

INTRODUCTION
In the fall of 2007, my new laboratory at Columbia was producing a wealth of interesting data. I resolved to cut down on travel, accept no invitations to write reviews, and to focus on getting these new data published. I briefly stuck to my guns, and was feeling quite noble about it, when Keith Lindor struck. In a letter informing me that I was a highly respected and internationally recognized authority in a field of liver disease, with a unique perspective, multiple contributions, and broad recognition, he invited me to submit an article to a series in Hepatology to be entitled “The Master’s Perspective”, providing my personal take “on an aspect of the field of liver disease and pathobiology that is near and dear to [my] heart, and which would help others understand how a true leader in the field approaches a problem” Well, flattery – especially of that magnitude - will get you almost anywhere, my resolve rapidly melted, and I began to work on the solicited article.
Query: Of what field was I such a master? While my work for 25 yrs had been on fatty acids, I felt I was still mainly regarded as a bilirubin maven. But the fatty acid work was more current and, in my view, more important, so fatty acids it will be. Lest readers wonder where the yellow went, I will briefly review the logical processes involved in transferring affection from one organic anion to another.
BACKGROUND
To begin at the beginning, my father, a general practitioner, was the best diagnostician I have ever known. He was also a subtle and devious man. He never said that he wanted me to become a physician. However, for my 10th birthday, he gave me his beautiful old brass Zeiss medical school microscope. Thereafter, every Wednesday evening, when he didn’t have evening office hours, he would bring home some blood smears from the office, set up the microscope on the dining room table, and we would sit together for hours reviewing the slides. I was ready to take the Hematology boards before I was ready for my Bar Mitzvah. A few years later, fascinated by a Scientific American article about mathematical modeling of cardiac function, I determined to be a cardiologist. That remained my goal through medical school. However, during my internship, stimulated by Paul Marks, Helen Anderson, and other hematologists at Columbia, I considered switching my allegiance back to Hematology.
Toward the end of that year my draft board began to take an interest in my future. I discussed the situation with my Chief of Medicine, Stanley Bradley. I remember our joking about how I was likely to end up at the NIH, by which he meant the mythical Ninth Infantry Hospital, allegedly located somewhere northwest of Danang. Dr. Bradley suggested I might want to consider the “other NIH”, and arranged an interview for me. The interview went very, very badly. In response to a question, I stumbled in attempting to describe the nature of the three principal projects that would be consuming NHLBI 100 years from that date and was summarily dismissed. Waiting for a bus back to National Airport, I met a friend who was already an NIH Clinical Associate. Knowing of my interest in mathematics, he suggested that I contact Dr. Nathaniel Berlin, Clinical Director of the National Cancer Institute, who was looking for a Clinical Associate with a mathematical background. At my friend’s urging, I called Dr. Berlin and explained the nature of my query to his administrative assistant, Pinky Ross, wife of a distinguished endocrinologist and possessor of a deeply southern accent straight out of Central Casting. She asked me to hold the phone while she went to speak with Dr. Berlin, returning to flummox me with an unexpected question for the second time that day, namely, “Do you like pastrami?” Dr. Berlin had just finished a luncheon meeting in his office, there was a leftover pastrami sandwich which he hated waste, and if I was willing to assist with its disposal, he was prepared to interview me on the spot. I immediately accepted his offer, although if I had known that the pastrami came on white bread with mayonnaise I might not have been so enthusiastic. In any case, I met with Dr. Berlin and ate the fateful sandwich. Dr. Berlin explained that a previous Clinical Associate, Peter Barrett, had developed a method for synthesizing [14 C]-bilirubin (1), and that his successor, Matt Menken, had administered it as an albumin complex to patients and defined the resulting plasma [14 C]-unconjugated bilirubin disappearance curves (2). What information, Dr. Berlin asked, could be obtained from those curves? After redrawing a couple of curves on semi-log paper, I replied that, over the 4 hour studies the curves appeared to have two exponential components; that the Y intercept defined the volume of bilirubin distribution, which, in view of albumin binding was likely to approximate the plasma volume; that the area under the curve was a measure of the fractional rate of bilirubin extraction; and that this, multiplied by the plasma volume and unconjugated bilirubin concentration, could provide a measure of daily bilirubin turnover. I also indicated that the 2-exponential plasma bilirubin disappearance curve was, in effect, the solution of a pair of differential equations defining a two compartment model of bilirubin metabolism, the parameters of which included estimates of bilirubin uptake into an extravascular compartment presumably representing the liver. I was just shifting into high gear when Dr. Berlin held up his hand to stop me. After a moment of meditation, he offered me a job. The rest is history. I forgot about my interest in cardiology for the next 42 years. More on that later.
BILIRUBIN
Dr. Berlin was interested in regulation of RBC production and destruction in cancer, and I was assigned to try to determine the RBC lifespan from the rate of bilirubin production. This initiated a much broader interest in bilirubin disposition, in which I was joined over the years by outstanding associates and Fellows. Indeed, my approach to Fellows was one of the most important things I learned from Nat Berlin: recruit the brightest Fellows you can, then stay out of their way once they arrive in the lab.
Building on the work of our predecessors, we extended the plasma 14C-bilirubin disappearance curve to 36 hrs and developed the mathematical tools for determining the rate of bilirubin turnover (BRT) from the curves (2, 3). BRT proved a close approximation of total bilirubin production (BRP) (4-6). Equations were also developed for calculating RBC lifespan (RBCLS) from BRT and the total circulating RBC volume (7). Since this took only 2 days, changes in RBCLS with therapy, e.g. steroids in autoimmune hemolysis (8) or splenectomy in spherocytosis (9), could be quantitated. A falling unconjugated bilirubin concentration accurately reflected a falling hemolytic rate with treatment while the reticulocyte count remained high until anemia was repaired (7-10). It was also possible to calculate hepatic bilirubin clearance (CBR), in ml/min, from the bilirubin curves (3,9,10). Not only was it a true quantitative test of a key liver function, but the derived relationship:
| [1] |
in which BR represents the plasma unconjugated bilirubin concentration, provided a basis for classifying unconjugated hyperbilirubinemias into those due to increased bilirubin production (mainly hemolysis), those due to reduced bilirubin clearance, and those in which both occur. This has proven useful not only in many studies of hereditary hyperbilirubinemias and hemolytic disorders (8,9), but – more recently – in interpreting changes in bilirubin metabolism due to antiviral agents used in the treatment of HIV (11).
What We Learned
Our most important contributions to the bilirubin world were the procedures just described for quantitation of BRT, CBR, and RBCLS in man (3,7), and interpretation of the data in terms of a model of bilirubin disposition (12,13). We used these methods to characterize the functional defects in bilirubin metabolism in Gilbert’s syndrome (14) and the Crigler-Najjar Syndromes (15-17), often leading to improved definitions of these disorders; to gain insights into bilirubin disposition in hemolytic anemias (7-9) and cholestatic liver diseases (18-20), and to clarify the sources of the early labeled peak of bilirubin synthesis (21-26), at the time a very hot topic which has subsequently cooled considerably. In particular, we established that Gilbert’s syndrome was characterized by a reduction of CBR to ~1/3 of normal, and that such a reduction, in the presence of normal LFTs and serum bile acids (27), served as an operational definition of the syndrome, previously a diagnosis of exclusion. Other studies found that CBR was increased in both GS and controls by phenobarbital or glutethimide (28). When CBR in patients with Crigler-Najjar syndromes, GS, normal subjects, and normal subjects on phenobarbital was plotted against hepatic bilirubin UDP-glucuronyl-transferase values, a highly significant correlation was observed (28) (Figure 1). Although Gilbert’s and the Crigler-Najjar syndromes shared phenotypic features, they had been considered to be genetically discrete entities with different modes of inheritance (e.g. 29, 30). We postulated that they all actually represented mutations of varying severity in a common gene. It took 15 years before this was confirmed by others based on molecular analyses (31-36).
Figure 1.
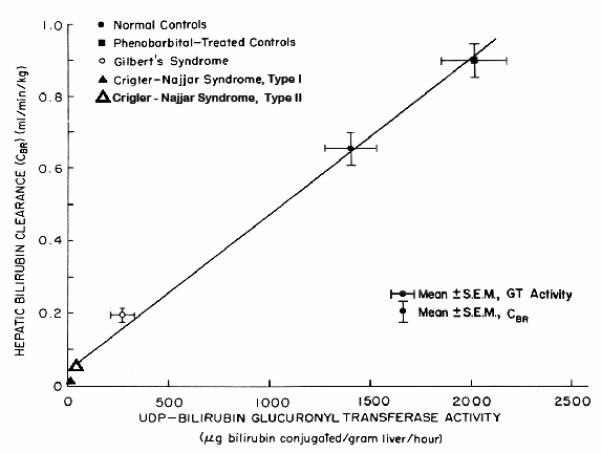
Comparison of mean values for hepatic bilirubin clearance (CBR) and bilirubin UDP-glucuronyl transferase (UGT) activity in liver biopsy specimens from patients with the Crigler-Najjar syndromes, Gilbert’s syndrome, and normal controls. For the control population, data are presented both for untreated and phenobarbital treated individuals. All values for CBR and approximately half of the UGT data are from the Berk laboratory. The remaining UGT values are from the published work of others, as cited (28). Modified from TF Blaschke et al (28) and reproduced with the permission of the publisher.
While the most innovative aspects of our bilirubin work involved humans, we also moved against the flow, from bedside back to bench, to investigate bilirubin disposition in animals. A study in 350 (!!) Sprague Dawley rats (37) showed that hepatic uptakes of bilirubin, BSP, and ICG were all saturable, exhibited mutual competitive inhibition, but were not inhibited by glycocholic acid, and all exhibited phenomena consistent with trans-stimulation and counter-transport. This provided the strongest evidence to that time that bilirubin uptake involved a specific transport process, shared at least in part with BSP and ICG but distinct from that for glycocholate. We were among the first to search for a plasma membrane bilirubin transporter, isolating a 54 kDa glycoprotein, BSP/bilirubin binding protein, as a candidate (38). Anti-BSP/BR-BP antibodies selectively inhibited BSP and bilirubin binding to liver plasma membranes (39) and their uptake by hepatocyte and hepatoma cell lines (40). However, efforts to clone it were unsuccessful. Indeed, despite many tries (e.g. 41-43), no agreed-upon BR transporter has yet been cloned.
Since albumin binding keeps unconjugated bilirubin in solution and determines its free concentration, which is the substrate for the uptake process, we studied the consequences of this binding relationship for its disposition (e.g. 44, 45). In 1982, a paper in Science reported that long chain fatty acid (LCFA) uptake in the perfused rat liver was mediated by an albumin receptor (46). A related abstract made the same claim for bilirubin uptake (47). Because of our interest in the role of albumin binding on bilirubin uptake, these reports immediately caught our attention.
FATTY ACIDS
We reproduced the kinetic observations on LCFA uptake with isolated hepatocytes (48, 49) (Figure 2). It was apparent that, despite publication in Science, the kinetic evidence for an albumin receptor was based on faulty interpretation of the data. If LCFA uptake was plotted as a function of the unbound rather than total LCFA, uptake was clearly a saturable function of the unbound LCFA concentration ([LCFAu]), consistent with conventional pharmacokinetic theory (49, 50) and suggestive of a facilitated uptake process. All experiments alleging competition for LCFA uptake by albumin could be explained by reductions in [LCFAu] produced by the added albumin (51). Studies of albumin binding to rat liver plasma membranes found no evidence for an albumin receptor (52). Because our argument against the receptor was controversial, we studied the issue by multiple approaches. The data strongly supported our initial interpretation (48, 53-57), and the albumin receptor hypothesis virtually disappeared from view. While the hypothesis was wrong, it was the biomedical equivalent of the forward fumble, the only football play in which I excelled during college, in that it unexpectedly led to significant advances.
Figure 2.
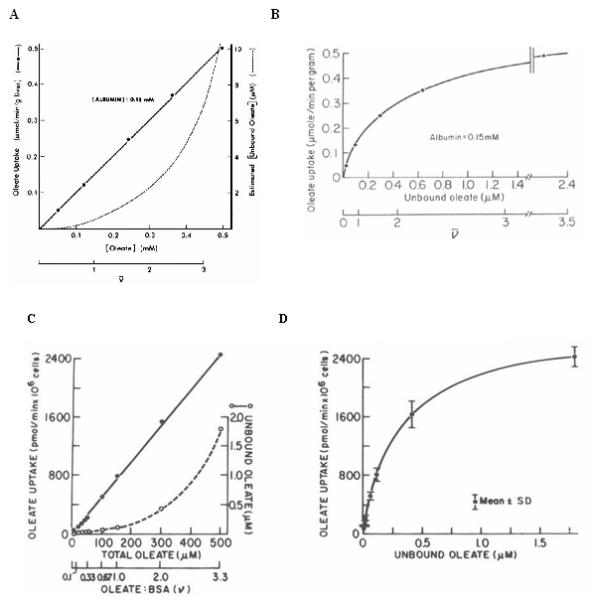
A Oleate uptake by the perfused rat liver as a function of different oleate:albumin molar ratios (ν). Uptake data plotted as a function of the total oleate concentration yield a straight line, and seem unrelated to the unbound oleate concentrations, which are indicated by the dashed line. Rproduced from Weisiger et al, Science (46), with the permission of the publisher. B. When the same uptake data are plotted against the corresponding unbound oleate concentrations in the perfusates, a hyperbolic curve results, suggesting that oleate uptake is, in fact, a saturable function of the unbound oleate concentration. Reproduced from Potter et al, Annu Rev Nutr (50), with the permission of the publisher. C. Oleate uptake by isolated rat hepatocytes. Initial uptake velocities of [3H]-oleate, determined in the presence of 150 μM BSA at oleate:albumin molar ratios ≤ 4:1, are plotted as a function of the total oleate concentration in the medium. As in the perfused liver studies (panel A), uptake could be described as a linear function of the total oleate concentration, unrelated to the equilibrium unbound oleate concentration ([Ou]) in the medium (- - -). However, as indicated in D., over the range of concentrations studied, the same results can equally well be expressed as a saturable function of [Ou]. Panels C and D reproduced from Berk and Stump, Molec Cell Biochem (66) with the permission of the publisher. Kinetic results similar to panels C and D were obtained when other LCFA-binding proteins such as β-lactoglubulin were substituted for albumin (48). These studies indicated that “albumin receptor” uptake kinetics did not require an intact lobular architecture and were not specific for albumin.
Cellular LCFA Uptake
Intrigued that hepatocellular LCFA uptake was saturable , we studied LCFA uptake by isolated rat hepatocytes (58-60) and their binding to liver plasma membranes (60, 61) in more detail. Studies were performed at physiologic pH, temperature, and physiologic or near physiologic albumin concentrations. Subsequent studies were conducted in rat adipocytes (62,63), jejunal enterocytes (64), and cardiac myocytes (65). In every case, LCFA uptake consisted of the sum of a saturable and a non-saturable component, each a function of [LCFAu], of the form:
| [2] |
where Vmax (pmol LCFA/50,000 cells/sec) and Km (nM) are, respectively, the maximal uptake rate of the saturable uptake component and the unbound LCFA concentration at half-maximal uptake velocity (nM), and kn (μl/50,000 cells/sec) is the rate constant for nonsaturable uptake (49,60,63,66). The equation describing binding to plasma membranes was a similar function of [LCFAu]. At physiologic values for [LCFAu], >90 % of uptake was via the saturable pathway; moreover, the saturable component of LCFA uptake was a linear function of the independently measured saturable component of binding (r>0.99!!!) (Figure 3). This implied that the limiting factor in LCFA uptake was availability of specific plasma membrane LCFA binding sites (60). Finally, based on studies with highly acidic, non-metabolizable LCFA analogues such as α2,β2,ω3-hepatofluorostearate, synthesized by Prof. Gerhart Kurz (67,68), we concluded that the saturable process reflected uptake of LCFA anions, while non-saturable uptake reflected passive trans-membrane flip-flop of the uncharged, protonated LCFA with which they were in equilibrium (66). We computed separate rate constants for transmembrane fluxes of LCFA by both the saturable (ks) and non-saturable (kns) uptake pathways from the combined kinetic constants for uptake and binding (60,63,66). In both hepatocytes and adipocytes, ks was ≥ 10 times faster than kns. Thus, the dominant LCFA uptake process under basal physiologic conditions was a selective uptake mechanism for LCFA anions exhibiting many of the kinetic properties (e.g. cis-inhibition, trans-stimulation (69-71)) of a regulatable, protein-mediated, transport process. We suggested that alterations in this process might play a role in the pathophysiology of diseases, e.g. obesity and fatty liver.
Figure 3.
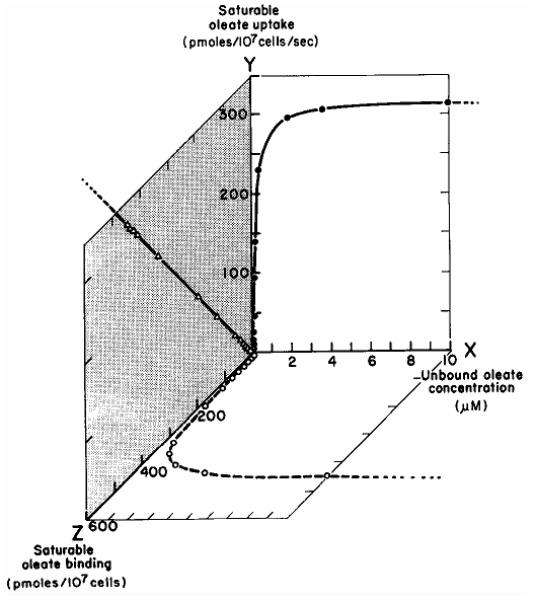
Relationships between the unbound oleate concentration [Ou] (X), saturable oleate uptake (Y) ( ) and saturable oleate binding to liver plasma membranes (Z) (
) and saturable oleate binding to liver plasma membranes (Z) ( ). Values for Y and Z represent values calculated by computer from the fits of the independent experimental measurements of total uptake (Eqn 2) and binding, respectively. Data points correspond to [Ou]s at which the actual experimental measurements of uptake were conducted. The relationship between saturable oleate binding and the saturable oleate uptake velocity is illustrated on the YZ plane (
). Values for Y and Z represent values calculated by computer from the fits of the independent experimental measurements of total uptake (Eqn 2) and binding, respectively. Data points correspond to [Ou]s at which the actual experimental measurements of uptake were conducted. The relationship between saturable oleate binding and the saturable oleate uptake velocity is illustrated on the YZ plane ( ). This indicates that saturable oleate uptake is a linear function of the amount of oleate bound to saturable binding sites in the membrane (r = 0.9997). Reproduced from Stump et al, J Hepatol (60) with permission of the publisher.
). This indicates that saturable oleate uptake is a linear function of the amount of oleate bound to saturable binding sites in the membrane (r = 0.9997). Reproduced from Stump et al, J Hepatol (60) with permission of the publisher.
These heretical observations were disputed by those who argued that LCFA uptake was exclusively a passive process, based on the finding of very rapid rates of LCFA influx into synthetic vesicles (e.g. 72-75,) and rat adipocytes (76) from media in which albumin was either lacking or present in miniscule concentrations. We again resorted to reanalysis of published data, finding e.g. that the rat studies (76) described a single influx process occurring at a rate identical to that denoted by kns in our studies (66). Due to the minimal amounts of albumin employed in those studies, [LCFAu] was extremely high and the corresponding rates of passive influx so great as to swamp the saturable process, making it undetectable. The existence of facilitated LCFA transport was confirmed subsequently by others (e.g.77-80), putting this controversy largely to rest.
Fatty Acid Transporters
The evidence for facilitated LCFA transport initiated a search for a transporter. We identified the first, a highly basic 43 kDa protein, in extracts of rat liver plasma membranes, in 1985 (81). An identical protein was purified from other cell types (62,64,65,82). Named plasma membrane fatty acid binding protein (FABPpm), it bound LCFA at a single high affinity site, was identifiable by immunofluorescence on plasma membranes of hepatocytes, adipocytes, jejunal enterocytes and cardiac myocytes, and appeared and progressively increased in quantity on the surface of 3T3-L1 pre-adipocytes during adipocyte differentiation (83,84). Monospecific anti-FABPpm antisera selectively inhibited LCFA uptake in all of the cell types enumerated. FABPpm was thus a promising candidate LCFA transporter. Three other putative transporters were subsequently described, including fatty acid translocase (FAT, or CD36) (85); the fatty acid transporting polypeptide (FATP) family (86-88), and caveolin -1 (89,90). As with FABPpm, there is evidence of a role in LCFA transport, but also unresolved issues that have raised persistent doubts, about each. While some or all almost certainly participate in LCFA uptake in different tissues under various conditions, it would not be surprising if entirely new LCFA transporters were to be discovered (91).
In 1990, the 35 N-terminal amino acids of FABPpm proved identical to those of mitochondrial aspartate aminotransferase (mAspAT) (92, 93), a well-studied protein never previously identified in extra-mitochondrial sites. This raised considerable doubt as to a role for FABPpm in LCFA transport. However, studies comparing mAspAT with FABPpm documented that each had a MW ~ 43 kDA; a basic pI ~ 9.1, with a characteristic pattern of multiple charge isomers; amino-terminal sequence identity; identical peptide maps; and immunological identity (92,93). Transfection of mAspAT message into 3T3 cells led to increased plasma membrane expression of a protein reactive with anti-FABPpm, and transfection and microinjection studies in 3T3 cells and Xenopus oocytes found that over-expression of mAspAT increased anti-FABPpm inhibitable LCFA uptake (84, 94,95) in both systems. Furthermore, as reviewed elsewhere (96), we have confirmed the presence of mAspAT on plasma membranes, much of it in coated pits, by multiple techniques including immuno-electron microscopy (97, 98); documented key steps in its trafficking, including its arrival at the plasma membrane and subsequent cellular export via a brefeldin-inhibitable vesicular pathway (99); shown the presence of a 500 Å3 hydrophobic cleft of appropriate size and hydrophobicity to serve as an LCFA binding site by molecular modeling of the protein’s published crystal structure (100); shown that arginine at position 201 was located precisely at the entrance to the cleft (a characteristic feature of most known LCFA binding sites in other proteins) and that mutation of this critical arginine to a non-charged threonine abolishes LCFA binding and the ability of the mutated protein to mediate LCFA uptake (101). We also have evidence that mAspAT may co-precipitate from plasma membranes linked with a second protein, and that it loses its affinity for LCFA at pH ≤ 5.5. Collectively, these data suggest that mAspAT might facilitate LCFA import into cells by an endocytic recycling process similar to that by which transferrin mediates Fe import (Figure 4). This remains a major area of interest in our lab.
Figure 4.
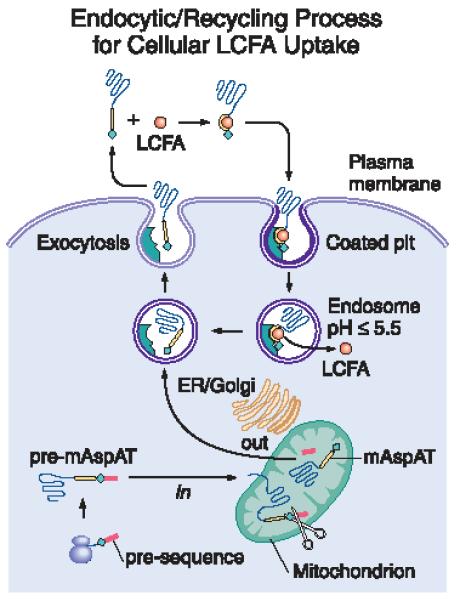
A proposed model of how mAspAT might function as transporter in the hepatocellular uptake of LCFA. Steps for which we have generated experimental evidence are indicated with an asterisk.

After synthesis on free ribosomes, pre-mAspAT is imported into mitochondria, where its leader sequence is excised (*). While the majority of the resulting mature protein remains in the mitochondria and carries out several well-known enzymatic functions, some of it exits the mitochondria, enters a brefeldin-inhibitable vesicular pathway (*) and is exported from the cell (*). mAspAT has a high affinity for LCFA (*). We postulate that the circulating mAspAT molecule binds a molecule of LCFA, and that the resultant conformational change results in its binding to a receptor within coated pits on the hepatocytes plasma membrane, where is has been observed by immuno-electron microscopy (*). Under mild conditions, detergent extraction of mAspAT from hepatocyte plasma membranes result in its co-extraction with a second protein (*), possibly an mAspAT receptor. After internalization, acidification of the resulting endosomal vesicle results in dissociation of the LCFA (*), which exit the endosome. We postulate that, after loss of its bound LCFA, the mAspAT molecule undergoes another conformational change that eliminates its affinity for the receptor, so that, when the vesicle migrates to and fuses with the plasma membrane, the mAspAT molecule is released into the plasma to bind a further LCFA molecule and repeat the cycle.
Obesity
Having found evidence for regulatable LCFA transport, we sought evidence of altered regulation of the process in relevant disease states, such as obesity. Obesity represents increased accumulation of fat (i.e. triglycerides, TG) in the body. TG are synthesized from glycerol and LCFA. Under most circumstances LCFA availability appears to be rate limiting in TG synthesis. The selective accumulation of TG at specific body sites in obesity suggests that some process other than the unregulated, passive diffusion of substrates across cell membranes must be involved. This made obesity an obvious starting point in a search for situations in which regulatable LCFA transport might be relevant.
We began these studies in Zucker fatty (fa/fa) rats (102) and ob/ob, db/db, fat and tubby mice, models with a variety of different obesity-causing mutations (103), including those in the genes for leptin (ob/ob) (104) and its receptor (db/db) (105,106); and dietary obesity models in the C57BL6/J mouse, and Sprague-Dawley and Osborne-Mendel rats (103,107). Patients undergoing bariatric surgery were also studied (108). The Vmax for saturable LCFA uptake was markedly up-regulated in adipocytes from all obesity models studied, including man (102,103,107,108). In addition, the sizes of adipocytes from obesity models in rats (102,107), mice (103,109), and in obese humans (108) were significantly increased. Vmax was significantly increased even when expressed per unit of adipocyte surface area (108,109), indicating that the increase in saturable LCFA uptake in obese adipocytes was not simply the result of increased cell size, but rather reflected up-regulation of a specific membrane transport process. In the Zucker rat (102) and several of the mouse models (103), the increase in Vmax was highly correlated with levels of expression of the mAspAT gene, and in several settings (102,103,110) with the gene for FAT, another putative LCFA transporter reportedly identical to the lipid scavenger receptor CD36 (85). By contrast, the rate constant k for non-saturable uptake, when expressed per unit surface area, did not differ between non-obese and much larger obese adipocytes, indicating that increases in non-saturable LCFA uptake observed in obesity were simply a reflection of the increased surface area of the enlarged obese cells (108). In pre-obese, weanling Zucker fa/fa rats (102) and both C57BL6/J mice (103) and Osborne-Mendel rats (107) starting a high fat diet, up-regulation of the Vmax for adipocyte LCFA uptake preceded weight gain and increased adipocyte size, whereas in leptin-infused ob/ob mice, down-regulation of Vmax preceded decreases in adipocyte size and weight-loss (109). These results suggest that regulation of adipocyte LCFA uptake is an important, previously unrecognized control point for body adiposity (Figure 5) (108,109, 111).
Figure 5.
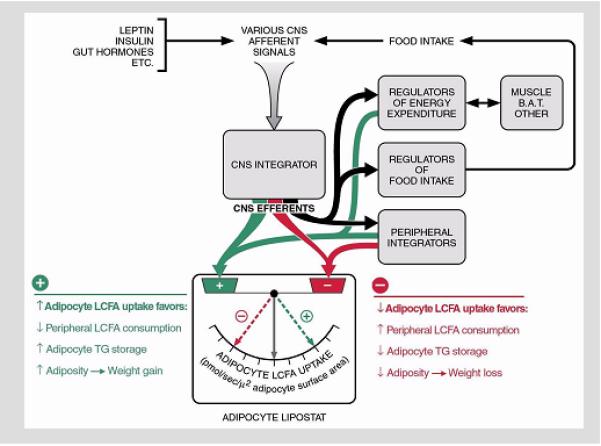
Regulation of adipocyte LCFA uptake appears to control body adiposity. All well-studied genetic and dietary animal models of obesity, as well as obese human subjects, exhibit selective up-regulation of facilitated LCFA by adipocytes. This suggests that regulation of adipocyte LCFA uptake represents a final, common pathway for control of body adiposity resulting from a diversity of primary causes. Reproduced from Bradbury and Berk, Clinics in Liver Disease (111) with the permission of the publisher.
Fatty Liver
NAFLD
We are in the midst of an obesity epidemic (112-116) linked, in turn, to an epidemic of hepatic disorders known collectively as non-alcoholic fatty liver disease (NAFLD) (117-123). NAFLD spans a spectrum (124-126) from simple hepatic steatosis (SHS), a benign and – to a point – reversible condition, through non-alcoholic steatohepatitis (NASH), a potentially progressive form of hepatic injury (127-129) histologically resembling alcoholic (steato)hepatitis (130,131), which may , in turn, may be complicated by development of fibrosis (132,133), and evolution in a limited proportion of patients to cryptogenic cirrhosis (134-137) and end-stage liver disease (ESLD), sometimes requiring transplantation (138). Although the proportion of patients progressing across this entire spectrum is small, the number of patients with SHS is very large. Hence, given declining numbers of new cases of hepatitis C and better treatment results for chronic infection, it is now projected that end-stage NAFLD will replace hepatitis C as the major indication for liver transplantation within 3-4 decades (139) .
Mechanisms Leading to Simple Hepatic Steatosis
The basis for frequent development of SHS in obesity is clear (e.g. 66, 111). Increased caloric intake in excess of expenditure leads sequentially to an increase, subtle at first, in plasma LCFA; increased adipocyte LCFA uptake, initially passive but subsequently also via an up-regulated facilitated transport process; and an increased adipocyte TG mass. This results in induction of adipocyte TNFα production. Increased TNFα causes an adipocyte-specific decrease in insulin receptor phosphorylation; decreased insulin receptor function; resistance to the antilipolytic effects of insulin; de-repression of adipocyte hormone sensitive lipase; increased, continuous lipolysis of the enlarged adipocyte TG mass; and a further increase in plasma LCFA. Within the portal circulation the increased LCFA leads to increased hepatocellular LCFA uptake and hepatic steatosis, while that in the general circulation causes glucoregulatory insulin resistance. These steps indicate, inter alia, that insulin resistance is more complex than generally appreciated. It may be substrate-specific (e.g. antilipolytic or gluco-regulatory) and tissue specific (adipose, liver, skeletal muscle), and the development of each variant may be dissociated in time. Conventional clinical measures, such as HOMA, principally reflect gluco-regulatory insulin resistance, which may not be the most critical component with respect to progression of SHS to NASH.
Not every case of SHS or its subsequent NAFLD consequences results from obesity. SHS can in theory result from any process that leads to increased TG input to or decreased TG output from the intrahepatocellular TG mass (111, 140). Virtually all of these processes (Figure 6) reportedly play a role in one or another model of hepatic steatosis (e.g. 141-144), but few studies assess several of these processes simultaneously, especially in man, so that their relative contributions remain largely unclear.
Figure 6.
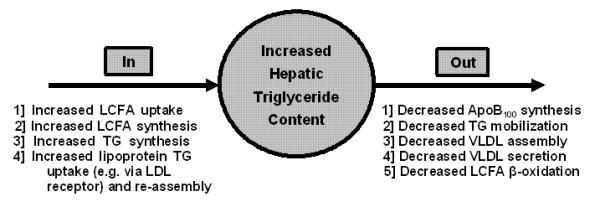
Processes that may contribute to the increased hepatic triglyceride content that characterizes hepatic steatosis and NASH. Those on the left contribute to an increased input of fatty acids and triglycerides. Those on the right increase hepatic triglyceride content by decreasing the normal levels of triglyceride output, principally in VLDL or through decreased oxidation. Modified from Bradbury and Berk, Clinics in Liver Disease (111) and reproduced with permission of the publisher.
Mechanisms of Steatohepatitis
Development of NASH is a “two hit” process in which the first “hit” is development of SHS (145). The second “hit” is, itself, multifactorial. In one model it begins with oxidative stress, leading to hepatocellular injury and apoptosis, followed by inflammation & cytokine cascades (e.g. 146-148). Further progression involves stellate cell activation, fibrosis, and – ultimately – progression to cirrhosis. Mechanisms of hepatocyte injury (149) and disease progression (150) have recently been reviewed elsewhere.
The linkage between obesity and NAFLD also reflects the fact that obesity results in enlargement of intra-abdominal visceral fat depots. While early SHS may develop without insulin resistance, a key step in further progression of NAFLD is the virtually universal development of insulin resistance. Adipocytes are normally intermittent importers of LCFA, sequestering them as TG post-prandially and then releasing them via lipolysis as needed. Insulin resistance, by de-repressing adipocyte hormone sensitive lipase, converts these cells into virtually continuous net LCFA exporters. In the intra-abdominal fat depots, the LCFA are released directly into the portal circulation and rapidly translocated to the liver. Reactive oxygen species (ROS), generated by hepatic LCFA oxidation, are precipitating factors in the cascade of events leading from SHS to NASH (146, 148-153), causing mitochondrial injury, including both morphologic changes and defective ATP repletion; lipid peroxidation with production of malondialdehyde (MDA) and hydroxynonenal (HNE); activation of the FAS system, and the release of specific cytokines, especially TNFα, TGFβ, and IL-6 (149,150). These result in the characteristic histologic features of NASH: apoptosis, Mallory’s hyaline, leukocyte infiltration, and fibrosis (127-129). ROS from EtOH oxidation lead to many of the same metabolites (148,154), which may explain histologic similarities between alcoholic and nonalcoholic steatohepatitis (130,131). However, as alcoholic steatohepatitis develops on a background of SHS, LCFA oxidation may also contribute to that condition (155).
Metabolic Syndrome
Type 2 diabetes mellitus, hypertension, specific dyslipidemias, and arteriosclerotic cardiovascular disease often occur together, comprising what is currently called the metabolic syndrome (e.g. 156,157). NAFLD and in particular NASH have been identified as its hepatic manifestations.(158-160). As in the liver, dysregulation of fatty acid disposition, with ectopic lipid accumulation in non-adipose cells, is a major factor contributing to other components of the syndrome. Some proponents of the metabolic syndrome concept have expressed doubts about its significance (161), but recent evidence suggest that its core components are linked by dysregulated LCFA disposition (e.g.162-166), with resultant cellular lipotoxicity (167-173). There are therefore solid reasons for hepatologists to study LCFA uptake by other cell types in obesity and NAFLD.
Early Studies
Culture of HepG2 cells in EtOH produces dose-dependent up-regulation of LCFA uptake that is highly correlated with mAspAT gene expression; increased synthesis and selective export to the medium of mAspAT protein, but not of other cytoplasmic or mitochondrial enzymes; and increased cellular accumulation of triglyceride (TG) (97). These changes may occur without ultrastructural evidence of mitochondrial injury. The amount of mAspAT exported, if extrapolated to the mass of the human liver, could account for the increased AST/ALT ratio typical of alcoholic liver disease, suggesting that changes in that ratio reflect pharmacologic up-regulation by EtOH of mAspAT gene expression and protein synthesis, rather than hepatocellular injury. EtOH-fed Wistar and obese Zucker fatty (fa/fa) rats both have appreciable hepatic steatosis (111, 174). Total LCFA uptake is increased ~ 3-fold in both. However, in EtOH-fed animals, increased LCFA uptake is mediated by up-regulation of mAspAT gene expression and the LCFA uptake Vmax. By contrast, in Zucker fa/fa animals, increased LCFA uptake is a passive consequence of an increased plasma LCFA concentration. Nevertheless, since generation of ROS from LCFA oxidation is an important pathogenetic factor for both alcoholic and non-alcoholic steatohepatitis, these data confirm that increased LCFA uptake, albeit by different mechanisms, is a common contributor to both obesity- and EtOH-related steatosis at least in specific models (84,111,174). In Zucker (fa/fa) rats (102) we also found that, in contrast to adipocytes, not only were there no obesity-associated changes in hepatocyte LCFA uptake Vmax , but also changes in LCFA uptake Vmax in cardiac myocytes were very small. This suggests that obesity is associated with altered LCFA partitioning, with an increased fraction of LCFA entering adipocytes for storage as fat and a relatively decreased fraction entering other cell types.
What Next?
As noted earlier, many processes could hypothetically contribute to accumulation of excess triglyceride in the liver (Figure 6). It would be desirable to assay all of them simultaneously. Several, such as hepatocellular LCFA uptake and oxidation, can be assayed directly. While direct quantitation of the others is more difficult, a first-order approximation could be obtained via gene expression studies. However, we estimate that the number of known genes potentially involved in SHS pathogenesis approaches 50. Assessment of all of their expression levels by Northern hybridization would simply be impractical. Techniques such as qRT-PCR are possible alternatives, but require considerable effort to optimize individual assays, and would not lead to identification of any relevant genes the role of which was a priori unsuspected. Finally, we have considered RNA expression microarray methods. An advantage is the potential to identify previously unknown genes or genes whose role in SHS is unsuspected. The costs, however, are prodigious, and identifying changes in gene expression, by whatever method, requires validation by an alternative technique and by directly measuring the expression of the encoded protein and/or the activity of the relevant pathway.
In going forward mice offer numerous advantages over rats. Both the literature and the advice of colleagues suggested that we incorporate into our program development of selected knockouts. This advice was so pervasive that I feel obliged to defend our decision not to pursue it. Recall that my background is in bilirubin. I used to be able to say that “Bilirubin is just there. It is produced, circulates, and gets excreted. Along the way it doesn’t regulate anything, and nothing regulates it”. In fact, I did say that, in innumerable lectures to students, house officers, and others. However, bilirubin has proven to have anti-oxidant properties, and there is now a literature suggesting that production of bilirubin from heme is regulated, thus regulating the intracellular antioxidant environment (e.g. 175-177). Even if true, in quantitative terms my earlier statement remains an accurate first approximation.
LCFA (even those that are not omega-3’s) are a different kettle of fish. The intermediary metabolism of fatty acids is tightly linked to that of carbohydrates by multiple levels of regulatory, counter-regulatory, and counter-counter-regulatory pathways. An experimental perturbation introduced into any part of that tightly linked system generates waves of responses, and the measured result depends not only on the perturbation introduced but the accessibility of that part of the system to all of the potential regulatory responses and the time between perturbation and measurement – in other words, how many rounds of regulatory responses have been allowed to occur. This was a major conclusion of a recent study of the acute effects of introduction of a high fat diet on adipocyte LCFA uptake in obesity-prone Osborne-Mendel rats (107). Heisenberg’s uncertainty principle states, in effect, that one can never know the precise location of a sub-atomic particle because the very photon introduced into the system to illuminate the location of the particle perturbs the system and moves the particle (178). Intermediary metabolism illustrates the biological equivalent of Heisenberg’s uncertainty principle. The observed responses to an experimental perturbation may be dramatically altered in a situation in which the accessibility of that part of the system to normal regulatory responses has been blocked by a gene knockout. This is not to say that knock-outs are not of value. They have led to important observations. But these observations may not exactly, or even accurately, reflect normal system responses, and some are of uncertain relevance to “real life”. Of many knock-outs with SHS identified in a recent Pub Med search, the gene knocked out often had no obvious relationship to SHS, and was chosen for knock-out for an entirely different purpose.
In any case, we began in 2007 to characterize several mouse models of obesity and SHS as substrates for future studies. Experimental groups include chow-fed C57BL6/J control mice; similar mice chronically fed a high fat diet (HFD) or 10%, 14%, or 18% EtOH in drinking water; ob/ob and db/db mice. For some purposes, we planned to compare data in the first five of these groups, designated the Functional Leptin Signaling Groups (FLSGs) with those in the Leptin Signalling Deficient Groups (LSDGs) (ob/ob and db/db). Since, in SHS, LCFA shuttle back and forth between liver and fat, we planned studies in adipose tissue as well as liver.
What we are finding, so far reported only in abstract form (e.g. 179-181), is the following. Liver weights and Vmax for saturable hepatocellular LCFA uptake are significantly increased in HFD mice and, in dose dependent fashion, in all of the EtOH consuming groups. Across all FLSGs Vmax is significantly correlated with liver weight and hepatic triglyceride content, indicating that saturable LCFA uptake is a major contributor to hepatic steatosis in mice with functioning leptin signaling systems. In ob/ob and db/db mice, by contrast, Vmax is at most minimally increased, and both liver weights and TG content greatly exceed values projected as a function of Vmax from regressions in FLSGs. Thus, processes besides LCFA uptake contribute appreciably to SHS in these strains. Increased FAS and SCD-1 gene expression suggests that enhanced LCFA synthesis is one such process.
Although Vmax for hepatocyte LCFA uptake was increased in mouse models of SHS associated with both dietary obesity and EtOH ingestion, the extent of these increases, ~3-fold, is still less than the ~8-fold increases in adipocyte LCFA uptake Vmax. Accordingly, our earlier conclusion about an obesity-associated alteration in fatty acid partitioning remains valid and seems a general property of obesity that protects against ectopic lipid accumulation, delaying onset of lipotoxicity and the metabolic syndrome.
We are particularly interested in the roles of leptin and insulin in regulating cellular LCFA uptake. Prior studies suggest that insulin up-regulates hepatocyte LCFA uptake. While a key modulator of overall lipid disposition (182), leptin’s role in SHS pathogenesis is unclear. The failure of hepatocyte LCFA uptake Vmax to increase appreciably in ob/ob and db/db mice compared with all FLSGs suggests that leptin is either an up-regulator of hepatocyte LCFA uptake or necessary for expression of the up-regulatory effects of insulin. By contrast, marked up-regulation of adipocyte LCFA uptake in ob/ob and db/db mice suggests that leptin is normally a down-regulator of LCFA uptake in these cells. The data suggest that leptin’s effects may indeed either parallel insulin’s or be counter-regulatory, depending on the tissue.
Our mouse studies are conducted in parallel with studies in obese bariatric surgery patients (e.g. 108), and we are immersed in a clinical obesity milieu. Ischemic heart disease is a major consequence of obesity. Less appreciated is a non-ischemic cardiomyopathy increasingly recognized in severely obese patients (e.g. 183-185). The average body weights of our HFD-fed, db/db, and ob/ob mice were 1.3, 1.9 and 2.3 time those of the control animals, corresponding in human terms to obesity, morbid obesity and super obesity. Studies of lipid-associated cardiomyopathy in mice (186-190) stimulated us, with the help of Prof. Sunichi Homma and his Research Fellow, Dr. Kotaro Arai, to examine the hearts of the mice whose livers we were studying. Hence, I returned to my interest in cardiology after a hiatus of more than 4 decades. Myocardial lipid accumulation was examined histologically and by biochemical assay. Possible impact of increased myocardial lipid on cardiac function was assessed with 2-D echocardiographic studies, performed with a special instrument designed for use in small animals which allows us to measure both fractional shortening of the left ventricular diameter during systole (FS) and the ejection fraction (EF). Each of the obesity groups (HFD, ob/ob, db/db) had significant myocardial lipid accumulation that was highly correlated with body weight, and a corresponding decrease in LV contractility. A dose-dependent increase in myocardial lipid and decrease in LV contractility was also seen in preliminary studies in EtOH-fed mice, but preliminary transmission electron microscopic data suggest that the mechanisms of myocardial injury might be different between obese and EtOH-fed mice. .
We will soon have a great deal more physiologic, biochemical, electron microscopic, and functional information about all of these models, including expression data on nearly 50 genes already linked to SHS. We will be initiating microarray studies in those settings in which pathogenesis of the steatosis is not already clear. Many implications of the data, such as the tissue-specific effects of leptin in regulating LCFA uptake, will lead to spin-off studies in which these implications can be tested directly. One thing my post-docs are not concerned about is having nothing to do.
SUMMING UP
My first paper was published 41 years ago. Since then our studies have made extensive use of recombinant and cell biologic technology; light and electron microscopy and their immunohistologic variants; monoclonal antibodies; detailed protein chemistry; and molecular modeling of 3-D protein structures. However, I do not consider myself a molecular or cell biologist. The core of our work has always been, and remains, solid 20th century cellular and whole animal physiology, to which these other disciplines have been ancillary tools. To some, what we do is old fashioned and out of date. Nevertheless, I am unapologetic. I believe that our broad-based overview of the fields in which we have worked has led to significant and seminal advances in understanding, and has pointed to way to further substantial advances by others.
ACKNOWLEDGEMENTS
It has been my pleasure over the years to work with outstanding Colleagues: Joe Bloomer, Tony Jones, Bob Howe, Michael Bradbury, Barry Potter, and Shengli Zhou; Fellows and House Officers: Terry Blaschke, Bruce Scharschmidt, James Martin, Allan Wolkoff, John Vierling, Bennett Blitzer, Juerg Reichen, Dario Sorrentino, Wolfgand Stremmel, Luis Isola, Rosa Nunez, William Schwieterman, Hiroaki Okuda, Fengxia Ge, Chunguang Hu, Xinqing Fan and Oana Petrescu. Guest Workers: Avi Zifroni and Joseph Brandes. Jeanne Waggoner, John Vergalla, Chi-Li Kiang, and Decherd Stump were superb laboratory technicians whose contributions always exceeded what could be expected from their job descriptions. James Darnell at Rockefeller and Richard Stockert at Einstein were gracious hosts for the two sabbaticals during which I learned whatever I know about molecular and cell biology.
My work has been supported continuously since 1979 by one or more of the following NIDDK grants: R01-DK26438, R01-DK52401, R01-DK 72526, and U01-DK-66667.
REFERENCES
- 1.Barrett PV, Mullins FX, Berlin NI. Studies on the biosynthetic production of bilirubin-C14: an improved method utilizing delta-aminolevulinic acid-4-C14 in dogs. J Lab Clin Med. 1966;68(6):905–12. [PubMed] [Google Scholar]
- 2.Barrett PVD, Berk PD, Menken M, Berlin NI. Bilirubin turnover studies in normal and pathologic states using bilirubin-14C. Ann Int Med. 1968;68:355–377. doi: 10.7326/0003-4819-68-2-355. [DOI] [PubMed] [Google Scholar]
- 3.Berk PD, Howe RB, Bloomer JR, Berlin NI. Studies of bilirubin kinetics in normal adults. J Clin Invest. 1969;48:2176–2190. doi: 10.1172/JCI106184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bloomer JR, Berk PD, Howe RB, Waggoner JG, Berlin NI. Comparison of fecal urobilinogen excretion with bilirubin production in normal volunteers and patients with increased bilirubin production. Clin Chim Acta. 1970;29:463–471. doi: 10.1016/0009-8981(70)90017-3. [DOI] [PubMed] [Google Scholar]
- 5.Berk PD, Rodkey FL, Blaschke TF, Collison HA, Waggoner JG. Comparison of plasma bilirubin turnover and carbon monoxide production in man. J Lab Clin Med. 1974;83:29–37. [PubMed] [Google Scholar]
- 6.Howe RB, Berlin NI, Berk PD. Berk PD, Berlin NI, editors. Estimation of bilirubin production in man. U.S. Department of Health, Education and Welfare, U.S. Government Printing Office; Washington: The Chemistry and Physiology of Bile Pigments. 1977
- 7.Berk PD, Bloomer RJ, Howe RB, Blaschke TF, Berlin NI. Bilirubin production as a measure of red cell life span. J Lab Clin Med. 1972;79:364–378. [PubMed] [Google Scholar]
- 8.Berlin NI, Berk PD. Quantitative aspects of bilirubin metabolism for hematologists: A review. Blood. 1981;57:983–999. [PubMed] [Google Scholar]
- 9.Berk PD, Berman MD, Blitzer BL, Chretien P, Martin JF, Scharschmidt BF, Vierling JM, Wolkoff AW, Vergalla J, Waggoner JG. Effect of splenectomy on hepatic bilirubin clearance in patients with hereditary spherocytosis: Implications for the diagnosis of Gilbert”s syndrome. J Lab Clin Med. 1981;98:37–45. [PubMed] [Google Scholar]
- 10.Berk PD, Martin JF, Blaschke TF, Scharschmidt BF, Plotz PH. Unconjugated hyperbilirubinemia: physiologic evaluation and experimental approaches to therapy. Ann Int Med. 1975;82:552–570. doi: 10.7326/0003-4819-82-4-552. [DOI] [PubMed] [Google Scholar]
- 11.Korenblat K, Berk PD. Hyperbilirubinemia in the setting of anti-viral therapy. Clin Gastroent Hepatol. 2005;3:303–310. doi: 10.1016/s1542-3565(05)00083-2. [DOI] [PubMed] [Google Scholar]
- 12.Jones EA, Carson ER, Berk PD. The role of kinetic analysis and mathematical modeling in the study of bilirubin metabolism in vivo. In: Heirwegh KPM, Brown SB, editors. Bilirubin, Vol. II. Metabolism. Vol. 133. CRC Press; Boca Raton, Florida: 1982. p. 172. [Google Scholar]
- 13.Jones EA, Carson ER, Berk PD. Quantitation of bilirubin metabolism in vivo: Kinetic studies and mathematical modeling. In: Ostrow JD, editor. Bilirubin, Bile Pigments and Jaundice. Marcel Dekker; New York: 1986. pp. 439–474. New York. [Google Scholar]
- 14.Berk PD, Bloomer JR, Howe RB, Berlin NI. Constitutional hepatic dysfunction (Gilbert’s syndrome): A new definition based on kinetic studies with unconjugated radio bilirubin. Am J Med. 1970;49:296–305. doi: 10.1016/s0002-9343(70)80020-1. [DOI] [PubMed] [Google Scholar]
- 15.Bloomer JR, Berk PD, Howe RB, Berlin NI. Bilirubin metabolism in congenital non-hemolytic jaundice. Ped Res. 1971;5:256–274. [Google Scholar]
- 16.Blaschke TF, Berk PD, Scharschmidt BF, Guyther JR, Vergalla JM, Waggoner JG. Crigler-Najjar Syndrome: An unusual course with development of neurologic damage at age 18. Ped Res. 1974;8:73–590. doi: 10.1203/00006450-197405000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Berk PD, Scharschmidt BF, Waggoner JG, White SC. The effect of repeated phlebotomy on bilirubin turnover, bilirubin clearance and unconjugated hyperbilirubinemia in the Crigler-Najjar syndrome and the jaundiced Gunn rat: Application of computers to experimental design. Clin Sci Molec Med. 1976;50:333–348. doi: 10.1042/cs0500333. [DOI] [PubMed] [Google Scholar]
- 18.Bloomer JR, Berk PD, Howe RB. Hepatic clearance of unconjugated bilirubin in cholestatic liver diseases. Am J Dig Dis. 1974;19:9–14. doi: 10.1007/BF01073348. [DOI] [PubMed] [Google Scholar]
- 19.Berk PD, Javitt N. Hyperbilirubinemia and cholestasis. Am J Med. 1978;64:311–326. doi: 10.1016/0002-9343(78)90061-x. [DOI] [PubMed] [Google Scholar]
- 20.Summerfield JA, Scott J, Berman M, Cameron G, Bloomer JR, Berk PD, Sherlock S. Benign recurrent intrahepatic cholestasis: Studies of bilirubin kinetics, bile acids, and cholangiography. Gut. 1980;21:154–160. doi: 10.1136/gut.21.2.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones EA, Shrager R, Bloomer JR, Berk PD, Howe RB, Berlin NI. Quantitative studies of the delivery of hepatic synthesized bilirubin to plasma utilizing aminolevulinic acid 4-14C and bilirubin-3H in man. J Clin Invest. 1972;51:2450–2458. doi: 10.1172/JCI107058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gisselbrecht C, Berk PD. Failure of phenobarbital to increase bilirubin production in the rat. Biochem Pharm. 1974;23:2895–2905. doi: 10.1016/0006-2952(74)90064-1. [DOI] [PubMed] [Google Scholar]
- 23.Berk PD, Blaschke TF, Scharschmidt BF, Waggoner JG, Berlin NI. A new approach to quantitation of the various sources of bilirubin in man. J Lab Clinical Med. 1976;87:767–780. [PubMed] [Google Scholar]
- 24.Jones EA, Bloomer JR, Berk PD, Carson ER, Owens D, Berlin NI. Berk PD, Berlin NI, editors. Quantitation of hepatic bilirubin synthesis in man. U.S. Department of Health, Education and Welfare, U.S. Government Printing Office; Washington: The Chemistry and Physiology of Bile Pigments. 1977
- 25.Blaschke TF, Berk PD. Berk PD, Berlin NI, editors. A new approach to estimation of early labeled peak bilirubin synthesis in man. U.S. Department of Health, Education and Welfare, U.S. Government Printing Office; Washington: The Chemistry and Physiology of Bile Pigments. 1977
- 26.Okuda H, Tavoloni N, Blaschke TF, Kiang CL, Jones MJT, Waggoner JG, Sardana MK, Sassa S, Shrager RI, Berk PD. Phenobarbital does not increase early labelling of bilirubin from 4-[14C]-aminolevulinic acid in man and rat. Hepatol. 1991;14:1153–1160. [PubMed] [Google Scholar]
- 27.Vierling JM, Berk PD, Hofmann AF, Martin JF, Wolkoff AW, Scharschmidt BF. Normal fasting state levels of serum cholyl conjugated bile acids in Gilbert’s syndrome: An aid to the diagnosis. Hepatol. 1982;2:340–343. doi: 10.1002/hep.1840020309. [DOI] [PubMed] [Google Scholar]
- 28.Blaschke TF, Berk PD, Rodkey FL, Scharschmidt BF, Collison HA, Waggoner JG. Drugs and the liver. I. Effects of glutethimide and phenobarbital on hepatic bilirubin clearance, plasma bilirubin turnover and carbon monoxide production in man. Biochem Pharm. 1974;23:2795–2806. doi: 10.1016/0006-2952(74)90053-7. [DOI] [PubMed] [Google Scholar]
- 29.Sherlock S. Diseases of the Liver and Biliary System. 8th Edition Blackwell; London: 1989. pp. 241–243. [Google Scholar]
- 30.Blanckaert N, Schmid R. Physiology and pathophysiology of bilirubin metabolism. In: Zakim D, Boyer TD, editors. Hepatology. W B Saunders; Philadelphia: 1982. Chapter 10. [Google Scholar]
- 31.Robertson KJ, Clarke D, Sutherland L, Wooster R, Coughtrie MW, Burchell B. Investigation of the molecular basis of the genetic deficiency of UDP-glucuronosyltransferase in Crigler-Najjar syndrome. J Inherit Metab Dis. 1991;14:563–579. doi: 10.1007/BF01797927. [DOI] [PubMed] [Google Scholar]
- 32.Bosma PJ, Chowdhury NR, Goldhoorn BG, Hofker MH, Oude Elferink RPJ, Jansen PL, Chowdhury JR. Sequence of exons and the flanking regions of human bilirubin-UDP-glucuronosyltransferase gene complex and identification of a genetic mutation in a patient with Crigler-Najjar syndrome, type I. Hepatol. 1992;15:941–947. doi: 10.1002/hep.1840150531. [DOI] [PubMed] [Google Scholar]
- 33.Ritter JK, Chen F, Sheen YY, Tran HM, Kimura S, Yeatman MT, Owens IS. A novel complex locus UGT1 encodes human bilirubin, phenol, and other UDP-glucuronosyltransferase isozymes with identical carboxyl termini. J Biol Chem. 1992;267:3257–3261. [PubMed] [Google Scholar]
- 34.Bosma PJ, Seppen J, Goldhoorn B, Bakker C, Oude Elferink RPJ, Chowdhury JR, Chowdhury NR, Jansen PLM. Bilirubin UDP-glucuronosyltransferase 1 is the only relevant bilirubin glucuronidating isoform in man. J Biol Chem. 1994;269:17960–17964. [PubMed] [Google Scholar]
- 35.Burchell B, Hume R. Molecular genetic basis of Gilbert’s syndrome. J Gastroenterol Hepatol. 1999;14:960–966. doi: 10.1046/j.1440-1746.1999.01984.x. [DOI] [PubMed] [Google Scholar]
- 36.Bosma PJ. Inherited disorders of bilirubin metabolism. J Hepatol. 2003;38:107–117. doi: 10.1016/s0168-8278(02)00359-8. [DOI] [PubMed] [Google Scholar]
- 37.Scharschmidt BF, Waggoner JG, Berk PD. Hepatic organic anion uptake in the rat. J Clin Invest. 1975;56:1280–1292. doi: 10.1172/JCI108204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reichen J, Berk PD. Isolation of an organic anion binding protein from rat liver plasma membrane fractions by affinity chromatography. Biochem Biophys Res Com. 1979;91:484–489. doi: 10.1016/0006-291x(79)91547-x. [DOI] [PubMed] [Google Scholar]
- 39.Stremmel W, Gerber MD, Glezerov V, Thung SN, Kochwa S, Berk PD. Physicochemical and immuno-histological studies of a sulfo brompthalein and bilirubin binding protein from rat liver plasma membranes. J Clin Invest. 1983;71:1796–1805. doi: 10.1172/JCI110935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stremmel W, Berk PD. Hepatocellular uptake of sulfobromophthalein (BSP) and bilirubin is selectively inhibited by an antibody to the liver plasma membrane BSP/bilirubin binding protein. J Clin Invest. 1986;78:822–826. doi: 10.1172/JCI112646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Briz O, Serrano MA, MacIas RI, Gonzalez-Gallego J, Marin JJ. Role of organic anion-transporting polypeptides, OATP-A, OATP-C and OATP-8, in the human placenta-maternal liver tandem excretory pathway for foetal bilirubin. Biochem J. 2003;371(Pt 3):897–905. doi: 10.1042/BJ20030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cui Y, König J, Leier I, Buchholz U, Keppler D. Hepatic uptake of bilirubin and its conjugates by the human organic anion transporter SLC21A6. J Biol Chem. 2001;276(13):9626–9630. doi: 10.1074/jbc.M004968200. [DOI] [PubMed] [Google Scholar]
- 43.Wang P, Kim RB, Chowdhury JR, Wolkoff AW. The human organic anion transport protein SLC21A6 is not sufficient for bilirubin transport. J Biol Chem. 2003;278(23):20695–20699. doi: 10.1074/jbc.M301100200. [DOI] [PubMed] [Google Scholar]
- 44.Bloomer JR, Berk PD, Vergalla J, Berlin NI. Influence of albumin on the hepatic uptake of unconjugated bilirubin. Clin Sci. 1973;45:505–516. doi: 10.1042/cs0450505. [DOI] [PubMed] [Google Scholar]
- 45.Bloomer JR, Berk PD, Vergalla J, Berlin NI. Influence of albumin on the extravascular distribution of unconjugated bilirubin. Clin Sci. 1973;45:517–526. doi: 10.1042/cs0450517. [DOI] [PubMed] [Google Scholar]
- 46.Weisiger R, Gollan J, Ockner R. Receptor for albumin on the liver cell surface may mediate uptake of fatty acids and other albumin-bound substances. Science. 1981;211(4486):1048–51. doi: 10.1126/science.6258226. [DOI] [PubMed] [Google Scholar]
- 47.Weisiger R, Gollan J, Ockner R. An albumin receptor on the liver cell may mediate hepatic uptake of sulfobromphthalein and bilirubin; bound ligand, not free, is the major uptake determinant. Gastroent. 1980;79:1065. [Abstract] [Google Scholar]
- 48.Nunes R, Kiang CL, Berk PD. “Albumin receptor” uptake kinetics do not require an intact lobular architecture and are not specific for albumin. J. Hepatology. 1988;7:293–304. doi: 10.1016/s0168-8278(88)80001-1. [DOI] [PubMed] [Google Scholar]
- 49.Nunes RM, Isola LM, Sorrentino D, Berk PD. Oleate uptake by isolated hepatocytes consists of two components, each driven by the unbound oleate concentration: Proceedings, 3rd Internat Congr, Math Model Liver Excr Process, Juntendo Univ. Press; Tokyo. 1990.pp. 312–316. [Google Scholar]
- 50.Potter BJ, Sorrentino D, Berk PD. Mechanisms of cellular uptake of free fatty acids. Annu. Rev. Nutr. 1989;9:253–270. doi: 10.1146/annurev.nu.09.070189.001345. [DOI] [PubMed] [Google Scholar]
- 51.Sorrentino D, Potter BJ, Berk PD. From albumin to the cytoplasm: the hepatic uptake of organic anions. Prog. Liv. Dis. 1990;IX:203–204. [PubMed] [Google Scholar]
- 52.Stremmel W, Potter BJ, Berk PD. Studies of albumin binding to rat liver plasma membranes: implications for the albumin receptor hypothesis. Biochim Biophys Acta. 1983;756:20–27. doi: 10.1016/0304-4165(83)90019-3. [DOI] [PubMed] [Google Scholar]
- 53.Sorrentino D, Robinson RB, Kiang CL, Berk PD. At physiologic albumin/oleate concentrations oleate uptake by isolated hepatocytes, cardiac myocytes and adipocytes is a saturable function of the unbound oleate concentration. Uptake kinetics are consistent with the conventional theory. J Clin Invest. 1989;84:1325–1333. doi: 10.1172/JCI114301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sorrrentino D, Zifroni A, Van Ness K, Berk PD. Unbound ligand drives hepatocyte taurocholate and BSP uptake at physiological albumin concentration. Am J Physiol. 1994;266:G425–G432. doi: 10.1152/ajpgi.1994.266.3.G425. [DOI] [PubMed] [Google Scholar]
- 55.Sorrentino D, Van Ness K, Berk PD. Oleate uptake kinetics in the perfused rat liver are consistent with pseudofacilitation by albumin. J Hepatol. 1994;21:551–559. doi: 10.1016/s0168-8278(94)80100-2. [DOI] [PubMed] [Google Scholar]
- 56.Sorrentino D, Berk PD. Free fatty acids, albumin and the sinusoidal plasma membrane: concepts, trends and controversies. In: Tavoloni N, Berk PD, editors. Hepatic Transport and Bile Secretion. Raven Press; New York: 1993. pp. 197–210. [Google Scholar]
- 57.Potter BJ, Berk PD. Liver plasma membrane fatty acid binding protein. In: Tavoloni N, Berk PD, editors. Hepatic Transport and Bile Secretion. Raven Press; New York: 1993. pp. 253–267. [Google Scholar]
- 58.Stremmel W, Berk PD. Hepatocellular influx of 14C-oleate reflects membrane transport rather than intracellular metabolism or binding. Proc Nat Acad Sci USA. 1986;83:3086–3090. doi: 10.1073/pnas.83.10.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stremmel W, Strohmeyer G, Berk PD. Hepatocellular uptake of oleate is energy dependent, sodium linked, and inhibited by an antibody to a hepatocyte plasma membrane fatty acid binding protein. Proc Nat Acad Sci USA. 1986;83:3584–3588. doi: 10.1073/pnas.83.11.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stump DD, Nunes RM, Sorrentino D, Berk PD. Characteristics of oleate binding to liver plasma membrane and its uptake by isolated hepatocytes. J Hepatol. 1992;16:304–315. doi: 10.1016/s0168-8278(05)80661-0. [DOI] [PubMed] [Google Scholar]
- 61.Stremmel W, Kochwa S, Berk PD. Studies of oleate binding to rat liver plasma membranes. Biochem Biophys Res Commun. 1983;112:88–95. doi: 10.1016/0006-291x(83)91801-6. [DOI] [PubMed] [Google Scholar]
- 62.Schwieterman W, Sorrentino D, Potter BJ, Rand J, Kiang CL, Stump D, Berk PD. Uptake of oleate by isolated rat adipocytes is mediated by a 40 kDa plasma membrane fatty acid binding protein closely related to that in liver and gut. Proc Nat Acad Sci USA. 1988;85:359–363. doi: 10.1073/pnas.85.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stump DD, Fan X, Berk PD. Oleic acid uptake and binding by rat adipocytes define dual pathways for cellular fatty acid uptake. J Lip Res. 2001;42:509–520. [PubMed] [Google Scholar]
- 64.Stremmel W, Lotz G, Strohmeyer G, Berk PD. Identification, isolation and partial characterization of a fatty acid binding protein from rat jejunal microvillous membranes. J Clin Invest. 1985;75:1068–1076. doi: 10.1172/JCI111769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sorrentino D, Stump D, Potter BJ, Robinson R, White R, Kiang CL, Berk PD. Oleate uptake by cardiac myocytes is carrier mediated and involves a 40 kDa plasma membrane fatty acid binding protein similar to that in liver, adipose tissue and gut. J Clin Invest. 1988;82:928–935. doi: 10.1172/JCI113700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berk PD, Stump DD. Mechanisms of cellular uptake of long chain free fatty acids. Mol Cell Biochem. 1999;192:17–31. [PubMed] [Google Scholar]
- 67.Stoll GH, Voges R, Gerok W, Kurz G. Synthesis of a metabolically stable modified long-chain fatty acid salt and its photolabile derivative. J Lipid Res. 1991;32(5):843–857. [PubMed] [Google Scholar]
- 68.Schmider W, Fahr A, Blum HE, Kurz G. Transport of heptafluorostearate across model membranes. Membrane transport of long-chain fatty acid anions I. J Lipid Res. 2000;41(5):775–787. [PubMed] [Google Scholar]
- 69.Sorrentino D, Zhou SL, Kokkoutou E, Berk PD. Sex differences in hepatic fatty acid uptake reflect a greater affinity of the membrane transport system in females. Am J of Physiol: GastrointestLiver Physiol. 1992;263:G380–G385. doi: 10.1152/ajpgi.1992.263.3.G380. [DOI] [PubMed] [Google Scholar]
- 70.Sorrentino D, Zhou SL, Van Ness K, Isola LM, Berk PD. The hepatocellular uptake of free fatty acids is selectively preserved during starvation. Gastroent. 1994;107:1415–1424. doi: 10.1016/0016-5085(94)90544-4. [DOI] [PubMed] [Google Scholar]
- 71.Sorrentino D, Van Ness K, Simard A, Andreas J, Schwab AJ, Stump D, Goresky CA, Berk PD. Oleate uptake by isolated hepatocytes and the perfused rat liver is competitively inhibited by palmitate. Am J Physiol. 1996;270:G385–G392. doi: 10.1152/ajpgi.1996.270.2.G385. [DOI] [PubMed] [Google Scholar]
- 72.Kamp F, Hamilton JA. pH gradients across phospholipids membranes caused by fast flip-flop of unionized fatty acids. Proc Nat Acad Sci USA. 1992;89:11367–11370. doi: 10.1073/pnas.89.23.11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kamp F, Westerhoff HV, Hamilton JA. Movement of fatty acids, fatty acid analogues, and bile acids across a phospholipids bilayer. Biochem. 1993;32:11074–11086. doi: 10.1021/bi00092a017. [DOI] [PubMed] [Google Scholar]
- 74.Kamp F, Zakim D, Zhang F, Noy N, Hamilton JA. Fatty acid flip-flop in phospholipids bilayers is extremely fast. Biochem. 1995;34:11928–11937. doi: 10.1021/bi00037a034. [DOI] [PubMed] [Google Scholar]
- 75.Daniels C, Noy N, Zakim D. Rates of hydration of fatty acids bound to unilamellar vesicles of phosphatidylcholine or to albumin. Biochem. 1985;24:3286–3292. doi: 10.1021/bi00334a032. [DOI] [PubMed] [Google Scholar]
- 76.Civelek VN, Hamilton JA, Tornheim K, Kelly KL, Corkey BE. Intracellular pH in adipocytes: effects of free fatty acid diffusion across the plasma membrane, lipolytic agonists, and insulin. Proc Nat Acad Sci USA. 1996:10139–10144. doi: 10.1073/pnas.93.19.10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luiken JJ, Turcotte LP, Bonen A. Protein-mediated palmitate uptake and expression of fatty acid transport proteins in heart giant vesicles. J Lipid Res. 1999;40(6):1007–1016. [PubMed] [Google Scholar]
- 78.Luiken JJ, Glatz JF, Bonen A. Fatty acid transport proteins facilitate fatty acid uptake in skeletal muscle. Can J Appl Physiol. 2000;25(5):333–352. [PubMed] [Google Scholar]
- 79.Kampf JP, Kleinfeld AM. Fatty acid transport in adipocytes monitored by imaging intracellular free fatty acid levels. J Biol Chem. 2004;279(34):35775–35780. doi: 10.1074/jbc.M403630200. [DOI] [PubMed] [Google Scholar]
- 80.Kleinfeld AM, Kampf JP, Lechene C. Transport of 13C-oleate in adipocytes measured using multi imaging mass spectrometry. J Am Soc Mass Spectrom. 2004 Nov;15(11):1572–1580. doi: 10.1016/j.jasms.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 81.Stremmel W, Strohmeyer G, Borchard F, Kochwa S, Berk PD. Isolation and partial characterization of a fatty acid binding protein in rat liver plasma membranes. Proc Nat Acad Sci USA. 1985;82:4–8. doi: 10.1073/pnas.82.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Potter BJ, Stump D, Schwieterman W, Sorrentino D, Jacobs LN, Kiang CL, Rand J, Berk PD. Isolation and partial characterization of plasma membrane fatty acid binding proteins from myocardium and adipose tissue and their relationship to analogous proteins in liver and gut. Biochem Biophys Res Commun. 1987;148:1370–1376. doi: 10.1016/s0006-291x(87)80283-8. [DOI] [PubMed] [Google Scholar]
- 83.Zhou SL, Stump D, Sorrentino D, Potter BJ, Berk PD. Adipocyte differentiation of 3T3 L1 cells involves augumented expression of a 43 kDa plasma membrane fatty acid binding protein. J Biol Chem. 1992;267:14456–14461. [PubMed] [Google Scholar]
- 84.Zhou SL, Stump DD, Isola LM, Berk PD. Mitochondrial aspartate aminotransferase expressed on the surface of 3T3 L1 adipocytes mediates saturable fatty acid uptake. Proc Soc Exper Biol Med. 1995;208:263–270. doi: 10.3181/00379727-208-43854. [DOI] [PubMed] [Google Scholar]
- 85.Abumrad NA, el-Maghrabi MR, Amri EZ, Lopez E, Grimaldi PA. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J Biol Chem. 1993;268:17665–17668. [PubMed] [Google Scholar]
- 86.Schaffer JE, Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994;79(3):427–436. doi: 10.1016/0092-8674(94)90252-6. [DOI] [PubMed] [Google Scholar]
- 87.Hirsch D, Stahl A, Lodish HF. A family of fatty acid transporters conserved from mycobacterium to man. Proc Natl Acad Sci U S A. 1998;95(15):8625–8629. doi: 10.1073/pnas.95.15.8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stahl A, Gimeno RE, Tartaglia LA, Lodish HF. Fatty acid transport proteins: a current view of a growing family. Trends Endocrinol Metab. 2001;12(6):266–273. doi: 10.1016/s1043-2760(01)00427-1. [DOI] [PubMed] [Google Scholar]
- 89.Trigatti BL, Anderson RG, Gerber GE. Identification of caveolin-1 as a fatty acid binding protein. Biochem Biophys Res Commun. 1999;255(1):34–39. doi: 10.1006/bbrc.1998.0123. [DOI] [PubMed] [Google Scholar]
- 90.Pohl J, Ring A, Stremmel W. Uptake of long-chain fatty acids in HepG2 cells involves caveolae: analysis of a novel pathway. J Lipid Res. 2002;43(9):1390–1399. doi: 10.1194/jlr.m100404-jlr200. [DOI] [PubMed] [Google Scholar]
- 91.Kampf JP, Parmley D, Kleinfeld AM. Free fatty acid transport across adipocytes is mediated by an unknown membrane protein pump. Am J Physiol:Endocrinol Metab. 2007;293(5):E1207–1214. doi: 10.1152/ajpendo.00259.2007. [DOI] [PubMed] [Google Scholar]
- 92.Berk PD, Wada H, Horio Y, Potter BJ, Sorrentino D, Zhou SL, Isola LM, Stump D, Kiang CL, Thung S. Plasma membrane fatty acid binding protein and mitochondrial glutamic oxaloacetic transaminase of rat liver are related. Proc Nat Acad Sci USA. 1990;87:3484–3488. doi: 10.1073/pnas.87.9.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stump DD, Zhou SL, Berk PD. Comparison of the plasma membrane FABP and mitochondrial isoform of aspartate aminotransferase from rat liver. Am J Physiol. 1993;265:G894–G902. doi: 10.1152/ajpgi.1993.265.5.G894. [DOI] [PubMed] [Google Scholar]
- 94.Zhou SL, Stump D, Isola LM, Berk PD. Constitutive expression of a saturable transport system for non-esterified fatty acids in Xenopus laevis oocytes. Biochem J. 1994;297:315–319. doi: 10.1042/bj2970315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Isola LM, Zhou S-L, Kiang C-L, Stump DD, Bradbury MW, Berk PD. 3T3 fibroblasts transfected with a cDNA for mitochondrial aspartate aminotransferase express plasma membrane fatty acid-binding protein and saturable fatty acid uptake. Proc Nat Acad Sci USA. 1995;92:9866–9870. doi: 10.1073/pnas.92.21.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bradbury MW, Berk PD. Cellular uptake of long chain free fatty acids: the structure and function of plasma membrane fatty acid binding protein. Adv Mol Cell Biol. 2004;33:47–81. [Google Scholar]
- 97.Zhou SL, Gordon RE, Bradbury M, Stump D, Kiang CL, Berk PD. Ethanol up regulates fatty acid uptake and plasma membrane expression and export of mitochondrial aspartate aminotransferase in HepG2 cells. Hepatol. 1998;27:1064–1074. doi: 10.1002/hep.510270423. [DOI] [PubMed] [Google Scholar]
- 98.Cechetto JD, Sadacharan SK, Berk PD, Gupta RS. Immunogold localization of mitochondrial aspartate aminotransferase in mitochondria and on cell surface in normal rat tissues. Histol Histopath. 2002;17:353–364. doi: 10.14670/HH-17.353. [DOI] [PubMed] [Google Scholar]
- 99.Berk PD, Delavega L, Stockert RJ. Mitochondrial aspartate aminotransferase reaches the plasma membrane and is exported from HuH7 cells via the golgi/endoplasmic reticulum. Hepatol. 1998;28:400A. [Google Scholar]
- 100.Guarnieri F, Stump DD, Roboz J, Yu Q, Berk PD. Mitochondrial aspartateaminotransferase contains a 500-Angstrom3 hydrophobic groove representing a putative fatty-acid-binding site. Hepatol. 1995;22:1269. [Google Scholar]
- 101.Bradbury M, Fan XQ, Stump DD, Odin JA, Guarnieri F, Berk PD. Mapping the fatty acid binding site in hepatic plasma membrane fatty acid binding protein with molecular modeling and mutagenesis. Hepatol. 34:256A. [Google Scholar]
- 102.Berk PD, Zhou SL, Kiang CL, Stump D, Bradbury M, Isola LM. Uptake of long chain free fatty acids is selectively up-regulated in adipocytes of Zucker rats with genetic obesity and non-insulin-dependent diabetes mellitus. J Biol Chem. 1997;272(13):8830–8835. doi: 10.1074/jbc.272.13.8830. [DOI] [PubMed] [Google Scholar]
- 103.Berk PD, Zhou S, Kiang C, Stump DD, Fan X, Bradbury MW. Selective up-regulation of fatty acid uptake by adipocytes characterizes both genetic and diet-induced obesity in rodents. J Biol Chem. 1999;274(40):28626–28631. doi: 10.1074/jbc.274.40.28626. [DOI] [PubMed] [Google Scholar]
- 104.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 105.Lee GH, Proenca R, Montez JM, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 379(6566):632–635. doi: 10.1038/379632a0. 996. [DOI] [PubMed] [Google Scholar]
- 106.Chua SC, Jr., Chung WK, Wu-Peng XS, et al. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science. 1996;271(5251):994–996. doi: 10.1126/science.271.5251.994. [DOI] [PubMed] [Google Scholar]
- 107.Petrescu O, Cheema AF, Fan X, Bradbury MW, Berk PD. Differences in Adipocyte Uptake of Long Chain Fatty Acids in Response to High Fat Diets in Osborne-Mendel and S5B/Pl Rats. Int J Obes. 2008;32:853–862. doi: 10.1038/sj.ijo.0803792. [DOI] [PubMed] [Google Scholar]
- 108.Petrescu O, Fan X, Gentileschi P, et al. Long-chain fatty acid uptake is upregulated in omental adipocytes from patients undergoing bariatric surgery for obesity. Int J Obes. 2005;29(2):196–203. doi: 10.1038/sj.ijo.0802868. [DOI] [PubMed] [Google Scholar]
- 109.Fan X, Bradbury MW, Berk PD. Leptin and insulin modulate energy efficiency and weight loss through selective regulation of long chain fatty acid uptake by adipocytes. J Nutr. 2003;133:2707–2715. doi: 10.1093/jn/133.9.2707. [DOI] [PubMed] [Google Scholar]
- 110.Ibrahimi A, Sfeir Z, Magharaie H, Amri EZ, Grimaldi P, Abumrad NA. Expression of the CD36 homolog (FAT) in fibroblast cells: effects on fatty acid transport. Proc Natl Acad Sci U S A. 1996;93(7):2646–2651. doi: 10.1073/pnas.93.7.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bradbury MW, Berk PD. Lipid metabolism in hepatic steatosis. Clin Liver Dis. 2004;8:639–671. doi: 10.1016/j.cld.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 112.Goverment Printing Office: U.S. Department of Health and Human Services; Washington DC: The Surgeon General’s Call to Action to Prevent and Decrease Overweight and Obesity - 2001. 2001 [PubMed]
- 113.Yanovski SZ, Yanovski JA. Obesity. N Engl J Med. 2002;346(8):591–602. doi: 10.1056/NEJMra012586. [DOI] [PubMed] [Google Scholar]
- 114.Flegal KM, Carroll MD, Ogden CL, Johnson CL. Prevalence and trends in obesity among US adults, 1999-2000. JAMA. 2002;288(14):1723–1727. doi: 10.1001/jama.288.14.1723. [DOI] [PubMed] [Google Scholar]
- 115.Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroent. 2007;132(6):2087–2102. doi: 10.1053/j.gastro.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 116.State-specific prevalence of obesity among adults--United States, 2005. MMWR Morb Mortal Wkly Rep. 2006 Sep 15;55(36):985–988. [PubMed] [Google Scholar]
- 117.Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatol. 1990;12(5):1106–1110. doi: 10.1002/hep.1840120505. [DOI] [PubMed] [Google Scholar]
- 118.Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated amino-transferase levels in the United States. Am J Gastroent. 2003;98(5):960–967. doi: 10.1111/j.1572-0241.2003.07486.x. [DOI] [PubMed] [Google Scholar]
- 119.Ruhl CE, Everhart JE. Determinants of the association of overweight with elevated serum alanine aminotransferase activity in the United States. Gastroent. 2003;124(1):71–79. doi: 10.1053/gast.2003.50004. [DOI] [PubMed] [Google Scholar]
- 120.Ruhl CE, Everhart JE. Epidemiology of nonalcoholic fatty liver. Clin Liver Dis. 2004;8(3):501–519. vii. doi: 10.1016/j.cld.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 121.Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatol. 2004;40(6):1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 122.Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroent. 2006;40(3 Suppl 1):S5–10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- 123.Kopelman PG. Obesity as a medical problem. Nature. 2000;404(6778):635–643. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- 124.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatol. 2005;41(6):1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 125.Bondini S, Kleiner DE, Goodman ZD, Gramlich T, Younossi ZM. Pathologic assessment of non-alcoholic fatty liver disease. Clin Liver Dis. 2007;11(1):17–23. vii. doi: 10.1016/j.cld.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 126.Yeh MM, Brunt EM. Pathology of nonalcoholic fatty liver disease. Am J Clin Pathol. 2007;128(5):837–847. doi: 10.1309/RTPM1PY6YGBL2G2R. [DOI] [PubMed] [Google Scholar]
- 127.Brunt EM. Nonalcoholic steatohepatitis: definition and pathology. Semin Liver Dis. 2001;21(1):3–16. doi: 10.1055/s-2001-12925. [DOI] [PubMed] [Google Scholar]
- 128.Brunt EM, Neuschwander-Tetri BA, Oliver D, Wehmeier KR, Bacon BR. Nonalcoholic steatohepatitis: histologic features and clinical correlations with 30 blinded biopsy specimens. Hum Pathol. 2004;35(9):1070–1082. doi: 10.1016/j.humpath.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 129.Brunt EM. Nonalcoholic steatohepatitis: pathologic features and differential diagnosis. Semin Diagn Pathol. 2005;22(4):330–338. doi: 10.1053/j.semdp.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 130.Thung SNGM. Alcoholic hepatitis vs nonalcoholic steatohepatitis. In: Thung SN, Gerber MA, editors. Differential Diagnosis in Pathology: Liver Disorders. Igaku-Shoin; New York: 1995. pp. 24–36. [Google Scholar]
- 131.Pinto HC, Baptista A, Camilo ME, Valente A, Saragoca A, de Moura MC. Nonalcoholic steatohepatitis. Clinicopathological comparison with alcoholic hepatitis in ambulatory and hospitalized patients. Dig Dis Sci. 1996;41(1):172–179. doi: 10.1007/BF02208601. [DOI] [PubMed] [Google Scholar]
- 132.Gramlich T, Kleiner DE, McCullough AJ, Matteoni CA, Boparai N, Younossi ZM. Pathologic features associated with fibrosis in nonalcoholic fatty liver disease. Hum Pathol. 2004;35(2):196–199. doi: 10.1016/j.humpath.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 133.Leclercq IAHY. Cell biology of NASH: fibrosis and cell proliferation. In: Farrell GC, George J, Hall P de la M, McCullough AK, editors. Fatty Liver Disease: NASH and Related Disorders. Blackwell; Oxford: 2005. pp. 143–158. [Google Scholar]
- 134.Clark JM, Diehl AM. Nonalcoholic fatty liver disease: an under-recognized cause of cryptogenic cirrhosis. JAMA. 2003;289(22):3000–3004. doi: 10.1001/jama.289.22.3000. [DOI] [PubMed] [Google Scholar]
- 135.Caldwell SH, Crespo DM. The spectrum expanded: cryptogenic cirrhosis and the natural history of non-alcoholic fatty liver disease. J Hepatol. 2004;40(4):578–584. doi: 10.1016/j.jhep.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 136.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatol. 2006;43(2 Suppl 1):S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 137.Charlton M, Kasparova P, Weston S, et al. Frequency of nonalcoholic steatohepatitis as a cause of advanced liver disease. Liver Transpl. 2001;7(7):608–614. doi: 10.1053/jlts.2001.25453. [DOI] [PubMed] [Google Scholar]
- 138.Burke A, Lucey MR. Non-alcoholic fatty liver disease, non-alcoholic steatohepatitis and orthotopic liver transplantation. Am J Transplant. 2004;4(5):686–693. doi: 10.1111/j.1600-6143.2004.00432.x. [DOI] [PubMed] [Google Scholar]
- 139.Charlton M. Nonalcoholic fatty liver disease: a review of current understanding and future impact. Clin Gastroenterol Hepatol. 2004;2(12):1048–1058. doi: 10.1016/s1542-3565(04)00440-9. [DOI] [PubMed] [Google Scholar]
- 140.Goldberg IJ, Ginsberg HN. Ins and outs modulating hepatic triglyceride and development of nonalcoholic fatty liver disease. Gastroent. 2006;130(4):1343–6. doi: 10.1053/j.gastro.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 141.Chirieac DV, Chirieac LR, Corsetti JP, Cianci J, Sparks CE, Sparks JD. Glucose-stimulated insulin secretion suppresses hepatic triglyceride-rich lipoprotein and apoB production. Am J Physiol: Endocr Metab. 2000;279(5):E1003–E1011. doi: 10.1152/ajpendo.2000.279.5.E1003. [DOI] [PubMed] [Google Scholar]
- 142.Diehl AM. Lessons from animal models of NASH. Hepatol Res. 2005 Oct;33(2):138–144. doi: 10.1016/j.hepres.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 143.Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res. 2008;49(5):1068–1076. doi: 10.1194/jlr.M800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Minehira K, Young SG, Villanueva CJ, Yetukuri L, Oresic M, Hellerstein MK, Farese RV, Jr, Horton JD, Preitner F, Thorens B, Tappy L. Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J Lipid Res. 2008 Jun 1; doi: 10.1194/jlr.M800248-JLR200. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroent. 1998;114(4):842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 146.McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroent. 2006;40(3 Suppl 1):S17–29. doi: 10.1097/01.mcg.0000168645.86658.22. [DOI] [PubMed] [Google Scholar]
- 147.Merriman RB, Aouizerat BE, Bass NM. Genetic influences in nonalcoholic fatty liver disease. J Clin Gastroent. 2006;40(3 Suppl 1):S30–33. doi: 10.1097/01.mcg.0000168643.16074.19. [DOI] [PubMed] [Google Scholar]
- 148.Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med. 2008;44(7):1259–1272. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gores G. Mechanisms of hepatocyte Injury and death in NAFLD. Semin Liv Dis. 2008;38 In Press. [Google Scholar]
- 150.Diehl A. Mechanisms of disease and progression in NAFLD. Semin Liv Dis. 2008;38 doi: 10.1055/s-0028-1091981. In Press. [DOI] [PubMed] [Google Scholar]
- 151.Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest. 2000;105(8):1067–1075. doi: 10.1172/JCI8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Robertson G, Leclercq I, Farrell GC. Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am J Physiol: Gastrointest Liver Physiol. 2001;281(5):G1135–1139. doi: 10.1152/ajpgi.2001.281.5.G1135. [DOI] [PubMed] [Google Scholar]
- 153.Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroent. 2001;120(5):1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 154.De Minicis S, Brenner DA. Oxidative stress in alcoholic liver disease: role of NADPH oxidase complex. J Gastroenterol Hepatol. 2008;23(Suppl 1):S98–103. doi: 10.1111/j.1440-1746.2007.05277.x. [DOI] [PubMed] [Google Scholar]
- 155.Verna EC, Berk PD. Role of fatty acids in the pathogenesis of obesity and fatty liver: Impact of bariatric surgery. Semin Liver Dis. 2008;28(4) doi: 10.1055/s-0028-1091985. In Press. [DOI] [PubMed] [Google Scholar]
- 156.Grundy SM, Brewer HB, Jr., Cleeman JI, Smith SC, Jr., Lenfant C. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Arterioscler Thromb Vasc Biol. 2004;24(2):e13–18. doi: 10.1161/01.ATV.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- 157.Reaven G. Metabolic syndrome: pathophysiology and implications for management of cardiovascular disease. Circulation. 2002;106(3):286–288. doi: 10.1161/01.cir.0000019884.36724.d9. [DOI] [PubMed] [Google Scholar]
- 158.Haynes P, Liangpunsakul S, Chalasani N. Nonalcoholic fatty liver disease in individuals with severe obesity. Clin Liver Dis. 2004;8(3):535–547. viii. doi: 10.1016/j.cld.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 159.Choudhury J, Sanyal AJ. Insulin resistance and the pathogenesis of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8(3):575–594. ix. doi: 10.1016/j.cld.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 160.Marceau P, Biron S, Hould FS, et al. Liver pathology and the metabolic syndrome X in severe obesity. J Clin Endocrinol Metab. 1999;84(5):1513–1517. doi: 10.1210/jcem.84.5.5661. [DOI] [PubMed] [Google Scholar]
- 161.Kahn R, Buse J, Ferrannini E, Stern M. The metabolic syndrome: time for a critical appraisal: joint statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2005;28(9):2289–2304. doi: 10.2337/diacare.28.9.2289. [DOI] [PubMed] [Google Scholar]
- 162.Reaven GM. The individual components of the metabolic syndrome: is there a raison d’etre? J Am Coll Nutr. 2007;26(3):191–195. doi: 10.1080/07315724.2007.10719601. [DOI] [PubMed] [Google Scholar]
- 163.Pagano G, Pacini G, Musso G, et al. Nonalcoholic steatohepatitis, insulin resistance, and metabolic syndrome: further evidence for an etiologic association. Hepatol. 2002;35(2):367–372. doi: 10.1053/jhep.2002.30690. [DOI] [PubMed] [Google Scholar]
- 164.Wisse BE. The inflammatory syndrome: the role of adipose tissue cytokines in metabolic disorders linked to obesity. J Am Soc Nephrol. 2004;15(11):2792–2800. doi: 10.1097/01.ASN.0000141966.69934.21. [DOI] [PubMed] [Google Scholar]
- 165.Kerner A, Avizohar O, Sella R, et al. Association between elevated liver enzymes and C-reactive protein: possible hepatic contribution to systemic inflammation in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2005;25(1):193–197. doi: 10.1161/01.ATV.0000148324.63685.6a. [DOI] [PubMed] [Google Scholar]
- 166.Ferroni P, Basili S, Falco A, Davi G. Inflammation, insulin resistance, and obesity. Curr Atheroscler Rep. 2004;6(6):424–431. doi: 10.1007/s11883-004-0082-x. [DOI] [PubMed] [Google Scholar]
- 167.McGarry JD, Dobbins RL. Fatty acids, lipotoxicity and insulin secretion. Diabetologia. 1999;42(2):128–138. doi: 10.1007/s001250051130. [DOI] [PubMed] [Google Scholar]
- 168.Unger RH, Orci L. Lipotoxic diseases of nonadipose tissues in obesity. Int J Obes Relat Metab Disord. 2000;24(Suppl 4):S28–32. doi: 10.1038/sj.ijo.0801498. [DOI] [PubMed] [Google Scholar]
- 169.Unger RH. Lipotoxic diseases. Annu Rev Med. 2002;53:319–336. doi: 10.1146/annurev.med.53.082901.104057. [DOI] [PubMed] [Google Scholar]
- 170.Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144:5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- 171.Schaffer JE. Lipotoxicity: when tissues overeat. Curr Opin Lipidol. 2003;14(3):281–287. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- 172.Abdul-Ghani MA, Muller FL, Liu Y, Chavez AO, Balas B, Zuo P, Chang Z, Tripathy D, Jani R, Molina-Carrion M, Monroy A, Folli F, Van Remmen H, Defronzo RA. Deleterious Action of FA metabolites on ATP synthesis: The link between lipotoxicity, mitochondrial dysfunction and insulin resistance. Am J Physiol Endocrinol Metab. 2008 July 1; doi: 10.1152/ajpendo.90287.2008. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 173.Duncan JG. Lipotoxicity: what is the fate of fatty acids? J Lipid Res. 2008;49:1375–1376. doi: 10.1194/jlr.E800010-JLR200. [DOI] [PubMed] [Google Scholar]
- 174.Berk PD, Zhou SL, Bradbury MW. Increased hepatocellular uptake of long chain fatty acids occurs by different mechanisms in fatty livers due to obesity or excess ethanol use, contributing to development of steatohepatitis in both settings. Trans Am Clin Clim Assoc. 2005;116:335–345. [PMC free article] [PubMed] [Google Scholar]
- 175.Stocker RR, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 176.Rigato I, Ostrow JD, Tiribelli C. Bilirubin and the risk of common non-hepatic diseases. Trends Mol Med. 2005;11(6):277–83. doi: 10.1016/j.molmed.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 177.Shekeeb Shahab M, Kumar P, Sharma N, Narang A, Prasad R. Evaluation of oxidant and antioxidant status in term neonates: a plausible protective role of bilirubin. Mol Cell Biochem. 2008 Jun 17; doi: 10.1007/s11010-008-9807-4. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 178.Heisenberg W. Über den anschaulichen Inhalt der quantentheoretischen Kinematik und Mechanik. Zeitschrift für Physik. 1927;43:172–198. [Google Scholar]
- 179.Zhou S, Twaddell WS, Carras E, Lefkowitch J, Berk PD. Dose-dependent effects of ethanol on adipocyte, hepatocyte and cardiac myocyte fatty acid uptake and triglyceride accumulation in C57BL/6J mice. Hepatology. 2007;46((4) Suppl. 1):325A. [Google Scholar]
- 180.Zhou S, Ge F, Berk PD. Comparison of plasma insulin and leptin concentrations with long chain fatty acid uptake in hepatocytes and adipocytes from mice with hepatic steatosis clarifies the hormonal regulation of fatty acid disposition. Gastroent. 2008;134((4) Suppl. 1):A–778. [Google Scholar]
- 181.Ge F, Arai K, Zhou S, Homma S, Berk PD. Histologic and echocardiographic evidence of lipid-associated cardiomyopathy in multiple mouse obesity models. Obesity. 2008;16(9) Meeting Abstract Supplement (In Press) [Google Scholar]
- 182.Unger RH. The physiology of liporegulation. Annu Rev Physiol. 2003;65:333–347. doi: 10.1146/annurev.physiol.65.092101.142622. [DOI] [PubMed] [Google Scholar]
- 183.Wong C, Marwick TH. Obesity cardiomyopathy: pathogenesis and pathophysiology. Nat Clin Pract Cardiovasc Med. 2007;4:436–443. doi: 10.1038/ncpcardio0943. [DOI] [PubMed] [Google Scholar]
- 184.Wong C, Marwick T. Obesity cardiomyopathy: diagnosis and therapeutic implications. Nat Clin Practice. 2007;4:480–490. doi: 10.1038/ncpcardio0964. [DOI] [PubMed] [Google Scholar]
- 185.McGavock JM, Victor RG, Unger RH, Szczepaniak LS. Adiposity of the heart, revisited. Ann Int Med. 2006;144:517–524. doi: 10.7326/0003-4819-144-7-200604040-00011. [DOI] [PubMed] [Google Scholar]
- 186.Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279:E1104–E1113. doi: 10.1152/ajpendo.2000.279.5.E1104. [DOI] [PubMed] [Google Scholar]
- 187.Chiu H-C, Kovacs A, Ford DA, Hsu F-F, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100(8):1208–1217. doi: 10.1161/01.RES.0000264104.25265.b6. [DOI] [PubMed] [Google Scholar]
- 189.Lee Y, Naseem RH, Park BH, Garry DJ, Richardson JA, Schaffer JE, Unger RH. Alpha-lipoic acid prevents lipotoxic cardiomyopathy in acyl CoA-synthase transgenic mice. Biochem Biophys Res Commun. 2006;344(1):446–452. doi: 10.1016/j.bbrc.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 190.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, Schaffer JE. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005;96(2):225–233. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]