

Abstract
Recent advances in the structural study of fatty acid synthase (FAS) and polyketide synthase (PKS) biosynthetic enzymes have illuminated our understanding of modular enzymes of the acetate pathway. However, one significant and persistent challenge in such analyses is resolution of the acyl carrier protein (ACP), a small (~9 kDa) protein to which biosynthetic intermediates are tethered throughout the biosynthetic cycle. Here we report a chemoenzymatic crosslinking strategy in which the installation of a historical suicide substrate scaffold upon the 4′-phosphopantetheine (PPant) arm of the ACP is used to capture the active site of acyl carrier protein dehydratase (DH) domains in FAS. Through the synthesis of a small panel of related probes we identify structural features essential for ACP–DH crosslinking, and apply gel-based assays to demonstrate the stability as well as purification strategies for isolation of the chemoenzymatically modified ACP. Applying these carrier protein crosslinking techniques to the structural analysis of FAS and PKS complexes has the potential to provide snapshots of these biosynthetic assembly lines at work.
Keywords: Mechanism-based inhibition, Polyketide biosynthesis, Fatty acid biosynthesis, Dehydratase, Protein, protein interaction
Recent advances in the structural study of fatty acid synthase (FAS) and polyketide synthase (PKS) biosynthetic enzymes have advanced our understanding of modular enzymes of the acetate pathway.1–3 However, one persistent challenge in such analyses is resolution of the dynamic acyl carrier protein (ACP), a small (~9 kDa) protein to which biosynthetic intermediates are tethered throughout the biosynthetic cycle.4 The ACP is inherently flexible, relying on specific protein–protein interactions to recognize and channel biosynthetic intermediates to the proper partner enzyme during each step of the iterative biosynthetic process. The transient nature of these interactions presents a challenge to traditional methods of structural analysis. To capture these interactions, we have pursued a strategy to install mechanism-based inhibitor moieties on the 4′-phosphopantetheine (PPant) arm of the ACP which react with the active site of partner proteins.5–7 This process results in the selective crosslinking of the ACP with a single partner domain. Initially applied to the study of ketosynthase domains, formation of the resulting crosslinked species is highly dependent on ACP-mediated protein–protein interactions, and has been applied as a tool in structural analysis and investigations of ACP-partner protein interactions.
In this study we extend the repertoire of chemoenzymatic ACP-crosslinking to study the interactions of ACP and dehydratase (DH) domains in FAS. DH domains catalyze the dehydration of β-hydroxyacyl-ACP substrates to α,β-unsaturated acyl-ACPs during the reductive steps of fatty acid and polyketide biosynthesis (Fig. 1a).8,9 In addition, DH domains play a key role in initiation of bacterial unsaturated fatty acid biosynthesis, isomerizing trans-2-decenoyl-ACP to cis-3-decenoyl-ACP. These reactions were first studied in the context of the Escherichia coli FAS, where they are catalyzed by the prototypical DH enzyme FabA.10,11 These studies also yielded the discovery of the first mechanism-based inhibitor of a fatty acid biosynthetic enzyme, 3-decynoyl-N-acetyl-cystamine (NAC). This suicide substrate undergoes α-deprotonation in the FabA active site to form an electrophilic allene, which then modifies the active site histidine of FabA to irreversibly inactivate the enzyme.12–14 Here we expand the use of this historical inhibitor scaffold to guide site-specific crosslinking of ACP and DH domains.
Figure 1.

General strategy for site-specific mechanism-based crosslinking of ACP and DH domains. (a) Dehydration and isomerization reactions catalyzed by the E. coli FAS DH enzyme FabA. (b) Reactive ACPs can be generated through the CoA biosynthetic enzymes (PanK, PPAT, and DPCK) and the PPTase Sfp. Upon addition of an ACP-partner protein, the native protein–protein interactions result in a transient interaction, which is captured by the electrophile. (c) Structures of electrophilic pantetheine analogues 1–8.
To this end, a small panel of pantetheine analogues was synthesized incorporating well-known inhibitor scaffolds of DH and other α-deprotonating enzymes (Fig. 1c, 1–8). These pantetheine analogues can be transformed into CoA analogues and site-specifically incorporated into ACPs using the one-pot chemoenzymatic method depicted in Figure 1b.15 In addition to the 3-decynoyl and 2,3-decadienoyl thioester inhibitors (1 and 2), we also examined 3-decynoyl-oxoesters and amides (3 and 4), a transition state analogue (5), 2-octynoyl thioesters and amides capable of forming reactive allenes upon γ-deprotonation (6 and 7), and a simple histidine reactive acyl-bromoacetamide affinity tag (8).16–18 These pantetheine analogues were assayed for their relative abilities to modify the active site of FabA by testing their ability to block labeling by fluorescent probe 9, a 3-decynoyl-NAC derivative which reacts with FabA in an active-site dependent manner (Fig. 2a). As expected, pre-incubation of FabA with the denaturing agent SDS or known inhibitor scaffolds 1 or 2 each efficiently blocked labeling by 9 (Fig. 2a). 3-Decynoyl-oxoester 3 and 2-octynoyl thioester 6 showed decreased active site modification, blocking fluorescent labeling by 9 to a lesser degree, while pantetheine analogues 4, 5, 7, and 8 showed no effect. This is consistent with previous studies on mechanism-based inhibition of FabA by 3-decynoic acid analogues, which found enzyme inactivation to be strongly dependent on the pKa of the α-proton of the suicide substrate (thioester > oxoester ≫ acid/amide) and lead us to focus on the use of thioesters 1, 2, and 6 as ACP–DH crosslinking reagents.12,13,19
Figure 2.

Evaluation of ACP–DH crosslinking agents 1–8. (a) Thioesters 1–2 and 6 and ester 3 demonstrate the ability to block the FabA active site from labeling by fluorescent FabA probe 9. (b) ACP–DH crosslinking of the E. coli FAS enzymes AcpP and FabA. Crosslinking is sensitive to inactivation of FabA by 9 and by pre-denaturation of the DH domain by heat (Δ). (c) ACP–DH crosslinking is dependent on the presence of 1, with less crosslinking observed as 1 is decreased. (d) Reaction of FabA with purified apo-ACP (lane 1), 2-octynoyl-ACP (lane 2), and 3-decynoyl-ACP (lane 3). (e) Reaction of FabA with purified apo-GFP AcpP (lane 1), 2-octynoyl GFP AcpP (lane 2), and 3-decynoyl GFP AcpP (lane 3).
First, the ability of analogues 1, 2, and 6 to modify the E. coli FAS ACP (AcpP) was demonstrated. Using the CoA biosynthetic enzymes PanK, PPAT, and DPCK along with the permissive PPTase Sfp, we were able to observe modification of AcpP by 1, 2, and 6 by SDS–PAGE, in which a characteristic gel-shift to lower molecular weight was observed upon AcpP-incorporation of fatty acyl pantetheines 1, 2, or 6 (Fig. 2b).20,21 Upon addition of FabA to AcpP modified by 1, a faint band appearing at ~45 kDa corresponding to a putative AcpP–FabA complex was observed (Fig. 2b). While this band co-migrated with a persistent FabA disulfide, it could be clearly visualized using strongly reducing SDS–PAGE conditions. This putative AcpP–FabA complex was observed to be dependent upon the presence and amount of 1 added to the reaction mix (Fig. 2c). ACP, PanK, and Sfp were each also judged to be necessary components for this crosslinking to occur (Fig. S1). In addition, complex formation was highly sensitive to the integrity of the FabA active site, and was not observed in reactions in which FabA had been pre-denatured by boiling or inactivated by high concentrations of 9 (Fig. 2b). Pantetheine analogue 2 resulted in approximately equivalent results, while analogue 6 produced noticeably reduced crosslinking (Fig. S2).
For unambiguous identification of the crosslinked species we applied an orthogonal purification strategy to isolate inhibitor modified (termed crypto) AcpP from the reaction mixture.22 The resulting crypto-ACPs were then incubated with FabA, allowing observation of a distinct gel-shift to higher molecular weight corresponding to formation of the ACP–DH complex. This shift was observed only in the presence of agents 1 or 6 (Fig. 2d). A similar result was seen when using a GFP-tagged AcpP, which facilitates visualization of the AcpP–FabA complex by removing it from the molecular weight region of the FabA disulfide (Fig. 2e). Both the GFP–AcpP and native AcpP–FabA complexes were excised, subjected to tryptic digest, and identified by MALDI-TOF/TOF and peptide mass fingerprinting (Fig. S3).
One consideration when applying Michael acceptors 2 and 6 to ACP–DH crosslinking of multidomain synthases is their potential reactivity with KS domains. Indeed, when crypto-AcpPs generated from 1, 2, and 6 were incubated with the E. coli KS enzyme FabB, each demonstrated ACP–KS crosslinking activity (Fig. S4). This could be avoided by pre-incubation of FabB with the KS-selective reagent cerulenin, a strategy which may prove useful for achieving ACP–DH specific crosslinking in multidomain synthases. The finding that 3-decynoyl thioester 1 promotes ACP–KS crosslinking suggests some non-enzymatic allenic isomerization of this agent occurs during AcpP loading of 1. This process may also contribute to the presence of two ACP–DH bands formed using 1 but not 6 (Fig. 2d), possibly due to differential crosslinking of the AcpP-bound diastereomeric allene following chemical isomer- ization.
Finally, we tested the ability of the ACP–DH crosslinking reaction to discriminate between native and non-native carrier protein–DH pairs.7 In addition to the FAS carrier protein AcpP, the PKS carrier proteins Otc and Fren, as well as the NRPS carrier protein EntB were post-translationally modified by 1 and incubated with FabA. While the limited progress of these crosslinking reactions necessitated high exposure times to discriminate CP–DH complexes from background, FabA appears to crosslink preferentially with 3-decynoyl ACPs from FAS and PKS systems compared to the PCP EntB (Fig. 3). This indicates that, as seen with ACP–KS crosslinking, the formation of ACP–DH complexes is driven by the inherent compatibility of the protein–protein interactions of the ACP–DH pair.
Figure 3.
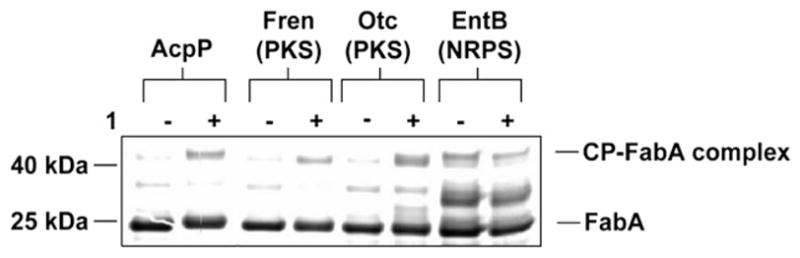
Effect of carrier protein identity on CP-FabA crosslinking. 3-decynoyl pantetheine 1 was used to modify the carrier proteins AcpP (FAS), Fren (PKS), Otc (PKS), and EntB (NRPS). Upon addition of FabA, crosslinking is observed only with AcpP, Fren, and Otc, indicating the preferential interaction of FabA with 3-decynoyl-acyl carrier proteins over peptidyl carrier proteins.
A limitation to this ACP–DH crosslinking approach is the modest yield of AcpP–FabA complex observed. While affinity chromatography was used to provide a partial purification for the purposes of this study, a further step of size-exclusion chromatography will be necessary to isolate crosslinked complex from unreacted FabA in sufficient purity for structural analysis. Despite the modest crosslinking yield, this approach may be immediately applicable to NMR analysis of ACP–DH interactions. For example, isolation of ~10 mg of an 15N-labeled AcpP–FabA complex would be necessary to acquire spectrum of 15N-AcpP in its FabA-bound confirmation and potentially provide residue specific information about the ACP–DH binding interface. Scale-up of the crosslinking reaction to allow isolation of such quantities is plausible given the high expression yields of E. coli AcpP and FabA. Also promising is the finding that little hydrolysis of the complex is observed after 24 h (Fig. S5). This phenomenon is supported by the crystal structure of FabA in complex with 3-decynoyl-NAC, wherein the fatty acyl inhibitor motif is efficiently sequestered from bulk solvent in the hydrophobic FabA active site.23 The lack of hydrolysis bodes well for the application of this technique to the structural analysis of ACP–DH interactions, as the complex is stable for extended periods of times necessary for isolation and characterization.
In seeking reasons the reaction did not proceed to completion, the finding that crosslinking yield was not dramatically improved by purification of 3-decynoyl crypto-ACP (Fig. 1d) could be an indication the inhibitor moiety of 1 is inactivated, most likely through non-enzymatic allenic isomerization and nucleophilic addition to the 2,3-didecenoic thioester. This is supported by the observation that purification of 3-decynoyl-AcpP prior to FabB addition resulted in lower ACP–KS crosslinking than the crude reaction (Fig. S3). A key challenge in the development of second generation ACP–DH crosslinking probes will be to develop 3-alkynoyl carbonyl scaffolds in which the acidity of the α-proton is sufficiently moderated to avoid non-enzymatic inactivation, while still facilitating protein–protein interaction mediated crosslinking.24 Alternatively, the low yield of ACP–DH crosslinking observed may reflect the unique equilibrium of the E. coli AcpP–FabA pair. In this respect it is important to note that while the current study focuses on ACP-crosslinking of the dual-activity dehydratase/isomerase FabA, E. coli and many other bacteria contain an alternative fatty acid biosynthetic enzyme with dehydratase activity, known as FabZ. FabZ is most active on short chain (C4–C6) acyl-ACP substrates and more closely resembles the DH domains commonly found in PKS enzymes,8,9 while FabA is responsible for initiation of long-chain unsaturated fatty acid biosynthesis and more closely resembles activities found in multidomain polyunsaturated fatty acid synthase (PUFA) enzymes.8,25,26 Interestingly, PUFA biosynthetic enzymes often contain tandemly duplicated ACPs which interact with partner proteins, including FabA-like DH domains.26 The application of this crosslinking technique to PUFA and FabZ-type ACP–DH pairs has the potential to yield new insights into ACP–DH recognition in these megasynthase systems, and is thus a topic of active research.
This study demonstrates the first site-selective protein crosslinking of ACP and DH domains from a FAS system. The AcpP–FabA complex formed by 1 is modest in yield, but is stable and isolable by affinity chromatography. The findings of this study should thus facilitate the preparation of large quantities of AcpP–FabA complex for NMR spectroscopy, and provide a starting point for expansion of this technique to ACP–DH pairs from more complex PKS biosynthetic systems. By providing new tools for the covalent immobilization of ACP in bound conformations, the development of carrier protein crosslinking techniques are a necessary first step for acquiring snapshots of these biosynthetic assembly lines at work.
Supplementary Material
Acknowledgments
This work was supported by NIH NIGMS Grant R01 GM086225 and R01 GM075797. J.M. was supported by NIH T32DK007233.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2010.06.028.
References and notes
- 1.Smith S, Tsai SC. Nat Prod Rep. 2007;24:1041. doi: 10.1039/b603600g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Annu Rev Biochem. 2007;76:195. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 3.Leibundgut M, Maier T, Jenni S, Ban N. Curr Opin Struct Biol. 2008;18:714. doi: 10.1016/j.sbi.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Mercer AC, Burkart MD. Nat Prod Rep. 2007;24:750. doi: 10.1039/b603921a. [DOI] [PubMed] [Google Scholar]
- 5.Worthington AS, Rivera H, Torpey JW, Alexander MD, Burkart MD. ACS Chem Biol. 2006;1:687. doi: 10.1021/cb6003965. [DOI] [PubMed] [Google Scholar]
- 6.Kapur S, Worthington A, Tang Y, Cane DE, Burkart MD, Khosla C. Bioorg Med Chem Lett. 2008;18:3034. doi: 10.1016/j.bmcl.2008.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Worthington AS, Hur GH, Meier JL, Cheng Q, Moore BS, Burkart MD. Chembiochem. 2008;9:2096. doi: 10.1002/cbic.200800198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heath RJ, Rock CO. J Biol Chem. 1996;271:27795. doi: 10.1074/jbc.271.44.27795. [DOI] [PubMed] [Google Scholar]
- 9.Wu J, Zaleski TJ, Valenzano C, Khosla C, Cane DE. J Am Chem Soc. 2005;127:17393. doi: 10.1021/ja055672+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kass LR, Brock DJ, Bloch K. J Biol Chem. 1967;242:4418. [PubMed] [Google Scholar]
- 11.Brock DJ, Kass LR, Bloch K. J Biol Chem. 1967;242:4432. [PubMed] [Google Scholar]
- 12.Endo K, Helmkamp GM, Jr, Bloch K. J Biol Chem. 1970;245:4293. [PubMed] [Google Scholar]
- 13.Helmkamp GM, Jr, Brock DJ, Bloch K. J Biol Chem. 1968;243:3229. [PubMed] [Google Scholar]
- 14.Schwab JM, Ho CK, Li WB, Townsend CA, Salituro GM. J Am Chem Soc. 1986;108:5309. [Google Scholar]
- 15.Worthington AS, Burkart MD. Org Biomol Chem. 2006;4:44. doi: 10.1039/b512735a. [DOI] [PubMed] [Google Scholar]
- 16.Powell PJ, Thorpe C. Biochemistry. 1988;27:8022. doi: 10.1021/bi00421a008. [DOI] [PubMed] [Google Scholar]
- 17.Freund K, Mizzer J, Dick W, Thorpe C. Biochemistry. 1985;24:5996. doi: 10.1021/bi00342a046. [DOI] [PubMed] [Google Scholar]
- 18.Haeffner-Gormley L, Cummings JG, Thorpe C. Arch Biochem Biophys. 1995;317:479. doi: 10.1006/abbi.1995.1191. [DOI] [PubMed] [Google Scholar]
- 19.Morisaki M, Bloch K. Biochemistry. 1972;11:309. doi: 10.1021/bi00753a001. [DOI] [PubMed] [Google Scholar]
- 20.Rock CO, Cronan JE., Jr Methods Enzymol. 1981;71(Pt C):341. doi: 10.1016/0076-6879(81)71043-7. [DOI] [PubMed] [Google Scholar]
- 21.Meier JL, Mercer AC, Rivera H, Jr, Burkart MD. J Am Chem Soc. 2006;128:12174. doi: 10.1021/ja063217n. [DOI] [PubMed] [Google Scholar]
- 22.Haushalter RW, Worthington AS, Hur GH, Burkart MD. Bioorg Med Chem Lett. 2008;18:3039. doi: 10.1016/j.bmcl.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leesong M, Henderson BS, Gillig JR, Schwab JM, Smith JL. Structure. 1996;4:253. doi: 10.1016/s0969-2126(96)00030-5. [DOI] [PubMed] [Google Scholar]
- 24.Mishra PK, Drueckhammer DG. Chem Rev. 2000;100:3283. doi: 10.1021/cr990010m. [DOI] [PubMed] [Google Scholar]
- 25.Metz JG, Roessler P, Facciotti D, Levering C, Dittrich F, Lassner M, Valentine R, Lardizabal K, Domergue F, Yamada A, Yazawa K, Knauf V, Browse J. Science. 2001;293:290. doi: 10.1126/science.1059593. [DOI] [PubMed] [Google Scholar]
- 26.Jiang H, Zirkle R, Metz JG, Braun L, Richter L, Van Lanen SG, Shen B. J Am Chem Soc. 2008;130:6336. doi: 10.1021/ja801911t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.