

Abstract
A proteomic approach was applied to liver cytosol from rats fed a diet consisting of high fat and ethanol to identify 4-hydroxy-2-nonenal (4-HNE)-modified proteins in vivo. Cytosolic Hsp72, the inducible variant of the Hsp70 heat shock protein family, was consistently among the proteins modified by 4-HNE. Despite 1.3-fold induction of Hsp72 in the livers of ethanol-fed animals, no increase in Hsp70-mediated luciferase refolding in isolated heptocytes was observed, suggesting inhibition of this process by 4-HNE. A 50% and 75% reduction in luciferase refolding efficiency was observed in rabbit reticulocyte lysate (RRL) supplemented with recombinant Hsp72 which had been modified in vitro with 10 and 100 μM 4-HNE, respectively. This observation was accompanied by a 25% and 50% decrease in substrate binding by the chaperone following the same treatment; however, no effect on complex formation between Hsp72 and its co-chaperone Hsp40 was observed. Trypsin digest and mass spectral analysis of Hsp72 treated with 10 and 100 μM 4-HNE consistently identified adduct formation at Cys267 in the ATPase domain of the chaperone. The role of this residue in the observed inhibition was demonstrated through the use of DnaK, a bacterial Hsp70 variant lacking Cys267. DnaK was resistant to 4-HNE inactivation. Additionally, Hsp72 was resistant to inactivation by the thiolunreactive aldehyde malondialdehyde (MDA), further supporting a role for Cys in Hsp72 inhibition by 4-HNE. Finally, the affinity of Hsp72 for ATP was decreased 32% and 72% following treatment of the chaperone with 10 and 100 μM 4-HNE, respectively. In a model of chronic alcoholic liver injury, induction of Hsp72 was not accompanied by an increase in protein refolding ability. This is likely the result of 4-HNE modification of the Hsp72 ATPase domain.
Introduction
Molecular chaperones are involved in events such as protein folding and repair, protein translocation, and protein degradation (1-4). Heat, oxidative stress, and other cellular insults induce the heat shock response, characterized by a rapid production of additional molecular chaperones, or heat shock proteins. The heat shock proteins (Hsp) provide a defense mechanism against the formation of insoluble protein aggregates and apoptosis of the cell (5, 6).
The Hsp70 family of molecular chaperones interact with proteins through formation of Hsp70–substrate complexes. Conserved within the Hsp70 family is an ATP-binding domain which exhibits low (e.g., 5 pmol of ATP/min/μg of protein) intrinsic ATPase activity (2, 7). The importance of this domain regarding protein substrate binding has been well characterized, establishing that the ADP-bound state demonstrates the highest affinity for substrate, followed by the ATP-bound state, and finally by the nucleotide-unbound state which demonstrates the least affinity for substrate (8). Many of the molecular co-chaperones that influence the function of Hsp70 do so through manipulation of the intrinsic ATPase activity. Included among the co-chaperones associated with the Hsp70 family of molecular chaperones are Hsp40 and HIP, which stimulate ATPase activity, promoting substrate refolding, and Bag-1 and HspBP, both of which have demonstrated inhibitory roles toward the ATPase activity and protein refolding (9-13).
In addition to induction of the heat shock response, oxidative stress also results in peroxidation of polyunsaturated fatty acids (14). Spontaneous scission of the C–C bond on either side of the hydroperoxy group yields multiple electrophilic aldehyde species (14, 15). Among these compounds, the α,β-unsaturated aldehyde 4-HNE has demonstrated reactivity against cellular nucleophiles, particularly Cys, His, and Lys residues, and through the formation of Michael addition and Schiff base products with these residues can disrupt protein function (15-19). 4-HNE is associated with many oxidative stressrelated disorders, such as alcoholic liver disease and reperfusion injury, and likely exacerbates existing conditions through modification of cellular targets (15). In fact, 4-HNE formed during nonalcoholic fatty liver disease has recently been proposed to play a key role in the progression of this disease to fibrosis (20).
In the present study, rats fed a high-fat/ethanol diet demonstrate a remarkable increase in the number and severity of 4-HNE-modified proteins over their pair fed controls. A proteomics-based approach consisting of two-dimensional gel electrophoresis, immunoblotting, in-gel digest, and peptide mass fingerprinting has been used to identify proteins modified by 4-HNE. Using this approach, the inducible member of the Hsp70 family, Hsp72, was consistently among the proteins identified as substrates for in vivo 4-HNE modification. Although little or no hepatic induction of Hsp72 by ethanol is usually observed in animal feeding studies (21-23), a slight (1.3-fold) induction of the chaperone was present in the ethanol-fed animals. Interestingly, no corresponding increase in Hsp70-mediated protein refolding was observed by hepatocytes isolated from these same animals. It was therefore hypothesized that Hsp72 may be susceptible to inactivation by reactive lipid aldehydes. Following in vitro modification of the inducible member of Hsp72 by 4-HNE, it was determined that the aldehyde is a potent inhibitor of the chaperone’s refolding ability. Further experimentation revealed a decrease in substrate binding by 4-HNE-modified Hsp72 and that Cys modification in the ATPase domain of the chaperone is indirectly responsible for this effect.
Materials and Methods
Reagents
Unless stated otherwise, all reagents were purchased from Sigma-Aldrich Chemical Co. (Saint Louis, MO). Untreated RRL was purchased through Promega Corp. (Madison, WI). 4-HNE, 4-ONE, and MDA were synthesized in our laboratory according to procedures described previously, and purity and concentration were confirmed by TLC and UV/vis spectrophotometry (16, 24, 25). Recombinant firefly luciferase and a luciferase assay system were purchased through Promega Corp. (Madison, WI). Human recombinant Hsp72 and Hsp40 and bacterial DnaK were purchased through Stressgen Biotechnologies (Victoria BC, Canada).
Animals
All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the University of Colorado and were performed in accordance with published National Institutes of Health guidelines. Male Harlan Sprague–Dawley rats were fed a diet consisting of 45% fat-derived calories, 35% ethanol-derived calories, and 16% protein for a period of 60 days. Each ethanol-fed animal was paired with a pair-fed nutritional control that received a diet containing equivalent sucrose-derived calories. Upon completion of the feeding period, animals were anesthetized by intraperitoneal sodium pentobarbital injection and euthanized by exsanguination. Blood samples were collected for determination of plasma ALT and ammonia concentration, which were measured using Sigma-Aldrich Infinity ALT Reagent and Ammonia (Procedure No. 161-UV) kits, respectively. Livers were removed, and 10% (w/v) homogenates were prepared in 0.25 M sucrose containing 1.0 mM EDTA and 1.0 mM BHA. The homogenization solution was supplemented with a panel of serine, cysteine, aspartic acid, and metallo-protease inhibitors. Subcellular fractionation of the homogenates was accomplished by the differential centrifugation procedures described elsewhere (26). Animals from which hepatocytes were isolated were anesthetized by intraperitoneal sodium pentobarbital, and hepatocytes were isolated following the collagenase perfusion method described by Hartley et al. (27). Protease inhibitors were also added to the isolated hepatocyte preparations. Hepatocytes used for luciferase refolding assays were lysed using Luciferase Cell Culture Lysis Reagent (Promega Corp., Madison, WI) according to the kit instructions. Protein was quantified using the Biorad Protein Assay reagent. Cells and subcellular fractions were stored at −80 °C until analysis.
Two-Dimensional Electrophoresis and In-Gel Digest
Isoelectric focusing was performed using 180 mm × 70 mm × 2 mm (ID) tube gels. Tube gels consisted of 9.2 M urea, 4.5% (v/v) acrylamide, 24 mM CHAPS, 1% (v/v) Biorad Biolyte 5/7 ampholyte (Hercules, CA), 4% (v/v) Biorad Biolyte 3/10 ampholyte, and 0.5% (v/ v) Igepal CA 630, and were polymerized with 2% (v/v) ammonium persulfate and 0.1% (v/v) TEMED. Protein from cytosolic fractions (200 μg) was loaded on each tube, and focusing was conducted over 20 h as follows: 200 V for 2 h, 500 V for 2 h, and 800 V for 16 h using 20 mM sodium hydroxide (anode buffer) and 5.9% (v/v) phosphoric acid (cathode buffer). At the completion of isoelectric focusing, each gel was extruded from its respective glass tube by water compression, followed immediately by SDS-PAGE separation. Slab gels (18 cm × 16 cm × 2 mm) used for the SDS-PAGE dimension consisted of a 6–15% linear gradient with a 5% stacking gel and were maintained at 4 °C for the duration of the process using a recirculating water bath.
Proteins were only considered for harvesting if they stained positive for 4-HNE adduction following immunoblot from no less than three ethanol-fed animals, while the pair-fed control consistently stained negative. Harvesting was performed by a modification of the procedures described by Shevchenko et al (28). Following two-dimensional gel electrophoresis and spot harvesting, proteins from ethanol-fed animals were fixed for 30 min in 1% (v/v) methanol, 0.7% (v/v) acetic acid. Proteins were visualized using a Pierce (Rockford, IL) GelCode E-Zinc Reversible Stain Kit and were excised and placed in a 1.5 mL microcentrifuge tube. Proteins were destained according to kit instructions, and the destain reagent was removed by suction. Spots were then equilibrated in 250 μL of 100 mM ammonium bicarbonate for 1 h at 25 °C, after which the ammonium bicarbonate was removed by suction and replaced with 100% acetonitrile (ACN) until the spot was completely dehydrated. The ACN was then removed by suction, and spots were allowed to air-dry for 1 h at 25 °C. Proteins were digested overnight (approximately 16 h) at 25 °C in the presence of 100 mM ammonium bicarbonate and 0.63 μg sequencing grade recombinant trypsin (Promega Corp.).
In Vitro Hsp72 Modification by 4-HNE and Tryptic Digest
Human recombinant Hsp72 or bacterial DnaK (5.75 μg, 1.6 μM final concentration) was incubated in the presence of 0, 10, or 100 μM 4-HNE in 50 mM sodium phosphate (pH 7.4) overnight (approximately 16 h) at 37 °C. For tryptic digest, Hsp72 was heat denatured at 100 °C for 5 min in the presence of 2 mM β-mercaptoethanol and cooled on ice prior to digest. Hsp72 was digested overnight at 37 °C in 10% (v/v) ACN, 50 mM ammonium bicarbonate, and 0.3 μg trypsin.
Mass Spectral Analysis
Peptides (8 μL) from each digest, or purified Hsp72 which had previously been treated with 4-HNE and subject to trypsin digest, were analyzed by liquid chromatography, tandem mass spectrometry (LC-MS/MS) using an Agilent (Palo Alto, CA) 1100 Series LC/ESI-MSD trap equipped with a Phenomenex (Torrance, CA) Jupiter C18 column (1 × 150 mm, 300 Å). The mobile phase consisted of 0.2% formic acid in water (A) and 0.2% formic acid in acetonitrile (B) with flow rate of 50 μL/min and gradient conditions as follows: 5% B at 0 min, 5% B at 5 min, 70% B at 35 min, 90% B at 38 min and held for 2 min, and 5% B at 42 min and held for 3 min. Mass spectrometric detection and analysis was accomplished using positive ion mode with a capillary voltage of 3.5 kV. Nebulizer pressure was set at 20 psi, and dry gas flow was set at 8 L/min with the temperature of the dry gas set to 350 °C. The scanning range for all analyses was 400–2000 m/z. Peptides within the mass range of 500–1500 Da were subject to MS/MS analysis. MS/MS ion search was performed on deconvoluted spectra using MASCOT (29). For in vitro modification experiments with Hsp72, tryptic peptides were calculated using the MS-Digest feature of Protein Prospector version 4.0.5 (http://prospector.ucsf.edu). Peptides modified by 4-HNE were identified on the basis of the presence of the parent peptide in both treated and untreated protein digests, in addition to a mass shift of the parent peptide equal to that of 4-HNE (156 D) in the treated protein. The identity of the aldehyde-modified peptide and the location of the adduct were determined via MS/MS analysis. Fragment ions were calculated using the MS-Product feature of Protein Prospector version 4.0.5.
Western Blotting
All gels used for Western blotting were transferred to PVDF membrane using a semidry transfer apparatus (BioRad, Hercules CA). All membranes were blocked in 5% (w/v) nonfat dry milk (NFDM) in TBS-T for 30 min at room temperature. Antibody dilutions were performed in 5% (w/v) NFDM in TBS-T. Horseradish peroxidase (HRP)-linked goat anti-rabbit secondary antibodies (Pierce) were used following a 1:5000 dilution for detection of primary antibodies generated in rabbit hosts. HRP-linked goat anti-mouse antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were diluted 1:4000 for detection of primary antibodies generated in mouse hosts. 4-HNE adducts were confirmed using custom antibodies generated in rabbit hosts against 4-HNE-modified keyhole limpet hemocyanin (27). Proteins were probed for 4-HNE adducts overnight at 4 °C using a 1:2000 dilution of the above-mentioned antibody. Antibodies against Hsp72 and Hsp40 (StressGen Biotechnologies) were used at dilutions of 1:2000 and 1:5000, respectively. Antibodies against luciferase were purchased from AbCam, Inc. (Cambridge, MA), and were used at a dilution of 1:7000. Membranes were treated with Amersham ECL-Plus (Piscataway, NJ) enhanced chemiluminescence reagent, and bands or spots were visualized using film and or a Molecular Dynamics STORM 860 (Sunnyvale, CA). Bands were quantified using the ImageQuant (Amersham Biosciences) software package.
Luciferase Refolding
Recombinant firefly luciferase was diluted to a concentration of 100 nM in 25 mM Tricine, pH 7.8, 8 mM magnesium sulfate, 10 mg/mL bovine serum albumin, and 10% (v/v) glycerol, and was stored in aliquots at −80 °C (13). Luciferase was denatured by heating the enzyme to 45 °C for 30 min and cooled on ice prior to the refolding assays. Luciferase refolding assays were conducted over the course of 60 min at 25 °C in RRL spiked with 600 nM recombinant Hsp72 or DnaK (control or 4-HNE treated), 120 nM recombinant Hsp40, 1.5 mM DTT, 2 mM ATP, and 7.5 nM heat-denatured luciferase. Hepatocyte lysates used for luciferase refolding were supplemented with 2 mM ATP and an ATP regenerating system consisting of 10 mM creatine phosphate and 0.1 mg/mL creatine phosphokinase. At the indicated time points, 5 μL aliquots were removed and added to 100 μL of luciferase assay reagent (Promega) in borosilicate glass tubes. Luminescence was immediately read using a Los Alamos Diagnostics 535 luminometer. For proteasome inhibition experiments, 50 μMMG-132 was included in the refolding system. Control experiments containing the equivalent volume of DMSO were also conducted, confirming that DMSO alone had no effect on refolding at the concentrations used.
Amino Acid Sequence Alignment
The amino acid sequences for Hsp72 and DnaK were obtained from NCBI (P08107 and P04475, respectively). Sequence alignment was performed using Clustal Version 1.8 (30).
Co-immunoprecipitation
Co-IP procedures were adapted from a previously published report (31). Briefly, reactions were assembled as in refolding experiments; however, 5 μg of mouse anti-Hsp72 (Stressgen Biotechnologies) was added to each reaction, and reactions were incubated overnight at 4 °C with gentle agitation. Next, 40 μL of protein A/G agarose (Calbiochem, San Diego CA) was added to each tube, and reactions were incubated for an additional 90 min at 4 °C with gentle agitation. Supernatant was removed following flash centrifugation (14 000g), and beads were washed three times using 50 mM Tris buffer (pH 7.8) containing 150 mM NaCl and 1% (v/v) Igepal CA 630. Next, 15 μL 6× SDS-PAGE loading buffer was added to each tube, followed by heating at 90 °C for 5 min. Beads were flash centrifuged following heating, and 15 μL of supernatant from each reaction was loaded and separated on a 10% SDS-polyacrylamide minigel. Western blots were conducted as described above. Antibody was stripped from membranes at 50 °C for 30 min using 100 mM β-mercaptoethanol in 50 mM tris buffer (pH 6.8) and 2% (v/ v) SDS, followed by thorough washing with TBS-T.
Hsp72 ATP Affinity
The affinity of recombinant Hsp72 for ATP was measured using ATP-linked agarose beads (Sigma). Briefly, Hsp72 was modified as described above in a 50 μL total volume to which 50 μL of agarose slurry was added. Binding was allowed to proceed for 1 h at 4 °C with gentle agitation. Reactions were flash centrifuged (14 000g), and supernatant was removed. The agarose beads were washed twice in 25 mM HEPES/KOH, pH 7.6, 50 mM KCl, 2.5 mM MgCl2, and 10 mM DTT (32). Next, 40 μL 6× SDS-PAGE loading buffer was added to each tube, and reactions were heated to 95 °C for 5 min. The extent of Hsp72 bound to the beads was determined by SDS-PAGE/immunoblot using antibodies against Hsp72.
Statistical Methods
Statistical analysis was performed using the software package GraphPad Prizm version 3.02 (GraphPad Software, San Diego, CA). Plasma ALT and ammonia, body-to-weight ratio, body weight, and Hsp72 induction in control animals were compared to their ethanol-fed pairs using a two-tailed t-test. The final time points from representative time courses were sampled no less than three times and were compared by one-way ANOVA with Bonferroni post-test. Co-IP experiments were performed in two independent sets, and densitometric data were pooled, normalized to Hsp72 signal, and compared by one-way ANOVA with Bonferroni post-test. Data from ATP affinity experiments were pooled from no less than two independent experiments and were compared using one-way ANOVA with Bonferroni post test. In all cases, mean differences were considered significant when p < 0.05 even though the level of significance was often much greater.
Results
Classic markers of liver injury include elevated blood ALT and ammonia, and an increase in liver-to-body weight ratio accompanied by a decrease in body weight. These markers were therefore used to provide evidence of liver injury following the high fat/ethanol feeding study described here. In this particular case, plasma ALT and ammonia values were elevated significantly (1.8 and 1.3-fold, respectively) in ethanol-fed animals, as was the liver-to-body weight ratio (1.2-fold), while body weight was significantly lower in ethanol-fed animals (1.1) as summarized in Table 1. Histopathologic evaluation (unpublished observation) of the livers obtained from ethanol-treated animals revealed primarily microsteatosis and, to a lesser extent, macrosteatosis. Hepatocellular necrosis and inflammation were not observed in the ethanol-treated rats. Steatosis, necrosis, and inflammation were not observed in liver sections prepared from isocaloric control rats. Collectively, the approximate 2-fold elevation of ALT values and detection of steatosis in the absence of necrosis and inflammation indicate that the ethanol treatment regimen used for this study resulted in mild liver injury.
Table 1.
Blood Ammonia and ALT, Body Weight, and Liver-to-Body Weight Ratio
| treatment | parameters |
|||
|---|---|---|---|---|
| body wt (g)a | liver:body (%)a | ALT (U/L)b | Ammonia (μM)b | |
| control | 441.9 (±8.7) | 3.04 (±0.11) | 19 (±3.0) | 176.6 (±22.38) |
| ethanol | 411.1 (±10.8)* | 3.62 (±0.07)* | 35 (±5.0)* | 226.2 (±23.26)* |
Body wt. and liver:body weight ± SEM (n = 8).
Ammonia and ALT ± SEM (n = 15).
Oxidative stress is central to alcoholic liver disease. Because lipid-derived aldehydes (e.g., 4-HNE) often accompany oxidative injury, a proteomics-based approach was used to identify proteins susceptible to 4-HNE modification in vivo. Following two-dimensional gel electrophoresis, 4-HNE-modified proteins were recognized by immunoblot, excised from the gel, and identified by in-gel trypsin digest, LC-MS/MS analysis, and MS/MS ion search. Hsp72, the inducible form of Hsp70, was found to be among the proteins consistently adducted by 4-HNE. Immunoblots against 4-HNE are shown in Figure 1, demonstrating an increase in the extent of 4-HNE modification of Hsp72 (arrowhead) following ethanol feeding. The appearance of immunoreactive spots in the control animals is attributed to low levels of oxidative stress induced in part by the high fat diet, as demonstrated via parallel experiments using animals fed standard rat chow (i.e., low fat diet) (unpublished observation). Several proteins in the immediate region of Hsp72 were used as landmarks (arrows) for locating this chaperone in liver cytosolic fractions from control (Figure 1A) and ethanol-treated (Figure 1B) rats. LC-MS/MS and subsequent MASCOT analysis following in-gel digest revealed 10 Hsp72 peptides (i.e., approximately 35% coverage). No other proteins were identified in this immunopositive spot.
Figure 1.
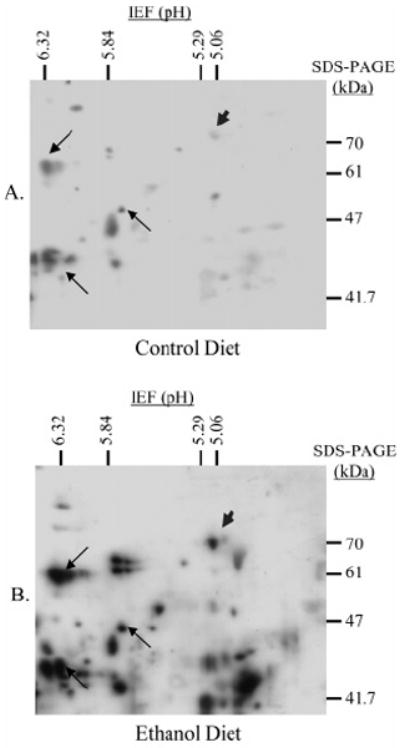
Two-dimensional electrophoresis and immunoblot against 4-HNE-modified proteins from control (A) and ethanol-fed (B) rat liver cytosolic fractions. Hsp72 was identified following in-gel digest and LC-MS/MS analysis interfaced with the MASCOT search engine, and is indicated (arrowhead) along with three spots used for alignment (arrows).
SDS-PAGE followed by Western blot against Hsp72 was performed to quantify Hsp72 induction by the ethanol diet. Figure 2A consists of a representative Western blot for Hsp72 in control animals (animals C1–C5; lanes 2–6) and their ethanol fed pairs (animals E1–E5; lanes 7–11). A positive control consisting of purified recombinant Hsp72 was included in lane 1 to aid in identification of the correct band. Densitometric analysis of Hsp72 in Figure 2B confirms an approximate 1.3-fold (p < 0.05) induction of the chaperone. It was anticipated that an increase in the amount of Hsp72 would enhance the hepatocellular ability to refold heat-denatured luciferase; however, this was not the case. No significant difference was observed in the ability to refold heat-denatured luciferase between hepatocytes from control and ethanol-treated animals, demonstrated by Figure 2C. The enzymatic activity of heat-denatured luciferase (HD Luc) was assessed after 1 h to control for spontaneous refolding, and the intrinsic luminescence of rat liver cytosol was also measured as a control against any background luminescence (cytosol). Taken together, the ethanol diet appears to have led to a subtle but significant induction of Hsp72, with no corresponding increase in the refolding ability when compared to pair-fed control animals.
Figure 2.
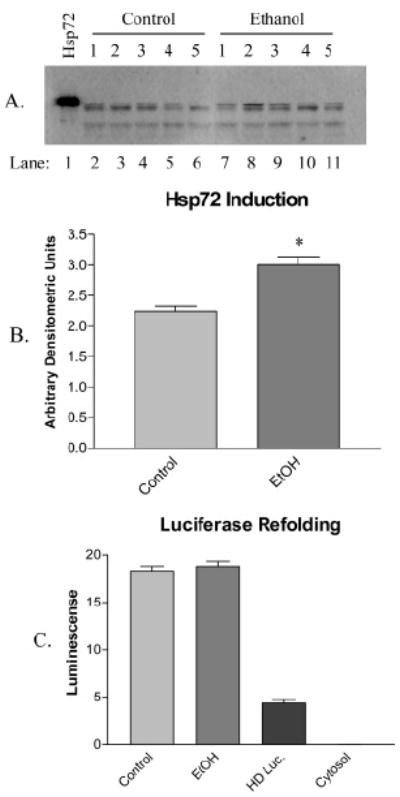
(A) Induction of Hsp72 is demonstrated in a representative immunoblot using antibodies against the chaperone in control (animals C1–C5; lanes 2–6) and ethanol-fed (animals E1–E5; lanes 7–11) rats. Protein identity was confirmed with recombinant Hsp72 (lane 1). (B) Densitometry confirms an approximate 1.3-fold induction (n = 4 ± SD). (C) Protein refolding by lysed hepatocytes from control and ethanol-fed animals using heat-denatured luciferase, with controls for spontaneous refolding (HD Luc.) and intrinsic luminescence (cytosol) (n = 3 ± SEM).
The importance of molecular chaperones in maintaining cellular homeostasis is widely accepted, as is the reactivity of lipid aldehydes toward protein modification. The consistent presence of 4-HNE-modified Hsp72 in ethanol-fed rat liver cytosol together with the absence of any enhanced refolding ability after induction of the chaperone led to a series of in vitro studies designed to assess the relationship between 4-HNE and Hsp72. Treatment of purified recombinant Hsp72 with concentrations of 4-HNE ranging from approximately 6 to 60-fold molar excess (10–100 μM) under physiological pH and temperature lead to a greater extent of adduct formation, as demonstrated by Figure 3A. Consistent gel loading was confirmed by stripping 4-HNE adduct-reactive antibodies from the membrane, and reprobing with antibodies against Hsp72 (Figure 3B). Trypsin digest of recombinant Hsp72 followed by LC-MS/MS peptide analysis typically yielded approximately 88% sequence coverage. This approach was therefore used to locate any site(s) of 4-HNE adduction. In Hsp72 modified by 10 or 100 μM 4-HNE, Cys267 in the ATPase domain of the protein was found to be adducted by the aldehyde. Figure 3C demonstrates modification of the peptide corresponding to amino acids 265–269 (TACER) by 4-HNE following treatment with 10 μM aldehyde. The presence of the adduct was confirmed by neutral loss of 4-HNE (156 D) following fragmentation of the peak at m/z 735.4 (Figure 3D). Furthermore, MS/MS analysis yielded the following fragment ions, thus confirming the identity of the peptide and location of the adduct: y2 (304.1), b3 (432.8), y3-H2O (545.3), y3-NH3 (546.3), b4 (561.1), y3 (563.3), y4-H2O (616.3), and y4 (634.3). Unfortunately, this peptide was not identified following in gel digest of Hsp72; thus the adduct could not be confirmed in vivo.
Figure 3.
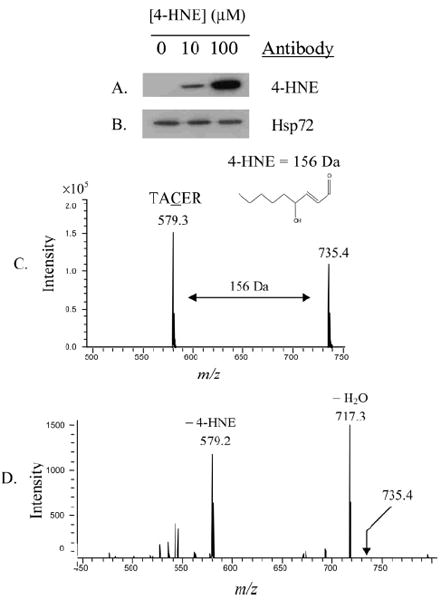
In vitro modification of recombinant Hsp72 by 4-HNE (A), and confirmation of consistent gel loading using antibodies against Hsp72 (B). Identification of 4-HNE adduct location on the Hsp72 peptide TACER, corresponding to amino acids 265–269. The peaks at m/z 579.2 and 735.4 represent the unmodified and 4HNE-modified peptide, respectively (C). Confirmation of Cys267 modification by 4-HNE was acquired fromMS/MS analysis of the peak at m/z 735.4, clearly demonstrating neutral loss of the 156 D 4-HNE adduct and fragmentation consistent with modification of Cys267 (D).
The effect of in vitro modification of Hsp72 by 4-HNE was assessed by measuring the extent of luciferase refolding by RRL supplemented with control or treated Hsp72 and Hsp40. Elevation of luciferase refolding by Hsp72/Hsp40 supplemented RRL over nonsupplemented RRL is presented by Figure 4A, which also clearly demonstrates inhibition of Hsp72-mediated luciferase refolding following pretreatment of Hsp72 with 4-HNE. Controls in which free 4-HNE was added to RRL yielded little inhibition (Figure 4B), suggesting that this effect is due to the Hsp72 pretreatment with 4-HNE. Interestingly, the refolding ability of the lysate was inhibited beyond the level of the RRL control.
Figure 4.
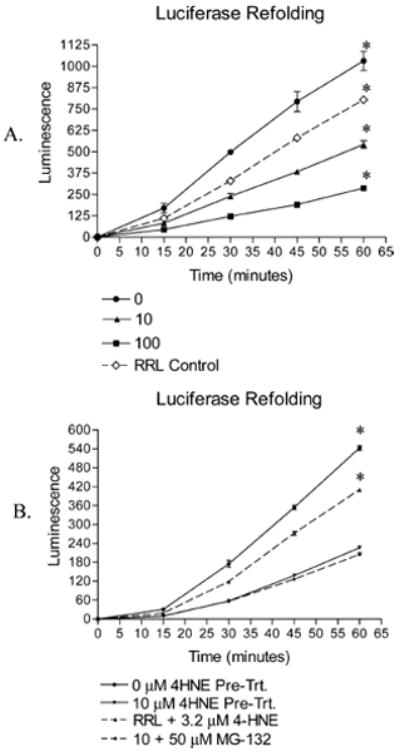
(A) Refolding of heat-denatured recombinant firefly luciferase by rabbit reticulocyte lysate alone (RRL control) or supplemented with Hsp40 and 4-HNE untreated (0 μM) or treated (10 μM, 100 μM) Hsp72. Inhibition of refolding correlates to 4-HNE pretreatment, and decreases refolding below that of the RRL control (n = 3 ± SD, * indicates significant difference from all groups). (B) Use of the proteasome inhibitor MG-132 has no effect on the recovery of luciferase activity, ruling out enhanced interaction between Hsp72 and the 26S proteasome. Free 4-HNE demonstrates inhibition of luciferase refolding, but is less pronounced than that following pretreatment of Hsp72 (B) (n = 3 ± SD, * indicates significant difference from all groups).
Several heat shock proteins, including members of the Hsp70 family, possess the ability to present their substrates to the 26S proteasome for degradation (3, 4). To rule this out as a possible explanation for the decrease in luciferase refolding, the proteasome inhibitor MG-132 was included in a series of luciferase refolding experiments at a final concentration of 50 μM (33). Because addition of the proteasome inhibitor did not restore luciferase activity (Figure 4B), interference with the refolding mechanism rather than more rapid degradation appears to be the explanation behind 4-HNE-mediated inhibition of luciferase refolding.
The role of 4-HNE modification at Cys267 regarding Hsp72-mediated protein refolding was further examined using the bacterial Hsp70 analogue DnaK, in which Cys267 is replaced with Ala (Figure 5A), which does not react with 4-HNE (16). Both Hsp72 and DnaK were pretreated with 0 μM or 10 μM 4-HNE and were added to RRL to assess their respective refolding abilities. Treatment of Hsp72 with 4-HNE led to the anticipated inhibition of luciferase refolding; however, DnaK treated the same way in all respects was completely resistant to inhibition by the aldehyde, demonstrated in Figure 5B. To provide further evidence of the importance of Cys modification in aldehyde-induced inhibition of Hsp72, the thiol-reactive aldehyde 4-oxononenal (4-ONE) (16) and the thiol nonreactive aldehyde malondialdehyde (MDA) (15) were used to treat Hsp72, and their respective effects on protein refolding were compared to that of 4-HNE. These results are demonstrated by Figure 5C, in which no inhibition of Hsp72-mediated luciferase refolding was observed following treatment of the chaperone with MDA, while treatment with the thiol-reactive aldehydes 4-HNE and 4-ONE inhibit refolding. When considered together, the results in Figure 5 strongly support the argument that Cys modification, specifically Cys267, plays a major role in 4-HNE inhibition of Hsp72.
Figure 5.
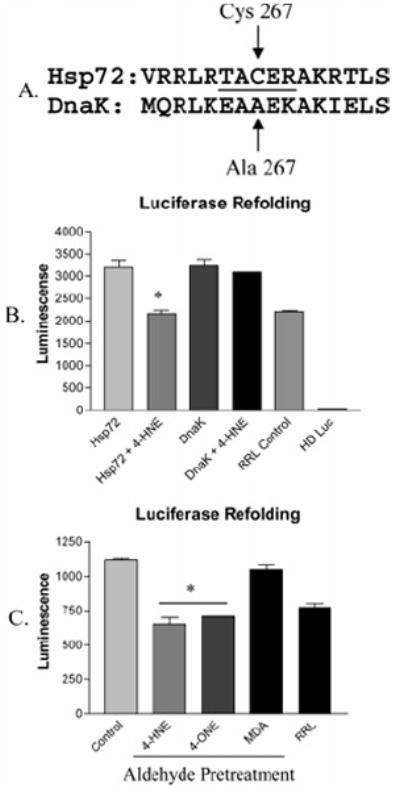
(A) CLUSTAL W amino acid alignment of Hsp72 (P08107) and bacterial DnaK (P04475), in which Cys267 is replaced with Ala. The peptide obtained from trypsin digest of Hsp72 is underlined. (B) Refolding of heat-denatured luciferase by rabbit reticulocyte lysate supplemented with control or 4-HNE-treated Hsp72 or DnaK, demonstrating resistance of DnaK to 4-HNE inhibition (n = 3 ± SEM). (C) Refolding of heat-denatured luciferase by rabbit reticulocyte lysate supplemented with Hsp72 pretreated with no aldehyde (control), Cys-reactive aldehydes (4-HNE, 4-ONE), or Cys nonreactive aldehyde (MDA), demonstrating a crucial role for Cys in aldehyde-mediated Hsp72 inhibition (n = 3 ± SEM).
Co-immunoprecipitation experiments were performed to compare the formation of Hsp72/luciferase and Hsp72/Hsp40 complexes following treatment of Hsp72 with 0, 10, or 100 μM 4-HNE in two independent series. Pooled data from each pair were then used for comparison between each treatment by one-way ANOVA. In control experiments, RRL was not supplemented with Hsp72 or Hsp40. The resulting amount of purified luciferase or Hsp40 was normalized to the amount of Hsp72 immunoprecipitated after stripping the membrane of antibodies and reprobing with antibodies against Hsp72. The results of these immunoblots are shown in Figure 6A–C, demonstrating that Hsp72 treated with 4-HNE has an impaired ability to bind substrate. Specifically, when the luciferase signal was normalized to Hsp72 signal, a 25% and 50% reduction in the ability of the chaperone to bind substrate is demonstrated following treatment with 10 and 100 μM 4-HNE, respectively (Figure 4D). The function of Hsp72 is dictated by its co-chaperones. Hsp40, which stimulates the intrinsic ATPase activity of Hsp72, is a demonstrated requirement for denatured luciferase refolding (9). Because the RRL was supplemented with Hsp40 in addition to Hsp72, co-immunoprecipitation was employed to measure any effect of 4-HNE on the formation of the Hsp72/Hsp40 complex. When normalized to the amount of Hsp72 immunoprecipitated, no apparent effect was observed regarding the interaction between Hsp40 and Hsp72 (Figure 6E). The significant difference demonstrated by the unsupplemented control (Figure 6E) likely represents an effect of the ratio of Hsp72 to Hsp40, as less Hsp72 was present in these lysates, rather than a greater affinity for the co-chaperone. Unfortunately, because Hsp70 typically exists in molar excess of its cochaperones (34), we were unable to assess complex formation with these other proteins.
Figure 6.
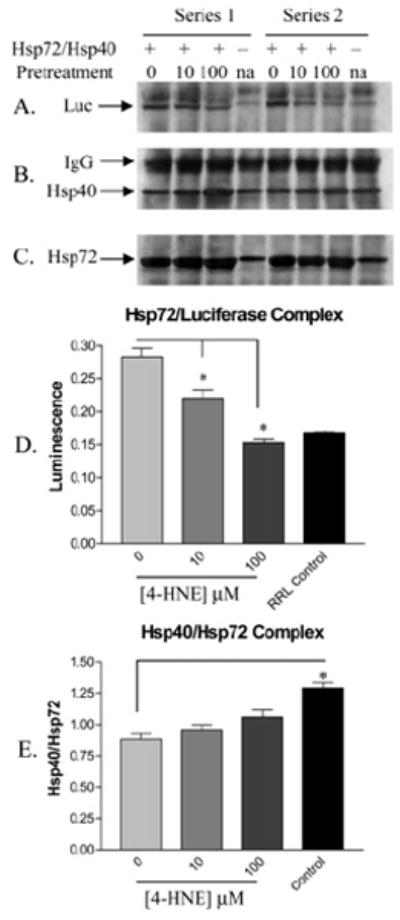
Co-immunoprecipitation of Hsp72 from rabbit reticulocyte lysate supplemented with Hsp40 and 4-HNE treated (10 μM, 100 μM) or untreated (0 μM) Hsp72 from a series of two independent experiments. Panels A–C demonstrate immunoreactivity against substrate (heat-denatured luciferase), co-chaperone (Hsp40), and Hsp72. (D) A decrease in substrate binding by 4-HNE pretreated Hsp72 is clearly demonstrated when luciferase signal is normalized to Hsp72 signal (n = 2 ± SD). (E) No change in complex formation between 4-HNE pretreated Hsp72 and Hsp40 following normalization of Hsp40 signal to Hsp72 signal (n = 2 ± SD).
ATP binding by Hsp72 is central to the ability of this chaperone to complex with and refold denatured proteins. Because Cys267 lies in proximity to the ATP binding site within the ATPase domain of Hsp72, the effect of 4-HNE adduction on the ability of Hsp72 to bind ATP was measured using ATP-linked agarose beads. Using this method, a significant (32% and 72%) decrease in ATP affinity by Hsp72 was observed following pretreatment of the chaperone with 10 and 100 μM 4-HNE, respectively, as demonstrated by the immunoblot in Figure 7A using antibodies against Hsp72. This was confirmed by densitometry presented in Figure 7B.
Figure 7.
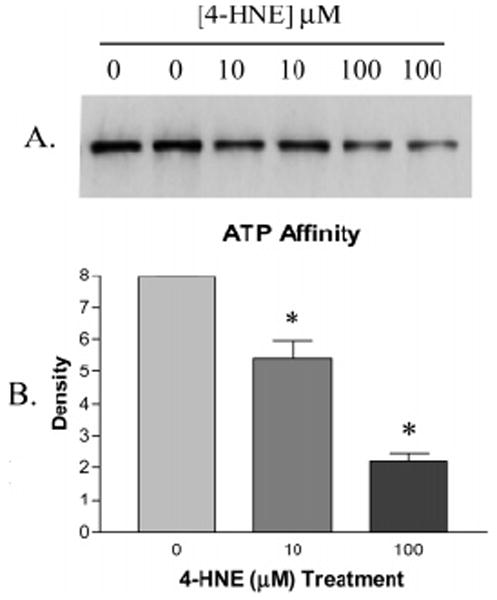
(A) Decreased affinity of 4-HNE pretreated Hsp72 for ATP following purification with ATP-linked agarose beads is demonstrated by immunoblot against Hsp72 and is confirmed by densitometry in panel B (n = 2 ± SD).
Discussion
Oxidative stress is often accompanied by the presence of lipid aldehyde-modified proteins (14). A proteomic-based approach was used to identify proteins susceptible to in vivo 4-HNE modification in rats fed a combination of high fat and ethanol. An increase in the number and severity of 4-HNE adducted proteins was clearly demonstrated in cytosolic fractions obtained from the ethanol-fed animals when compared to their respective pair-fed controls, which received isocaloric sucrose in place of ethanol. The inducible variant of Hsp70, Hsp72, was one of several proteins consistently modified by 4-HNE.
Members of the Hsp70 family of molecular chaperones are found throughout the cell in cytosol, mitochondria, and microsomes, and the Hsp72 variant is induced as part of the heat shock response. Functions performed by these chaperones include protein trafficking, stabilization, refolding, and degradation (1-4). The latter three roles constitute an important part of the protective measures taken by heat shock proteins to stabilize the cell under conditions of chemical or environmental insult (5, 6). Slight induction of Hsp72 was observed in hepatocytes isolated from ethanol-fed animals when compared to their respective control animals; however, no corresponding increase in the ability of these hepatocytes to refold heat-denatured luciferase was observed. Given the established importance of the heat shock response, further experimentation was conducted to assess the susceptibility of Hsp72 to inhibition by 4-HNE.
In vitro modification of recombinant Hsp72 by 6 and 60-fold molar excesses of 4-HNE (10 and 100 μM) yielded a corresponding increase in the number of aldehyde adducts detected by immunoblot. While the logical explanation for this observation is that more sites on the protein are modified as the concentration of aldehyde is increased, this was not supported by data obtained from trypsin digest of 4-HNE-modified protein and HPLC-MS/MS analysis of the peptides. This procedure typically resulted in approximately 88% coverage of the protein sequence; however, the only identified 4-HNE adduct was consistently found at Cys267 in the ATPase domain of the protein, regardless of the concentration of aldehyde. It is likely that a greater number of Hsp72 molecules are modified at this Cys residue, which would explain both our immunoblotting (Figure 3A,B) and mass spectral data (Figure 3C,D), rather than more amino acids modified per protein.
The effect of 4-HNE on the refolding ability of Hsp72 was assessed using heat-denatured recombinant firefly luciferase. This particular substrate was chosen due to the ease with which restoration of its activity can be measured and is in fact widely used for this purpose (13). The function of Hsp72 revolves around its ATPase activity, which is in turn regulated by an array of co-chaperones. RRL was therefore used as a source of co-chaperones, and was supplemented with 4-HNE treated or untreated Hsp72, allowing for the measurement of 4-HNE-mediated inhibition of Hsp72. While the refolding efficiency of RRL supplemented with nontreated Hsp72 significantly exceeded that of nonsupplemented (e.g., control) RRL, the refolding efficiency of RRL supplemented with 4-HNE-modified Hsp72 significantly decreased correspondingly with the concentration of aldehyde pretreatment. It has thus been demonstrated that 4-HNE is a potent inhibitor of Hsp72-mediated protein refolding.
Because adduction at Cys267 was consistently found on Hsp72, the importance of this particular residue in 4-HNE-mediated inhibition of the chaperone was further explored. All members of the Hsp70 family share a high degree of sequence homology in the ATPase domain (2). This holds true for DnaK, the bacterial variant of Hsp70, except that Cys267 is replaced with an Ala residue, which is not reactive with 4-HNE (12, 16). Following treatment of both Hsp72 and DnaK with a 6-fold molar excess of 4-HNE, the refolding ability of Hsp72 was inhibited as expected; however, DnaK was completely resistant to the effect of the aldehyde. Further evidence supporting a role for Cys in the demonstrated inhibition of Hsp72 by 4-HNE was obtained by treating the chaperone with 4-ONE, which is reportedly reactive with thiols, and MDA, which does not react with thiols (15, 16). The results of this study demonstrated complete resistance of Hsp72 toward MDA inhibition, while both 4-HNE and 4-ONE significantly decreased the refolding ability of the chaperone. Together, these experiments demonstrate reactivity with Cys is required for aldehyde-mediated inhibition of Hsp72.
Further insight into the mechanism of 4-HNE-mediated inhibition of Hsp72 was provided by co-immunoprecipitation experiments, in which a significant decrease in luciferase bound by the chaperone was observed. Cytosolic Hsp70 has a demonstrated role in the presentation of substrates to the 26S proteasome for degradation (3). To rule out the possibility that modification of Hsp72 leads to enhanced degradation of its substrate, rather than inhibition of refolding ability, refolding experiments were performed in the presence of the proteasome inhibitor MG-132. Inhibition of the proteasome did not restore luciferase activity, and it was thus determined that a decrease in complex formation between luciferase and Hsp72 is likely responsible for inhibition of Hsp72-mediated protein refolding. Interference with co-chaperone interaction could also lead to inhibition of Hsp72. Unfortunately, Hsp70 typically exists in molar excess to its co-chaperones (11, 31), making this impossible to measure using our methods, with the exception of Hsp40, with which the RRL was also supplemented. 4-HNE did not lead to any significant change in Hsp72 affinity for Hsp40, and in fact interference with co-chaperone binding is not supported by the data, which demonstrate that the refolding ability of RRL supplemented with 4-HNE treated Hsp72 decreases below nonsupplemented RRL. This suggests rather that the treated Hsp72 binds co-chaperones with an affinity equal to nontreated Hsp72, but is incapable of refolding substrate. Hsp72 modified by 4-HNE would thus sequester co-chaperones, preventing them from interacting with viable Hsp70. In this scenario, the Hsp72 inhibition by 4-HNE would be amplified, which is indeed supported by the data.
The affinity of the Hsp70 family for protein substrates is reportedly a function of nucleotide (e.g., ATP, ADP) present at the ATPase domain (2). Specifically, Hsp70 demonstrates its highest substrate affinity in the presence of ADP, and the affinity decreases correspondingly in the presence ATP or no nucleotide (8). Because Cys267 lies within the ATPase domain of Hsp72, it is unlikely that it directly inhibits binding of Hsp72 to luciferase. Alternatively, it is conceivable that modification of Cys267 indirectly inhibits substrate binding through interference with the nucleotide state. The affinity of Hsp72 for ATP was measured by comparing the amount of Hsp72 purified using ATP-linked agarose beads following treatment of the chaperone with 4-HNE. Immunoblot against purified Hsp72 demonstrated a significant decrease in the affinity of Hsp72 for ATP following treatment with 4-HNE. Because of the proximity of Cys267 to bound nucleotide, a possible mechanism explaining a decrease in ATP affinity by Hsp72 may simply be steric inhibition of nucleotide binding. Whatever the exact mechanism, the loss of affinity by Hsp72 for ATP suggests that 4-HNE modification of the chaperone forces either an ATP-bound state, or a state in which no nucleotide is bound, both of which would result in less efficient protein substrate binding.
Relationships between the Hsp70 family of heat shock proteins and alcoholic liver disease (ALD) have previously been proposed, citing induction of this chaperone in sections from alcoholic livers (35). Hallmark features of ALD include accumulation of insoluble cytokeratins, or Mallory bodies, impairment of the 26S proteasome, and accumulation of lipid aldehyde species such as 4-HNE (14, 35, 36). As noted, liver injury observed in the present study consisted primarily of fat accumulation or steatosis, which is considered to be an early event in ethanolinduced liver damage (37, 38). The recovery of 4-HNE-adducted Hsp72 from the livers of ethanol-treated rats suggests that this stress response protein is predisposed as a target for adduction by aldehydic products of lipid peroxidation. Because the involvement of Hsp72 in the prevention of insoluble protein aggregates and presentation of proteins to the proteasome for degradation has been well established (3-5), impairment of this family of heat shock proteins may contribute significantly to the pathogenesis of ALD.
In rats fed a diet consisting of high fat and ethanol, 4-HNE modification of cytosolic Hsp72 was consistently observed, along with induction of Hsp72. A corresponding increase in Hsp72 function was not observed, however, leading to the hypothesis that 4-HNE inhibits the protein refolding function of this chaperone. In vivo studies assessing the effect of 4-HNE on Hsp72-mediated protein refolding clearly demonstrate inhibition of refolding ability, likely through covalent modification of Cys267. Co-immunoprecipitation experiments have demonstrated a decreased affinity of Hsp72 for substrate, and experiments measuring ATP affinity and ATPase activity following modification of the chaperone with 4-HNE suggest an altered state of nucleotide binding as the mechanism behind this observation. A mechanism explaining the observed decrease in refolding efficiency by RRL supplemented with 4-HNE-modified Hsp72 below that of nonsupplemented RRL remains elusive. It is very likely, however, that, while Hsp72 is directly inhibited by 4-HNE, its ability to interact with its co-chaperones is unchanged. Because Hsp72 typically exists in molar excess of its co-chaperones, the direct result of this would be amplification of Hsp72 inhibition by 4-HNE.
Acknowledgments
This work was supported in part by grants NIH/NIAAA RO1AA09300 and NIH/NIEHS RO1ES09410 (D.R.P.), NIH/NIEHS F32 ES11937 (J.A.D.) and NIH/NIAAA F31 AA014308 (D.L.C.).
References
- 1.Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
- 2.Fink AL. Chaperone-mediated protein folding. Physiol Rev. 1999;79:425–442. doi: 10.1152/physrev.1999.79.2.425. [DOI] [PubMed] [Google Scholar]
- 3.Bercovich B, Stancovski I, Mayer A, Blumenfeld N, Laszlo A, Schwartz AL, Ciechanover A. Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J Biol Chem. 1997;272:9002–9010. doi: 10.1074/jbc.272.14.9002. [DOI] [PubMed] [Google Scholar]
- 4.Goasduff T, Cederbaum AI. CYP2E1 degradation by in vitro reconstituted systems: role of the molecular chaperone Hsp90. Arch Biochem Biophys. 2000;379:321–330. doi: 10.1006/abbi.2000.1870. [DOI] [PubMed] [Google Scholar]
- 5.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 6.Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 7.Sadis S, Hightower LE. Unfolded proteins stimulate molecular chaperone Hsc70 ATPase by accelerating ADP/ATP exchange. Biochemistry. 1992;31:9406–9412. doi: 10.1021/bi00154a012. [DOI] [PubMed] [Google Scholar]
- 8.Schmid D, Baici A, Gehring H, Christen P. Kinetics of molecular chaperone action. Science. 1994;263:971–973. doi: 10.1126/science.8310296. [DOI] [PubMed] [Google Scholar]
- 9.Michels AA, Kanon B, Bensaude O, Kampinga HH. Heat shock protein (Hsp) 40 mutants inhibit Hsp70 in mammalian cells. J Biol Chem. 1999;51:36757–36763. doi: 10.1074/jbc.274.51.36757. [DOI] [PubMed] [Google Scholar]
- 10.Hohfeld J, Minami Y, Hartl FU. Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell. 1995;83:589–598. doi: 10.1016/0092-8674(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 11.Nollen EAA, Brunsting JF, Song J, Kampinga HH, Morimoto RI. Bag1 functions as a negative regulator of Hsp70 chaperone activity. Mol Cell Biol. 2000;20:1083–1088. doi: 10.1128/mcb.20.3.1083-1088.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petersen G, Hahn C, Gehring U. Dissection of the ATP-binding domain of the chaperone hsc70 for interaction with the cofactor Hap46. J Biol Chem. 2001;276:10178–10184. doi: 10.1074/jbc.M006967200. [DOI] [PubMed] [Google Scholar]
- 13.Raynes DA, Guerriero V., Jr Inhibition of Hsp70 ATPase activity and protein renaturation by a novel Hsp70-binding protein. J Biol Chem. 1998;273:32883–32888. doi: 10.1074/jbc.273.49.32883. [DOI] [PubMed] [Google Scholar]
- 14.Esterbauer H, Zollner H, Schaur RJ. Aldehydes formed by lipid peroxidation: mechanisms of formation, occurrence, and determination. In: Vigo-Pelfreys C, editor. Membrane Lipid Oxidation. Vol. 1. CRC Press; Boca Raton, FL: 1989. pp. 239–268. [Google Scholar]
- 15.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malondialdehyde, and related aldehydes. Free Radical Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 16.Doorn JA, Petersen DR. Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem Res Toxicol. 2002;15:1445–1450. doi: 10.1021/tx025590o. [DOI] [PubMed] [Google Scholar]
- 17.Uchida K, Stadtman ER. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase: a possible involvement of intra- and intermolecular cross-linking reaction. J Biol Chem. 1993;268:6388–6393. [PubMed] [Google Scholar]
- 18.Luckey SW, Tjalkens RB, Petersen DR. Mechanism of inhibition of rat liver Class 2 ALDH by 4-hydroxynonenal. Adv Exp Med Biol. 1999;463:71–77. doi: 10.1007/978-1-4615-4735-8_9. [DOI] [PubMed] [Google Scholar]
- 19.Alderton AL, Faustman C, Liebler DC, Hill DW. Induction of redox instability of bovine myoglobin by adduction with 4-hydroxy-2-nonenal. Biochemistry. 2003;42:4398–4405. doi: 10.1021/bi0271695. [DOI] [PubMed] [Google Scholar]
- 20.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nanji AA, Griniuviene B, Yacoub LK, Sadrzadeh SMH, Levitsky S, McCully JD. Heat-shock gene expression in alcoholic liver disease in the rat is related to the severity of liver injury and lipid peroxidation. Proc Soc Exp Biol Med. 1995;210:12–19. doi: 10.3181/00379727-210-43918. [DOI] [PubMed] [Google Scholar]
- 22.Calabrese V, Renis M, Calderone A, Russo A, Barcellona ML, Rizza V. Stress proteins and SH-groups in oxidant-induced cell damage after acute ethanol administration in rat. Free Radical Biol Med. 1996;20:391–397. doi: 10.1016/0891-5849(95)02095-0. [DOI] [PubMed] [Google Scholar]
- 23.Tunici P, Schiaffonati L, Rabellotti E, Tiberio L, Perin A, Sessa A. In vivo modulation of 73 kDa heat shock cognate and 78 kDa glucose-regulating protein gene expression in rat liver and brain by ethanol. Alcohol: Clin Exp Res. 1999;23:1861–1867. [PubMed] [Google Scholar]
- 24.Mitchell DY, Petersen DR. Inhibition of rat hepatic mitochondrial aldehyde dehydrogenase-mediated acetaldehyde oxidation by trans-4-hydroxy-2-nonenal. Hepatology. 1991;13:728–734. doi: 10.1016/0270-9139(91)92572-p. [DOI] [PubMed] [Google Scholar]
- 25.Requena JR, Fu MX, Ahmed MU, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR. Quantification of malondialdehyde and 4-hydroxynonenal adducts to lysine residues in native and oxidized human low-density lipoprotein. Biochem J. 1997;322:317–325. doi: 10.1042/bj3220317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Little RG, Petersen DR. Subcellular distribution and kinetic parameters of HS mouse liver aldehyde dehydrogenase. Comp Biochem Physiol. 1983;74c:271–279. doi: 10.1016/0742-8413(83)90101-9. [DOI] [PubMed] [Google Scholar]
- 27.Hartley DP, Kroll DJ, Petersen DR. Prooxidant-initiated lipid peroxidation in isolated rat hepatocytes: detection of 4-hydroxynonenal- and malondialdehyde-protein adducts. Chem Res Toxicol. 1997;10:895–905. doi: 10.1021/tx960181b. [DOI] [PubMed] [Google Scholar]
- 28.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 29.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 30.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-gap penalties, and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anwar A, Siegel D, Kepa JK, Ross D. Interaction of the molecular chaperone Hsp70 with human NAD(P)H:Quinone oxidoreductase 1. J Biol Chem. 2002;277:14060–14067. doi: 10.1074/jbc.M111576200. [DOI] [PubMed] [Google Scholar]
- 32.Wu X, Yano M, Washida H, Kido H. The second metal-binding site of the 70 kDa heat-shock protein is essential for ADP binding, ATP hydrolysis, and ATP synthesis. J Biochem. 2004;378:793–799. doi: 10.1042/BJ20031680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siegel D, Anwar A, Winski SL, Kepa JK, Zolman KL, Ross D. Rapid polyubiquitination and proteasomal degradation of a mutant form of NAD(P)H:Quinone oxidoreductase 1. Mol Pharmacol. 2001;59:263–268. doi: 10.1124/mol.59.2.263. [DOI] [PubMed] [Google Scholar]
- 34.Murphy PJM, Kanelakis KC, Galigniani MD, Morishima Y, Pratt WB. Stoichiometry, abundance, and functional significance of the hsp90/hsp70-based multiprotein chaperone machinery in reticulocyte lysate. J Biol Chem. 2001;276:30092–30098. doi: 10.1074/jbc.M103773200. [DOI] [PubMed] [Google Scholar]
- 35.Omar R, Pappolla M, Saran B. Immunocytochemical detection of the 70-kd heat shock protein in alcoholic liver disease. Arch Pathol Lab Med. 1990;114:589–592. [PubMed] [Google Scholar]
- 36.Donohue TM, Jr, Zetterman RK, Zhang-Gouillon Z, French SW. Peptidase activities of the multicatalytic protease in rat liver after voluntary and intragastric ethanol administration. Hepatology. 1998;28:486–491. doi: 10.1002/hep.510280228. [DOI] [PubMed] [Google Scholar]
- 37.Diehl AM. Nonalcoholic fatty liver disease: implications for alcoholic liver disease. Alcohol: Clin Exp Res. 2001;25:8S–14S. doi: 10.1097/00000374-200105051-00004. [DOI] [PubMed] [Google Scholar]
- 38.Tsukamoto H, Shelly CL. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 2001;15:1335–1349. doi: 10.1096/fj.00-0650rev. [DOI] [PubMed] [Google Scholar]