SW. A reproducible model of acute hepatic failure by transient ischemia in the pig. J Surg Res. 1978;42:92–100. doi: 10.1016/0022-4804(87)90070-9. [DOI] [PubMed] [Google Scholar]
- 5.Cuervas MV, Golitsin A, Cienfuegos JA, et al. Hepatectomia del 70% y anastomosis portocava termino-lateral como modelo experimental de insuficiencia hepatica fulminante. Rev Esp Ap Enf Digest. 1983;64:389–94. [PubMed] [Google Scholar]
- 6.Rappaport AM, McDonald MH, Borowy ZJ. Hepatic coma following ischemia of the liver. Surg Gynecol Obstet. 1953;97:748. [PubMed] [Google Scholar]
- 7.Battersby C, Hickman R, Sauders SJ, et al. Liver function in the pig: 1. The effects of 30 minutes normothermic ischemia. Br J Surg. 1974;64:27–32. doi: 10.1002/bjs.1800610108. [DOI] [PubMed] [Google Scholar]
- 8.Fredlund PE, Ockerman PA, Vang JD. Acidosis and increased plasma levels of β-D glucosidase and β-D galactosidase after hepatic inflow occlusion in the pig. Acta Chir Scand. 1974;140:234. [PubMed] [Google Scholar]
- 9.Nodlinger B, Douvin D, Javaudin L, et al. An experimental study of survival after two hours of normothermic hepatic ischemia. Surg Gynecol Obstet. 1980;150:859–64. [PubMed] [Google Scholar]
- 10.Harris KA, Wallace AC, Wall WJ. Tolerance of the liver to ischemia in the pig. J Surg Res. 1982;33:524–30. doi: 10.1016/0022-4804(82)90072-5. [DOI] [PubMed] [Google Scholar]
- 11.Walker RM, Racz WJ, McElligott TF. Acetaminophen-induced hepatotoxicity in mice. Lab Invest. 1980;42:181. [PubMed] [Google Scholar]
- 12.Mitchell JR, Jollow DJ, Potter WZ, et al. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Ther. 1973;187:185–94. [PubMed] [Google Scholar]
- 13.Jollow DJ, Mitchell JR, Potter WZ, et al. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J Pharmacol Exp Ther. 1973;187:195–202. [PubMed] [Google Scholar]
- 14.Potter WZ, Davis DC, Mitchell JR, et al. Acetaminophen-induced hepatic necrosis. III. Cytochrome P450 mediated covalent binding in vitro. J Pharmacol Exp Ther. 1973;187:203–10. [PubMed] [Google Scholar]
- 15.Mitchell JR, Jollow DJ, Potter WZ, et al. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther. 1973;187:211–7. [PubMed] [Google Scholar]
- 16.Ortega L, Landa Garcia JI, Torres Garcia A, et al. Acetaminophen-induced fulminant hepatic failure in dogs. Hepatology. 1985;5:673–6. doi: 10.1002/hep.1840050425. [DOI] [PubMed] [Google Scholar]
- 17.Gazzard BG, Hughes RD, Mellon PJ, et al. A dog model of fulminant hepatic failure produced by paracetamol administration. Br J Exp Pathol. 1975;56:408. [PMC free article] [PubMed] [Google Scholar]
- 18.Miller DJ, Hickman R, Fratter R, et al. An animal model of fulminant hepatic failure: a feasibility study. Gastroenterology. 1976;71:109–13. [PubMed] [Google Scholar]
- 19.Prescott LF, Roscoe P, Wright N, et al. Plasma-paracetamol half-life and hepatic necrosis in patients with paracetamol overdosage. Lancet. 1971;i:519. doi: 10.1016/s0140-6736(71)91125-1. [DOI] [PubMed] [Google Scholar]
- 20.Routh JI, Shane NA, Arredondo EG, et al. Determination of N-acetyl-p-aminophenol in plasma. Clin Chem. 1968;14:882–99. [PubMed] [Google Scholar]
- 21.Bontempo FA, Lewis JH, Van Thiel DH. The relation of preoperative coagulation findings to diagnosis, blood usage, and survival in adult liver transplantation. Transplantation. 1985;39:532–6. doi: 10.1097/00007890-198505000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis JH. Coagulation defects. JAMA. 1961;178:1014. doi: 10.1001/jama.1961.73040490010008. [DOI] [PubMed] [Google Scholar]
- 23.Lewis JH. Hemostasis and hemorrhage. Sci Clin. 1971;1:1–66. [Google Scholar]
- 24.Lewis JH, Spero JA, Hasiba U. Bleeding disorders. Garden City, N.Y: Medical Examination Publishing; 1978. Diagnostic methods: laboratory tests; p. 22. [Google Scholar]
- 25.Van Assendelfp OW. Spectrophotometry of hemoglobin derivatives. CC Thomas; 1970. pp. 128–30. [Google Scholar]
- 26.Volpicelli NA. Hepatotrophic effects of insulin and glucagon and their potential role in the treatment of hepatic failure. In: Picazo J, editor. Glucagon in gastroenterology. Baltimore: University Park Press; 1978. pp. 121–34. [Google Scholar]
- 27.Redeker AG, Yamahiro US. Controlled trial of exchange transfusion therapy in fulminant hepatitis. Lancet. 1973;i:3–6. doi: 10.1016/s0140-6736(73)91220-8. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi Y, Shimizu M, Kosaka M. Nationwide statistics of severe hepatitis (fulminant hepatitis) Sashin Igaku. 1979;34:2285–8. [Google Scholar]
- 29.Makowka L, Falk RE, Rotstein LE, et al. Reversal of experimental acute hepatic failure in the rat. J Surg Res. 1980;29:479–87. doi: 10.1016/0022-4804(80)90016-5. [DOI] [PubMed] [Google Scholar]
- 30.Makowka L, Falk RE, Rotstein LE, et al. Cellular transplantation in the treatment of experimental hepatic failure. Science. 1980;210:901–18. doi: 10.1126/science.7001630. [DOI] [PubMed] [Google Scholar]
- 31.Makowka L. PhD thesis. University of Toronto; Toronto, Ontario, Canada: 1982. Studies into the reversal of experimental acute hepatic failure in the rat by hepatocyte transplantation. [Google Scholar]
- 32.Francavilla A, DiLeo A, Polimeno L, et al. The effect of hepatic stimulatory substance, isolated from regenerating hepatic cytosol, and 50,000 and 300,000 subfractions in enhancing survival in experimental acute hepatic failure in rats treated with D-galactosamine. Hepatology. 1986;6:1346–51. doi: 10.1002/hep.1840060621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blitzer BL, Waggoner JG, Jones AE, et al. A model of fulminant hepatic failure in the rabbit. Gastroenterology. 1978;74:664–71. [PubMed] [Google Scholar]
- 34.Chiu S, Bhakthan NMG. Experimental acetaminophen-induced hepatic necrosis: biochemical and electron microscopic study of cysteamine protection. Lab Invest. 1978;39:193. [PubMed] [Google Scholar]
- 35.Evelyn KA, Malloy HT. Microdetermination of oxyhemoglobin and sulphemoglobin in a single sample of blood. J Biol Chem. 1938;126:655. [Google Scholar]
- 36.McLean S, Murphy BP, Starmer GA, et al. Methemoglobin formation induced by aromatic amines and amides. J Pharm Pharmacol. 1967;19:146. doi: 10.1111/j.2042-7158.1967.tb08056.x. [DOI] [PubMed] [Google Scholar]
- 37.Levenson DJ, Skorecki KL, Narins RG. Acute renal failure associated with hepatobiliary disease. In: Brenner BM, Lazarus JM, editors. Acute renal failure. Philaddphia: WB Saunders; 1983. pp. 467–98. [Google Scholar]
- 38.Cobden I, Record CO, Wark MK, et al. Paracetamol-induced acute renal failure in the absence of fulminant liver damage. Br Med J. 1982;284:21–2. doi: 10.1136/bmj.284.6308.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leonard TB, Morgan DG, Dent JG. Ranitidine-acetaminophen interaction: effects on acetaminophen-induced hepatotoxicity in Fischer 344 rats. Hepatology. 1985;5:480–7. doi: 10.1002/hep.1840050323. [DOI] [PubMed] [Google Scholar]
- 40.Rendie S, Kajfez F, Ruf HH. Characterization of cimetidine, ranitidine, and related structures’ interaction with cyto-chrome P-450. Drug Metab Dispos. 1983;11:137–42. [PubMed] [Google Scholar]
- 41.Rende S, Alebic Kolbah T, Kajfez F, et al. Interaction of ranitidine with liver microsomes. Xenobiotica. 1982;12:9–17. doi: 10.3109/00498258209052450. [DOI] [PubMed] [Google Scholar]