

SUMMARY
Sequence-dependent recognition of dsDNA-binding proteins are well understood, yet sequence-specific recognition of dsRNA by proteins remains largely unknown, despite their importance in RNA maturation pathways. Adenosine deaminases that act on RNA (ADARs) recode genomic information by the site-selective deamination of adenosine. Here, we report the solution structure of the ADAR2 double-stranded RNA-binding motifs (dsRBMs) bound to a stem-loop pre-mRNA encoding the R/G editing site of GluR-2. The structure provides a molecular basis for how dsRBMs recognize the shape, and also more surprisingly, the sequence of the dsRNA. The unexpected direct readout of the RNA primary sequence by dsRBMs is achieved via the minor groove of the dsRNA and this recognition is critical for both editing and binding affinity at the R/G site of GluR-2. More generally, our findings suggest a solution to the sequence-specific paradox faced by many dsRBM-containing proteins that are involved in post-transcriptional regulation of gene expression.
INTRODUCTION
ADARs convert adenosine-to-inosine (A-to-I) by hydrolytic deamination in numerous mRNA and pre-mRNA transcripts (Bass, 2002; Nishikura, 2006). Due to the similar base-pairing properties of both nucleosides, inosine is interpreted as guanosine by cellular machineries during the processes of translation and splicing. In this way, editing-mediated alterations in sequence can alter codon identity or base-pairing interactions within higher-order RNA structures (Bass, 2002; Nishikura, 2006). As a result, ADARs can create protein isoforms or regulate gene expression at the RNA level (Bass, 2002; Nishikura, 2006; Valente and Nishikura, 2005). ADARs are widely expressed in most cell types, yet their expression and activity in neuronal tissues has been shown to be important for proper nervous system function (Higuchi et al., 2000; Palladino et al., 2000). Recent high-throughput sequencing analysis of A-to-I editing identified over 55 editing sites within the coding regions of mRNAs, with 38 of these sites involving a codon change that specifies an alternative amino acid. Many of these changes involve RNA transcripts encoding proteins that are critical for nervous system function (Li et al., 2009).
ADARs from all characterized species have a modular domain organization consisting of one-to-three dsRBMs followed by a conserved C-terminal catalytic adenosine deaminase domain. The structures of the two dsRBMs and of the isolated catalytic domain of ADAR2 have been determined in their free states (Macbeth et al., 2005; Stefl et al., 2006). Among the best-studied ADAR substrates are pre-mRNAs encoding subunits of the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)-subtype of ionotropic glutamate receptor (GluR-2, GluR-3 and GluR-4; Higuchi et al., 2000; Higuchi et al., 1993; Melcher et al., 1996) that contain one or both of two highly edited and functionally relevant sites, namely the R/G and Q/R editing sites (Aruscavage and Bass, 2000; Lomeli et al., 1994; Melcher et al., 1996).
ADARs can edit RNA substrates either specifically or non-specifically depending upon the structures of the RNA substrates (Bass, 2002). In vitro studies have shown editing of up to 50% of the adenosine residues in both strands using synthetic dsRNAs that are perfectly complementary (Cho et al., 2003; Lehmann and Bass, 2000). Such non-specific editing can be explained by the presence of dsRBMs which are thought to bind dsRNA in a sequence-independent manner (Tian et al., 2004), yet it remains unclear how certain RNA substrates are edited in a site-specific fashion. Several studies have suggested that the presence of non-canonical elements in these dsRNAs–such as mismatches, bulges, and loops–could be important for site-selective A-to-I conversion (Bass, 2002; Stefl et al., 2006; Tian et al., 2004).
The dsRBMs of ADARs are not only essential for editing (Stefl et al., 2006; Valente and Nishikura, 2007), but the dsRBM also represents the second most abundant family of RNA recognition motifs. In addition to RNA editing, dsRBMs are involved in numerous post-transcriptional regulatory processes and most prominently in micro RNA (miRNA) biogenesis and function and RNA export (Dreyfuss et al., 2002; Tian et al., 2004). The few solved structures of dsRBM-containing proteins bound to short, synthetic RNA duplexes have suggested that dsRBMs recognize the A-form helix of dsRNA in a sequence-independent manner, since the majority of dsRBM-RNA interactions involve direct contact with the 2'-hydroxyl groups of the ribose sugars and direct or water-mediated contacts with non-bridging oxygen residues of the phosphodiester backbone (Gan et al., 2006; Ramos et al., 2000; Ryter and Schultz, 1998; Wu et al., 2004), and that a subclass of dsRBMs prefer stem-loops over A-form helices (Ramos et al., 2000; Wu et al., 2004).
We previously determined that each of the two dsRBMs of ADAR2 bind to a distinct location on the GluR-2 RNA encompassing the R/G editing site and that the interdomain linker (amino acids 147–231) is unstructured both in the free protein and in the complex (Stefl et al., 2006). To better understand RNA substrate recognition by ADAR2, we have determined the solution structure of the RNA helix surrounding the editing site and the solution structure of the two dsRBMs of ADAR2 bound to the GluR-2 R/G site.
RESULTS
Structure of the GluR-2 R/G RNA helix surrounding the editing site
The GluR-2 R/G site (A8) is embedded within a 71 nt RNA stem-loop containing three base-pair mismatches and capped by a 5’-GCUAA-3’ pentaloop (Figure 1A). We previously determined the structure of the apical part of the stem-loop and showed that the pentaloop is structured and adopts a fold reminiscent of a UNCG-type family of tetraloops (Stefl and Allain, 2005). Here, we have investigated the structure of the RNA helix surrounding the editing site that contains two A–C mismatches, one at the editing site (A8) and a second one ten base-pairs downstream (A18, Figure 1B). Monitoring adenine C2 chemical shifts (a sensitive probe to monitor the protonation state of N1) during a pH titration, we observed that A8 and A18 are fully protonated below pH 6.5, partially protonated between pH 6.5–8.5, and unprotonated above pH 8.5 (Figure 1H and 1I). The pKa for the adenosines N1 can be estimated between 7 and 7.5 at 310 K which is 3.3 units higher than the value determined for an isolated AMP (pKa of 4.0; Legault and Pardi, 1994). Using 863 nOe-derived distance restraints, we solved the structure of the free RNA in the protonated state (pH 6.2). The structure is well defined, even for the A–C mismatches (Figure 1E and Table 1) that are stacked inside the stem. Therefore, at pH 6.2, the R/G site has a regular A-form helix structure (Figure 1D) containing two A+–C base-pairs adopting a wobble conformation, stabilized by two hydrogen-bonds each (Figure 1F and 1G).
Figure 1.

Secondary structures of the RNAs and solution structure of GluR-2 R/G LSL RNA (A) Secondary structure of GluR-2 R/G RNA. The indicated binding regions for the dsRBMs were proposed previously (7). (B) Secondary structure of the GluR-2 R/G lower stem-loop (LSL). (C) Secondary structure of the GluR-2 R/G upper stem-loop (USL). (D) Stereo view of the most representative structure of GluR-2 R/G LSL RNA. The A+–C wobble base-pairs are highlighted in bold sticks. (E) Overlay of the 20 lowest energy structures of GluR-2 R/G LSL. The A+–C wobble base-pairs A18-C54 (F) and A8-C64 (G) are shown. (H) H2-C2 region of adenines in the 13C-1H-HSQC spectra of the GluR-B R/G LSL is shown at pH 4.7 (green peaks), 6.6 (blue peaks), 7.9 (orange peaks) and 8.9 (red peaks). The two adenines involved in the A+–C wobble base-pair showed drastic perturbation. (I) Diagram showing the pH-dependence of 13C chemical shift changes of adenine C2's.
Table 1.
NMR and refinement statistics for the gluR-B R/G upper stem-loop (USL) RNA bound to ADAR2 dsRBM1, the free gluR-B R/G lower stem-loop RNA (LSL) and its complex with ADAR2 dsRBM2, and the RDC-reconstructed complex of the full-length gluR-B R/G stem-loop (SL) RNA bound to ADAR2 dsRBM12.
| USL RNA – dsRBM1 complex | LSL RNA | LSL – dsRBM2 complex | SL RNA – dsRBM12 complex | ||||
|---|---|---|---|---|---|---|---|
| USL RNA |
dsRBM1 | LSL RNA | dsRBM2 | SL RNA | dsRBM12 | ||
| NMR distance & dihedral constraints & RDCs | |||||||
| Distance restraints | |||||||
| Total NOE | 645 | 927 | 781 | 702 | 1054 | 1252 | 1981 |
| Intra-residue | 309 | 201 | 389 | 365 | 216 | 620 | 417 |
| Inter-residue | 336 | 726 | 392 | 337 | 838 | 631 | 1564 |
| Sequential (|i–j| = 1) | 270 | 252 | 352 | 306 | 241 | 555 | 493 |
| Non-sequential (|i–j| > 1 ) | 66 | 474 | 40 | 31 | 597 | 76 | 1071 |
| Hydrogen bonds | 35 | 64 | 81a | 75 | 62 | 132 | 126 |
| Protein–RNA intermolecular | 36 | 36 | 72 | ||||
| Total dihedral angle restraints | 180 | 252 | 267 | ||||
| RNA | |||||||
| Sugar pucker | 34 | 84 | 84 | ||||
| Backboneb | 146 | 168 | 183 | ||||
| RDC restraints | 45d | ||||||
| Structure statisticsc | |||||||
| Violations (mean and s.d.) | |||||||
| Number of distance restraint violations > 0.2 Å | 8.45 ± 2.50 | 0 | 1.10 ± 1.25 | 14.31 ± 3.86 | |||
| Number of dihedral angle restraint violations > 5° | 0.7 ± 0.47 | 0 | 0 | 5.30 ± 3.32 | |||
| Max. dihedral angle restraint violation (°) | 5.82 ± 1.22 | 3.28 ± 0.77 | 2.69 ± 1.12 | 15.51 ± 2.36 | |||
| Max. distance constraint violation (Å) | 0.29 ± 0.03 | 0.16 ± 0.01 | 0.23 ± 0.06 | 0.32 ± 0.05 | |||
| Deviations from idealized geometryd | |||||||
| Bond lengths (Å) | 0.0042 ± 0.00007 | 0.0046 ± 0.00005 | 0.0041 ± 0.00005 | 0.0048 ± 0.00005 | |||
| Bond angles (°) | 1.989 ± 0.011 | 2.137 ± 0.017 | 1.903 ± 0.011 | 1.995 ± 0.008 | |||
| RDCs violations | |||||||
| Absolute RDC violations (Hz) | 1.12 ± 0.82 | ||||||
| Average pairwise r.m.s.d (Å)c | |||||||
| Protein (79–142) for dsRBM1; (221–282) for dsRBM2 | |||||||
| Heavy atoms | 1.11 ± 0.17 | 1.01 ± 0.12 | 1.60 ± 0.36 | ||||
| Backbone atoms | 0.59 ± 0.14 | 0.37 ± 0.08 | 1.22 ± 0.42 | ||||
| RNA | |||||||
| All RNA heavy atoms | 0.60 ± 0.16 | 1.15 ± 0.35 | 1.48 ± 0.51 | 1.30 ± 0.40 | |||
| Complex | |||||||
| All complex heavy atoms | 1.01 ± 0.15 | 1.49 ± 0.39 | 1.75 ± 0.31 | ||||
In the final structure calculations of the free RNA, H-bond restraints were applied in the two A–C mismatches. This is based on initial structures and on the protonation state of A8/A18. For the structures of the RNA in complex no H-bond restraints for the two A–C mismatches have been applied.
Based on A-form geometry derived from high-resolution crystal structures: α(270°–330°), β(150°–210°), γ(30°–90°), δ(50°–110°), ε(180°–240°), and ζ(260°–320°). These restraints were used only for the double-helical region. No angle restraints were imposed on the two A–C mismatches and the loops.
Calculated for an ensemble of the 20 lowest energy structures.
16 RDCs of dsRBM1 and 29 RDCs of dsRBM2.
Structure of ADAR2 dsRBMs bound to their respective RNA targets
Considering the distinct RNA binding location found previously for each dsRBM (Stefl et al., 2006) and the high molecular weight (over 50 kDa) of the complex formed between the two dsRBMs of ADAR2 and the GluR-2 R/G substrate (Figure 1A), we adopted a modular approach to solve the structure of this complex in solution. To this end, we first solved the structure of dsRBM1 in complex with a modified GluR-2 upper stem–loop (USL, Figure 1C, and Figure S1) and then the structure of dsRBM2 bound to the GluR-2 lower stem-loop that contains the editing site (LSL, Figure 1B, and Figure S2). The use of a GluR-2 R/G USL mutant to determine the structure of dsRBM1 in complex with RNA was dictated by the poor data quality that we obtained with the wild-type (WT) sequence. In changing the loop sequence to that found in the GluR-3 USL (Aruscavage and Bass, 2000), we obtained a smaller and more stable RNA which provided NMR data of higher quality.
A total of 1707 and 1929 nOe-derived distance restraints (including 36 intermolecular ones for each complex) for ADAR2 dsRBM1–GluR-2 R/G USL mutant and ADAR2 dsRBM2–GluR-2 R/G LSL complexes, respectively, were used to obtain well-defined structures (Figure 2, Table 1). The two dsRBM-RNA complexes are stabilized by a combination of hydrophobic interactions, hydrogen-bonding and electrostatic contacts. In both dsRBM–RNA complexes, the dsRBMs adopt the expected αβββα topology in which the two α-helices are packed along the three-stranded antiparallel β-sheet. The entire interaction surface spans 12–14 base-pairs covering two minor grooves and a major groove (Figure 2). In both complexes, three distinct regions of the dsRBMs are involved in interaction with RNA. The first region is the helix α1, which interacts with the first minor groove of the RNA. The second region is a well-conserved KKNAK-motif, located at the amino-terminal tip of helix α2 and the preceding loop, that contact the RNA with non-sequence specific contacts between lysine side-chains and the phosphate oxygens across the major groove of the RNA (Lys127, 128, and 131 for dsRBM1 and Lys281, 282, 285 for dsRBM2, Figure 2). In addition, the dipole moment of helices α2 creates a positive charge in the N-terminal tip of these helices that interacts with the negatively charged phosphate backbone. This second set of interactions are mediated by the main-chain amides of K127 and K281, which are hydrogen bonded with the phosphates oxygen of A24 and U11, respectively (Figure 2). The third region of contact is the β1–β2 loop which interacts with the second minor groove of the RNA. The overall architecture of these two complexes resembles other previously determined dsRBM–RNA structures (Blaszczyk et al., 2004; Gan et al., 2008; Gan et al., 2006; Ramos et al., 2000; Ryter and Schultz, 1998; Stefl et al., 2005a; Wu et al., 2004). However, a detailed inspection of the interaction regions revealed striking differences between the two complexes and other dsRBM-RNA complexes, particularly in the first and the third regions where both dsRBMs present unexpected sequence-specific contacts to the RNA minor grooves (Figure 2).
Figure 2.
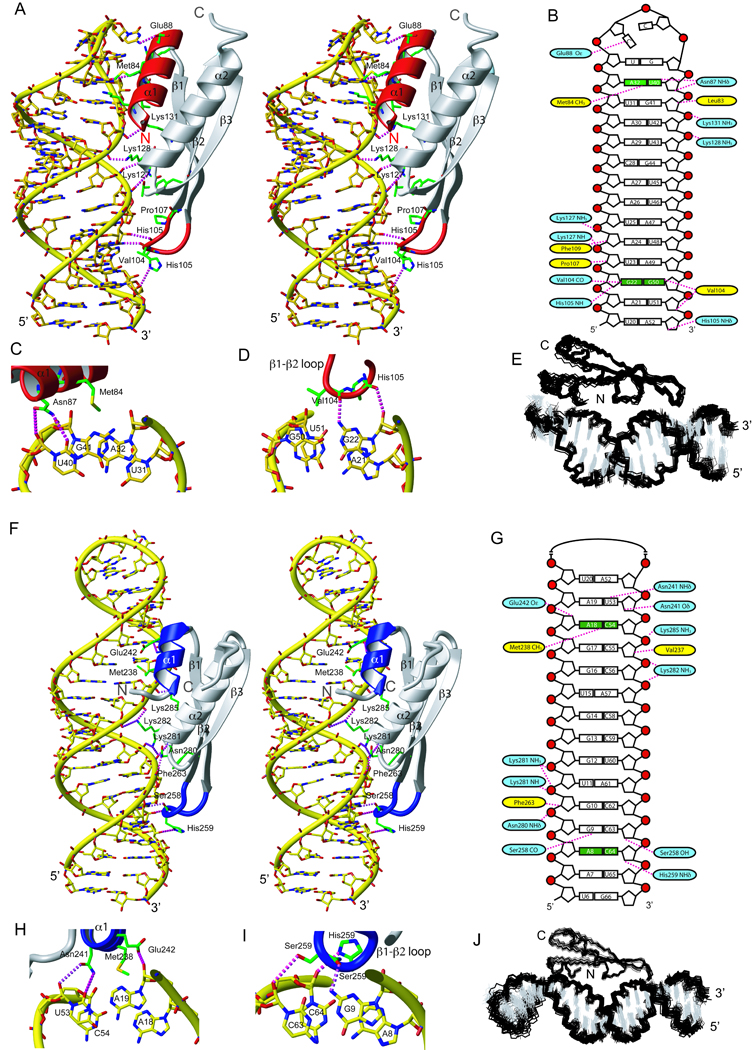
RNA recognition by ADAR2 dsRBM1 and dsRBM2. (A) Stereo view of the most representative structure of dsRBM1 bound to USL RNA. The RNA is represented as a yellow stick model and the protein is shown as a ribbon model with residues that contact the RNA shown in green. Helix α1 and the β1–β2 loop that mediate the sequence-specific contacts are colored in red. Hydrogen-bonds are indicated by magenta dotted lines (B) Scheme showing contacts between dsRBM1 and the USL RNA. Protein residues that form hydrogen-bonds to the RNA are shown in blue and the one having hydrophobic interactions are in yellow. Close-up view of minor groove sequence-specific recognitions mediated by helix α1 (C) and the β1–β2 loop (D) of dsRBM1. (E) Overlay of the 20 lowest energy structures of the dsRBM1-USL complex. (F) Stereoview of the most representative structure of the dsRBM2 bound to LSL RNA. Helix α1 and the β1–β2 loop that mediate the sequence-specific contacts are colored in blue. (G) Scheme showing contacts between dsRBM2 and the LSL RNA. Close-up view of the minor groove sequence-specific recognitions mediated by helix α1 (H) and the β1–β2 loop (I). (J) Overlay of the 20 lowest energy structures of the dsRBM2-LSL complex. For NMR data of these two complexes, see also Figure S1 and S2.
Sequence-specific recognition by ADAR2 dsRBM1
In the ADAR2 dsRBM1–RNA complex, contacts from helix α1 are centered at the A32-U40 base-pair below the UCCG tetraloop (Figure 2A and 2C). Met84 makes a sequence-specific hydrophobic contact with H2 of A32 and Asn87 contacts the 2’-hydroxyl and O2 of U40. The Oε of Glu88 is hydrogen-bonded to the amino group of the first cytosine of the tetraloop. In addition, Leu83 makes hydrophobic contacts with the sugar of G41. The entire helix α1 is tightly inserted in the minor groove created by the UCCG tetraloop and two adjacent base-pairs (Figure 2A). The β1–β2 loop of dsRBM1 binds the following minor groove of the RNA. This minor groove is widened as it has to accommodate base-pairing of two guanosines that make an N1 symmetrical G22–G50 mismatch (Figure 2A and 2D) that are the center of this interaction. Val104 side-chain contacts the H8 of G50 (that adopts a syn conformation) and a sequence-specific hydrogen-bond is formed between the main-chain carbonyl of V104 and the amino group of G22. The widened minor groove accommodates additional interactions between three side-chains (Phe109, Pro107, His105) and the sugars of the base-pairs above and below. Altogether, dsRBM1 binds the RNA stem-loop at a single register via two sequence-specific contacts at two consecutive RNA minor grooves: a hydrogen-bond to the amino-group of the G22 in the GG mismatch via the β1–β2 loop and an hydrophobic contact to the adenine H2 of A32 via Met84 in helix α1.
Sequence-specific recognition by ADAR2 dsRBM2
The dsRBM2 of ADAR2 is adjacent to the deaminase domain and is essential for A-to-I editing at the R/G site (Stefl et al., 2006; Xu et al., 2006). In the ADAR2 dsRBM2–GluR-2 R/G LSL complex, Asn241, Glu242, Met238, Val 237 of helix α1 contact the minor groove region centered at the A18-C54 mismatch (Figure 2F and 2H). At pH 7.6, where the protein-RNA complex has been determined, this mismatch is unprotonated and Met238 makes a sequence-specific hydrophobic contact with A18 H2. Contacts to the base-pair above and below by Asn241 and Glu242, and by Val 237, respectively, further stabilize the interaction of helix α1 in this region (Figure 2H). The β1–β2 loop of dsRBM2 interacts with the second minor groove. The contacts are centered at the G9-C63 Watson-Crick base-pair located above the A8-C64 mismatch containing the editing site. A sequence-specific hydrogen-bond is formed between the main-chain carbonyl of Ser258 and the amino of G9 (Figure 2F and 2I). Additionally, non-sequence specific contacts between the side-chains of Ser 258, His 259 and Phe 263 and the G9-C63 base-pair and the base-pairs above and below increase the stability of the interaction with the RNA minor groove (Figure 2G). In the vicinity of the editing site, dsRBM2 contacts C63, while A8 is not contacted by any residue from the β1–β2 loop therefore making A8 accessible to the deaminase domain. Altogether, dsRBM2 similar to dsRBM1, recognizes the RNA helix via two sequence-specific contacts at two consecutive RNA minor grooves: a hydrogen-bond to the amino-group of the G9 at the GC 3’ to the editing site via the β1–β2 loop and a hydrophobic contact to the adenine H2 of A18 via Met238 in helix α1. In the NMR spectra (data not shown), we could observe intermolecular nOes corresponding to dsRBM2 being positioned at a second binding register one base-pair above (although with only 20% occupancy). In this case the β1–β2 loop contact G10 and Met 238 contact A19. Although two consecutive binding sites for dsRBM2 are observed here, they both confirm the sequence-specific nature of the dsRBM2-RNA interaction.
Structure of ADAR2 dsRBM12 in complex with GluR-2 R/G RNA
Next, we determined the structure of ADAR2 dsRBM12 in complex with GluR-2 R/G RNA (Figures 3A and 3B). To calculate an atomic model of this complex, we used the distance constraints measured in the two sub-complexes described above (Figure 3C). This strategy could be used considering (i) the distinct RNA binding location for each dsRBMs, with no mutual interactions (Stefl et al., 2006), (ii) the flexible unstructured linker connecting dsRBM1 and dsRBM2 in the complex (Stefl et al., 2006) and (iii) an overlap in the RNA sequence of the joint region of the subcomplexes (Figure 1). Long-range structural constraints for this elongated complex were derived from residual dipolar couplings (RDCs) measured with a deuterated protein on the full-length complex (dsRBM12 bound to GluR-2 R/G RNA, Figure 1A). The pentaloop which is not contacted by dsRBM1 was modeled using the structure that was determined previously (Stefl and Allain, 2005). With this strategy, we could then determine a precise solution structure of this 50 kDa complex using 45 15N-1H RDCs (Figure 3D, Table 1). In the structure, the two dsRBMs bind one face of the RNA covering approximately 120 degrees of the space around the RNA helix (Figure 3B). This suggests that the binding of an additional molecule of ADAR2 would be sterically possible, consistent with studies indicating that ADAR2 dimerization is necessary for RNA editing (Chilibeck et al., 2006; Cho et al., 2003; Gallo et al., 2003; Valente and Nishikura, 2007).
Figure 3.
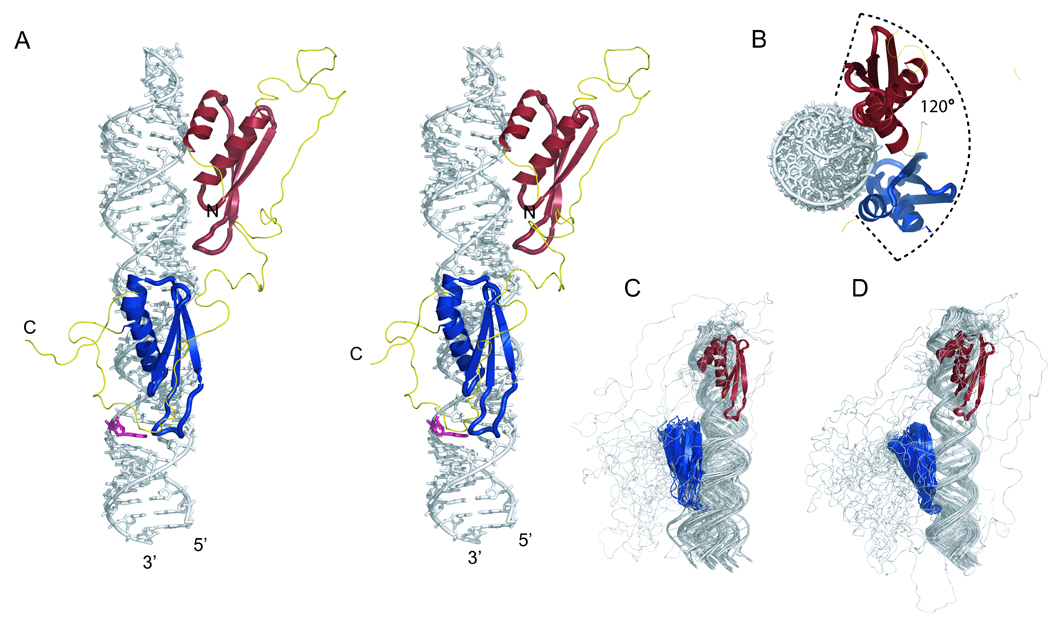
Structure of ADAR2 dsRBM12 bound to GluR-2 R/G. (A) Stereo view of the most representative RDC-reconstructed structure of the ADAR2 dsRBM12 bound to GluR-2 R/G. The RNA is represented as a stick model (in grey; the edited adenosine is highlighted in pink) and the protein is shown as a ribbon model (dsRBM1 in red; dsRBM2 in blue; linker in yellow). (B) Top view of the complex. Overlay of the 20 lowest energy structures calculated without (C) and with RDCs (D).
Sequence-specific contacts of ADAR2 dsRBMs are important for binding affinity
To confirm the ADAR2 dsRBMs sequence-specific preference in a quantitative solution binding assay, we performed fluorescence anisotropy (FA) experiments by titrating dsRBM1 and dsRBM2 against a labeled USL and LSL RNAs, respectively. Unlabeled wild-type and mutant RNAs (Figure S3) were used for competition experiments as described in Experimental procedures. The equilibrium dissociation constants were calculated from the displacement of the binding curves (Figure 4). We designed two sets of mutations, one set was designed to change the recognition sequence of USL and LSL RNAs (Figures 4A and 4C and Figure S3) and a second set was designed to maintain the recognition sequence, but change the RNA shape via mismatches of USL and LSL RNAs into Watson-Crick base-pairs (Figures 4B and 4D and Figure S3) to measure their effect on overall binding affinity. In mutating any of the bases that are recognized in a sequence-specific manner by dsRBM1 in USL (G22, A32 or C34), the apparent affinity is reduced compared to the wild-type (Figures 4A and 4B). However when the G22–G50 mismatch is replaced by a Watson-Crick G22–C50 pair, the affinity is almost identical to wild-type RNA, confirming that dsRBM1 recognizes the sequence rather than the shape of the RNA helix (note that G41 was mutated in the first RNA mutant to prevent the sequence-specific recognition of G41 by dsRBM1). Similarly for the LSL, when G9 or A18 are mutated, dsRBM2 binding is reduced more than five-fold (Figure 4C), yet when the two AC mismatches are replaced by Watson-Crick AU pairs, the affinity is only reduced by two-fold (Figure 4D). In this latter context, the sequence-specific contacts are the same for the WT and mutant RNAs, but the presence of a more deformable A18-C54 base-pair in the WT structure could explain the higher affinity of dsRBM2 to the WT RNA (note that additional mutations were introduced in the first two RNA mutants of LSL to abolish the two binding registers found in the wild-type LSL). Altogether, the FA data strongly support the idea that the sequence-specific interactions observed in the structures of ADAR2 dsRBMs-dsRNA are important for the affinity of both dsRBMs and that they finely tune the preferential binding to these recognition motifs.
Figure 4.

ADAR2 dsRBMs bind preferentially to RNAs that contains their sequence-specific recognition motifs. (A) ADAR2 dsRBM1 was titrated with fluorescently labeled USL and binding was measured by fluorescence anisotropy (black circles; fluorescein labeled reference, Flc-USL). The same experiment was then carried out in the presence of competing unlabeled USL wt (○), USL G22A/G50U/G41A mutant (▼), and USL A32G mutant (△). Equilibrium dissociation constants (Kd) were calculated from the best fit to the data as described in Experimental procedures. (B) The same assay as shown in (A) but for USL C34U mutant (▼) and USL G50C mutant (△). (C) ADAR2 dsRBM2 was titrated with fluorescently labeled LSL and binding was measured by fluorescence anisotropy (●; fluorescein labeled reference, Flc-LSL). The same experiment was then carried out in the presence of competing unlabeled LSL wt (○), LSL G9A/G10A/C62U/C63U mutant (▼), and LSL A18G/A19G/U53C mutant (△) (D). The same assay as shown in (C) but for LSL C54U/C64U mutant (▼).Wild-type and mutant sequences are shown in Figure S3.
Sequence-specific contacts of ADAR2 dsRBMs are important for editing
To test the functional importance of the four sequence-specific contacts identified in the ADAR2 dsRBM12-GluR-2 R/G RNA complex, single amino acid mutants in helix α1 (M84 or M238) were mutated to alanine or double mutants in the β1–β2 loop in either dsRBM1 or dsRBM2 were evaluated for their ability to edit the wild-type GluR-2 R/G site (Figure 5A). It was necessary to generate double mutants around the carbonyls of V104 in dsRBM1 and S258 in dsRBM2 to change the structure of the main-chain of this loop. All four mutants showed a significant decrease in RNA editing ranging from a near ablation of editing (S258A,H259A in the β1–β2 loop of dsRBM2), to 20% editing (V104A,H105A in the β1–β2 loop of dsRBM1 and M84A in helix α1 of dsRBM1), to 30% editing (M238A in helix α1 of dsRBM2) of that demonstrated by the wild-type protein. These data clearly show that the loss of the sequence-specific contacts of any of the two dsRBMs strongly decreases editing at the R/G site with the contact mediated by the β1–β2 loop of dsRBM2 more strongly affecting editing than the other contacts. In agreement with deletion studies of ADAR2 (Macbeth et al., 2004; Stefl et al., 2006), the S258A,H259A mutations have a stronger effect, likely due to the binding of the β1–β2 loop of dsRBM2 near the editing site.
Figure 5.

Sequence-specific contacts of ADAR dsRBMs are important for editing activity. (A) Quantitative analysis of in vitro editing efficiency for ADAR2 dsRBM double mutants; all mutants were assayed in duplicate for in vitro editing activity at the GluR-2 R/G site using three independent nuclear extracts (mean ± SEM; *p<0.05, **p<0.005; ***p<0.001). (B) Kinetic analysis of wild-type ADAR2 editing with GluR-2 R/G mutants. Increasing concentrations of GluR-2 RNAs (see Figure S4; wild-type ●; mut 1 ■, mut 2 ○, mut 3 ▲, mut 4 □) were incubated with wild-type rat ADAR2 protein as described above; all mutant RNAs were assayed in triplicate for determination of in vitro reaction velocity (mean ± SEM). Non-linear fitting of kinetic curves corresponded to a model of substrate inhibition (R2 = 0.91–0.98 for all RNAs) with Vmax values corresponding to 3.92, 3.84, 2.08, 1.20 and 1.29 fmol/min for wild-type, mut1, mut2, mut3 and mut4, respectively. Wild-type and mutant sequences are shown in Figure S4.
Converse experiments in which mutations in the sequence-specific recognition motifs of dsRBM2 (mut1 and mut2), dsRBM1 (mut4) or both (mut3) within the GluR-2 RNA (Figure S4) were assessed for their ability to affect R/G editing by wild-type ADAR2 revealed a significant decrease in maximal editing rates (Vmax) for all RNA mutants tested (Figure 5B) providing further support for the functional significance of these contacts. Best-fit kinetic curves for wild-type and mutant RNAs corresponded to a model of substrate inhibition, consistent with previously observed kinetic models for ADARs in which the formation of a ternary complex containing an ADAR dimer and RNA substrate is required for efficient adenosine deamination (Hough and Bass, 1994; Jaikaran et al., 2002).
DISCUSSION
In solving the structure of ADAR2 dsRBMs bound to the GluR-2 R/G site, we demonstrated that despite forty-four possible binding sites on the GluR-2 R/G RNA stem-loop (considering a 32 base-pair stem, a 10 base-pair register between the two sequence-specific contacts and two possible orientations for the dsRBM), each dsRBM binds at a very specific register on this large RNA molecule. This binding is achieved by a direct readout of the RNA sequence in the minor groove of the A-form helix. The two dsRBMs of ADAR2 use helix α1 and the β1–β2 loop as molecular rulers to find their binding register in the RNA minor groove of the GluR-2 R/G RNA. Through the β1–β2 loop, the carbonyl oxygens of Val104 in dsRBM1 and Ser258 in dsRBM2 contact the amino-groups of base-paired guanines, G22 and G9 respectively. The same type of sequence-specific RNA recognition of GC or GU base-pairs in the minor groove of RNA helices have been observed in several ribosomal proteins of the large subunit (Klein et al., 2004) and in some tRNA synthetases bound to RNA (Rould et al., 1989) although the fold of these proteins and the overall binding mode are different from a dsRBM. Through helix α1, the side-chain methyl groups of Met84 in dsRBM1 and of Met238 in dsRBM2 are in contact with the H2s of A32 and A18, respectively. Recognition of these two anchoring points in the minor-groove, separated by 9 and 8 base-pairs for dsRBM1 and dsRBM2, respectively, illustrates how the two dsRBMs find their sequence-specific binding registers, demonstrating that these dsRBMs have more sequence-specificity than previously thought. Interestingly, in each complex, one of the two anchoring points involves a mismatched base-pair (the G22–G50 base-pair for dsRBM1 and the A18-C54 base-pair for dsRBM2). It is therefore possible that the highly exposed amino or C2H2 groups of these mismatches in the minor groove further assist the dsRBMs of ADAR2 to find their binding register, supporting earlier findings that these two mismatches are important for positioning ADAR2 at the R/G site (Ohman et al., 2000). In addition to sequence-specific interactions between ADAR2 dsRBMs and its GluR-2 target, K127 (dsRBM1) and K281 (dsRBM2) make contacts with phosphate oxygens across the major groove of the RNA (Figure 2). These basic amino acid moieties are conserved in the loop between the β3 and α2 regions for all dsRBMs (Tian et al, 2004) and mutation of these residues in PKR and Staufen have been shown to ablate dsRNA-binding activity (McMillan et al., 1995; Ramos et al., 2000), indicating the importance of both sequence-specific and sequence–independent recognition of the RNA substrate for site-specific adenosine deamination.
Prior to this work, the structures of only four dsRBM-containing proteins in complex with RNA had been determined by X-ray crystallography (XlrbpA and Aquifex aeolicus (Aa) RNAseIII) or NMR spectroscopy (Staufen and Rnt1p; Gan et al., 2006; Ramos et al., 2000; Ryter and Schultz, 1998; Wu et al., 2004). In the two solution structures, the dsRBMs appear to recognize primarily the loop of the RNA while in the two crystal structures the dsRBMs are found bound across the junction between coaxially stacked helices. Lack of clear sequence-specific contacts led to the general opinion that dsRBMs are shape-specific rather than sequence-specific RNA binding domains (Stefl et al., 2005a). The two dsRBM-RNA complexes of ADAR2 reported here have revealed that dsRBMs recognize not only the shape of the RNA (a stem-loop for dsRBM1 and an A-form helix for dsRBM2), but also more surprisingly the sequence of the RNA. Interestingly, in a recent crystal structure of an Aa RNAseIII dsRBM bound to a stem-loop, sequence-specific contacts in the minor groove via helix α1 and the β1–β2 loop have been observed (Gan et al., 2008). The helix α1 in Aa RNAseIII is elongated by one turn compared to the helix α1 of the dsRBMs of ADAR2 and a Gln side-chain recognizes a guanine by two sequence-specific hydrogen-bonds (Figure S5). The contact mediated by the β1–β2 loop in Aa RNAseIII are similar to the dsRBMs in ADAR2. The β1–β2 loop has the same length (six amino-acids) and the main-chain carbonyl of the third residue of the loop is hydrogen-bonded to a guanine amino of a GU base-pair. Despite similarities in the mode of binding, the three dsRBMs recognize different sequences and different register lengths. The dsRBM of Aa RNAseIII preferentially recognizes an RNA helix containing a G-X10-G sequence while the dsRBM1 and dsRBM2 of ADAR2 preferentially recognize G-X9-A and G-X8-A sequences, respectively (Figure 6). The length and the positioning of helix α1 relative to the dsRBM fold appear to be the key structural elements that determine the register length of the different dsRBMs (Figure 6C).
Figure 6.
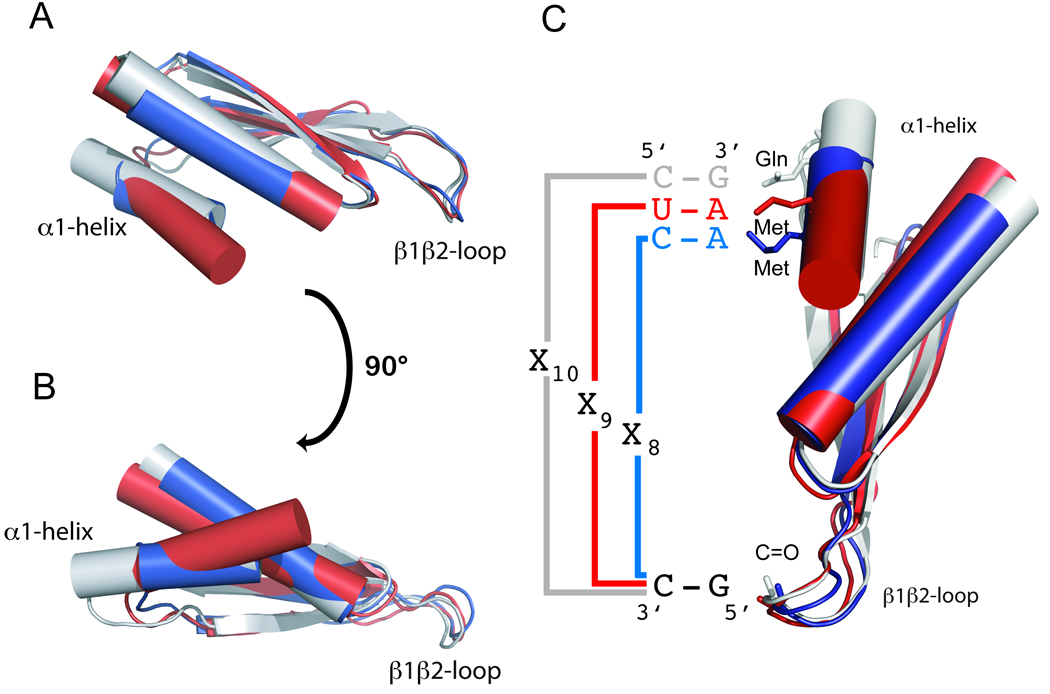
RNA recognition code of various dsRBMs. (A and B) Overlay of the ADAR2 dsRBM1 (in blue), ADAR2 dsRBM2 (in red), and Aquifex aeolicus RNAseIII dsRBM (in grey) structures highlights the variability of helix α1 within the dsRBM fold and its importance for the determination of the register length between the two specific contacts on the RNA helix (C). For Aquifex aeolicus RNAseIII dsRBM–dsRNA interactions, see also Figure S5 and for sequence alignments of different dsRBMs, see also Figure S6.
Our findings regarding the RNA binding specificity of dsRBMs have important implications for the sequence-specificity paradox of ADAR2, but also of many other dsRBM-containing proteins that continue to puzzle investigators (Tian et al., 2004). Apparent differences in the sequences of dsRBMs between mammalian ADAR2 and ADAR1 (Figure S6), where ADAR1 dsRBMs appears to have a longer helix α1 and lack the ADAR2 equivalent of Met 84 and Met 238, could explain why ADAR1 and ADAR2 have different substrate specificities (Bass, 2002; Lehmann and Bass, 2000). Furthermore, our structure shows how dsRBM2 of ADAR2 binds the GluR-2 R/G site near the editing site in recognizing the amino group of the guanosine 3’ to the edited A. This would explain the strong preference for a guanosine moiety 3’ to the edited adenosine that is found in a great majority of substrates selectively edited by ADAR2 (Bass, 2002; Lehmann and Bass, 2000; Li et al., 2009; Riedmann et al., 2008) and more recently in long double-stranded RNA (Eggington and Bass, personal communication). This sequence preference disappears when the dsRBMs are deleted from ADAR2 (Eggington and Bass, personal communication) further supporting that this sequence requirement is due to dsRBM binding. Finally, in interacting with the guanosine 3’ to the edited adenosine and to the nucleotide that base-pairs with the editing site, dsRBM2 not only brings the deaminase domain in close proximity to the editing site, but also does not prevent access of the adenosine to the deaminase domain. When this precise positioning is impaired, specific editing is nearly abolished (see the effect of the S258A, H259A mutant) which emphasizes the functional importance of sequence-specific recognition of RNA by dsRBMs for A-to-I editing.
The sequence-specific contacts that we observed with the dsRBMs ADAR2 are interesting when comparing sequence alignments of several dsRBM structures that have been determined (Figure S6). This alignment reveals a surprisingly high variability in the length and amino-acid sequence composition of the two regions of the dsRBMs mediating the sequence-specific interactions with the RNA, namely the helix α1 and the β1–β2 loop. This strongly suggests that dsRBMs are likely to have different binding specificity in agreement with reports indicating that dsRBMs from different proteins are not functionally interchangeable (Liu et al., 2000; Parker et al., 2008). Similar to ADAR2, many dsRBM-containing proteins involved in miRNA and siRNA processing and function are likely to bind RNA in a sequence-specific manner, that would modulate their target selection and mechanism of action. For example, DICER was shown to compete with ADARs for the same RNA substrates (Kawahara et al., 2007; Yang et al., 2006). Interestingly, ADARs modulate the processing of miRNA precursors not only by A-to-I modifications that alter the secondary structure of pri-miRNA (Kawahara et al., 2007; Tonkin and Bass, 2003; Yang et al., 2006), but also simply by RNA-binding alone to pri-miRNAs, as recently shown with catalytically inactive ADARs (Heale et al., 2009). This latter function for ADARs, as regulators of pri-miRNA processing, closely resemble that found for single-stranded sequence-specific RNA-binding proteins such as Lin28, hnRNP A1 or KSRP (Guil and Caceres, 2007; Heo et al., 2008; Heo et al., 2009; Michlewski et al., 2008; Newman et al., 2008; Trabucchi et al., 2009). Furthermore, RNAi activity has been shown to coincide with siRNA sequence motifs (Katoh and Suzuki, 2007). Altogether it is becoming clear that sequence-specific recognition mediated by dsRBMs is functionally important for dsRBM containing proteins. We have demonstrated here with ADAR2 how such sequence-specific recognition is mediated in dsRBMs and how this is relevant for RNA editing. Future work will be required to elucidate the variations in dsRNA-binding specificity and their functional relevance for numerous other members of the dsRBM-containing protein family.
EXPERIMENTAL PROCEDURES
Preparation of proteins
Details on cloning, expression and purification of the ADAR2 dsRBM1, ADAR2 dsRBM2, and ADAR2 dsRBM12 constructs have been described previously (Stefl et al., 2005b; Stefl et al., 2006).
NMR spectroscopy
All NMR spectra were acquired at 310 K. Spectra were recorded at 500, 600 and 900 MHz Bruker spectrometers. All spectra were processed with XWINNMR or Topspin1.3/2.0 (Bruker BioSpin) and analyzed with Sparky 3.0 (Goddard T.G. and Kellner D.G., University of California, San Francisco). The 1H, 13C and 15N chemical shifts of the protein in complex, were assigned by standard methods (Sattler et al., 1999). The 1H-15N HSQC and 1H-13C HSQC spectra of dsRBM1 and dsRBM2 in free and bound forms are shown in Figures S1 and S2. All distance restraints were derived from 3D 15N,13C-edited NOESYs and 2D 1H-1H NOESY (tm=150 ms) collected at 900 MHz. RNA exchangeable proton resonances were assigned using 1H-1H NOESY spectrum (tm=200 ms) at 278 K. Non-exchangeable proton resonances were assigned using 1H-1H, NOESY, 1H-1H TOCSY, 1H-13C HSQC, 3D 13C-edited NOESY, 2D 1H-1H double-half-filtered NOESY (tm=150ms) (Peterson et al., 2004) and 3D 13C F1-edited, F3-filtered NOESY-HSQC spectrum (tm=150ms) (Zwahlen et al., 1997) in 99.99% 2H2O (v/v). The NOEs were semi-quantitatively classified based on their intensities in the 2D and 3D NOESY spectra. Hydrogen-bond distance restraints were used for base-pairs, when the imino-protons were observed experimentally. The assignments of intermolecular NOEs were based on 3D 13C F1-edited, F3-filtered NOESY-HSQC spectrum (tm=150ms), 2D 1H-1H F1-13C-filtered F2-13C-edited NOESY (tm=150ms) on the protein-RNA complexes with either the protein or the RNA 13C-15N labeled. In case of dsRBM2–GluR-2 R/G LSL RNA complex, we observed an extra set of five weaker intermolecular nOes, which were discarded from structure calculation. These intermolecular restraints cannot be explained with the presented structure of dsRBM2-GluR-2 R/G LSL RNA complex. They originate from a minor conformation in which the protein is shifted up by one base pair towards the UUCG tetraloop.
Structure calculation and refinement
Distance constraints for the proteins bound to RNA where generated by the ATNOS/CANDID package (Herrmann et al., 2002). The accuracy of the list of automatically generated distance constraints was manually checked. Distance constraints for the free and bound RNAs as well as for the intermolecular NOEs were assigned manually. Preliminary structures of the free RNA and the protein-RNA complexes were obtained by a simulated annealing protocol in CYANA (Guntert et al., 1997; Herrmann et al., 2002). To impose better convergence of the ensemble, an artificial torsion angles for the canonical dsRNA regions were used as described previously (Oberstrass et al., 2006). Additional angle restraints to maintain proper local geometries were used (Tsui et al., 2000). The final refinement of all structures was performed using a 20 ps simulated annealing protocol in AMBER (Case, 2002) as described in supplementary material. From 40 refined structures, the twenty conformers with the lowest AMBER energy were selected to form the final ensemble of structures. Structural quality was assessed using PROCHECK (Laskowski et al., 1996). Figures were prepared with MOLMOL (Koradi et al., 1996) and Pymol (DeLano, 2002)
Fluorescence anisotropy
Fluorescence anisotropy was measured on a FluoroMax-4 spectrofluorometer (Horiba Jobin-Yvon, USA) equipped with a thermostated cell holder and an automatic titrator. All measurements were conducted in 50mM sodium phosphate buffer (pH 7.0) and 100mM NaCl at 10°C. To avoid any effects caused by 5’-end labeling of RNAs, the experiments were designed as a competition assay. At first, a reference measurement was carried out in which 1400 µl of 10nM fluorescein labeled wild type RNA was titrated by the protein. Then, the same titration experiment was repeated in the presence of 500nM unlabeled RNA (either wild type or mutants; Vasiljeva et al., 2008). Total volume of protein added to each reaction was 33 µl. The fitting was performed using DynaFit software (Kuzmic, 1996, 2006). Initially, the Kd for the reference protein–labeled RNA complex was determined. The obtained Kd value was then used as a fixed parameter when fitting the competition data. A 1:1 binding stoichiometry was assumed in all cases. The data were normalized for visualization purposes.
Quantitative analysis of in vitro RNA editing
For in vitro editing reactions, a 116 nt RNA encoding a portion of the mouse GluR-2 pre-mRNA with the complete R/G duplex was transcribed in vitro (Stefl et al., 2006) and incubated with wild-type or mutant ADAR2 proteins derived from nuclear extracts obtained from transiently transfected HEK293 cells (Sansam et al., 2003). Equivalent amounts of wild-type and mutant ADAR2 protein, as determined by Western blotting, were incubated with 40 ng of the R/G transcript at 30°C for 20 minutes. These incubation conditions were determined empirically by performing time-course analyses with wild-type ADAR2 protein to ensure that the assay was in the linear range (data not shown). The reaction was stopped and the R/G transcript isolated by direct addition of TRI Reagent (Molecular Research Center) at the end of the incubation period. For quantification of RNA editing, the in vitro reaction product was reverse transcribed using AMV Reverse Transcriptase (Promega) and an antisense primer (5’-CGGCCAATCGTACGTACCTCCGGCCGAATTCTACAAACCGTTAAGAGTCTTA-3’) with a unique 5’-extension (underlined). The resulting amplicon was diluted 1:1000 and 1 µl was subsequently amplified by PCR using sense (5’-CCGGGAGCTCATCGCCACACCTAAAGGATCC-3’) and antisense (5’-CGGCCAATCGTACGTACCTCC-3’) primers corresponding to GluR-2 and the unique 5’-extension sequences, respectively. PCR amplicons were purified using the Wizard SV PCR and Gel Cleanup System (Promega) and digested with Mse I (New England Biolabs) to generate 100 and 70 bp products representing edited and non-edited transcripts, respectively. The resulting digestion products were resolved on a 4% Agarose gel and editing efficiency was quantified by phosphorimager analysis (GE Healthcare).
In vitro editing reactions using GluR-2 R/G mutant RNAs were performed as described above with equivalent amounts of wild-type ADAR2 protein derived from nuclear extracts obtained from transiently transfected HEK293 cells (Sansam et al., 2003). Wild-type and mutant transcripts were trace labeled with [α-32P]-UTP and concentrations of in vitro transcribed RNAs were determined using a Perkin-Elmer Tri-Carb 2800TR scintillation spectrometer based upon the calculated specific activity for each transcript.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the Swiss National Science Foundation (Nr. 3100A0-118118) and the SNF-NCCR structural biology to F.H.T.A and the National Institutes of Health (R01 NS33323) to R.B.E. R.S. is supported by the Ministry of Education of the Czech Republic (MSM0021622413, Ingo LA08008), GACR (204/08/1212, 305/10/1490), GAAV (IAA401630903), HHMI/EMBO start-up grant, and HFSP Career Development Award. M.Z. and C.H. are supported by GACR (204/08/H054) and by the Ministry of Education of the Czech Republic (MSM0021622415). M.Z. is in receipt of a Brno City Scholarship for Talented Ph.D. Students. The coordinates of the structures of GluR-2 LSL RNA, ADAR2 dsRBM1 bound to GluR-2 USL RNA, ADAR2 dsRBM2 bound to GluR-2 LSL RNA and ADAR2 dsRBM12 bound to GluR-2 have been deposited in the Protein Data Bank with accession codes 2l2j, XXX, 2l2k, and XXX, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL DATA
Supplemental data include six Supplemental Figures and supplemental experimental procedures that can be found with this article online at http://www.cell.com.
References
- Aruscavage PJ, Bass BL. A phylogenetic analysis reveals an unusual sequence conservation within introns involved in RNA editing. RNA. 2000;6:257–269. doi: 10.1017/s1355838200991921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczyk J, Gan J, Tropea JE, Court DL, Waugh DS, Ji X. Noncatalytic assembly of ribonuclease III with double-stranded RNA. Structure. 2004;12:457–466. doi: 10.1016/j.str.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Case DA, Pearlman DA, Caldwell JW, Cheatham TE, III, Wang J, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Gohlke H, Radmer RJ, Duan Y, Pitera J, Massova I, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER. Vol. 7. San Francisco: University of California; 2002. [Google Scholar]
- Chilibeck KA, Wu T, Liang C, Schellenberg MJ, Gesner EM, Lynch JM, MacMillan AM. FRET analysis of in vivo dimerization by RNA-editing enzymes. J Biol Chem. 2006;281:16530–16535. doi: 10.1074/jbc.M511831200. [DOI] [PubMed] [Google Scholar]
- Cho DS, Yang W, Lee JT, Shiekhattar R, Murray JM, Nishikura K. Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. J Biol Chem. 2003;278:17093–17102. doi: 10.1074/jbc.M213127200. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- Gallo A, Keegan LP, Ring GM, O'Connell MA. An ADAR that edits transcripts encoding ion channel subunits functions as a dimer. EMBO J. 2003;22:3421–3430. doi: 10.1093/emboj/cdg327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan J, Shaw G, Tropea JE, Waugh DS, Court DL, Ji X. A stepwise model for double-stranded RNA processing by ribonuclease III. Mol Microbiol. 2008;67:143–154. doi: 10.1111/j.1365-2958.2007.06032.x. [DOI] [PubMed] [Google Scholar]
- Gan J, Tropea JE, Austin BP, Court DL, Waugh DS, Ji X. Structural insight into the mechanism of double-stranded RNA processing by ribonuclease III. Cell. 2006;124:355–366. doi: 10.1016/j.cell.2005.11.034. [DOI] [PubMed] [Google Scholar]
- Guil S, Caceres JF. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat Struct Mol Biol. 2007;14:591–596. doi: 10.1038/nsmb1250. [DOI] [PubMed] [Google Scholar]
- Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- Heale BS, Keegan LP, McGurk L, Michlewski G, Brindle J, Stanton CM, Caceres JF, O'Connell MA. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009 doi: 10.1038/emboj.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let -7 precursor MicroRNA. Mol Cell. 2008;32:276–284. doi: 10.1016/j.molcel.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Heo I, Joo C, Kim YK, Ha M, Yoon MJ, Cho J, Yeom KH, Han J, Kim VN. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Single FN, Kohler M, Sommer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell. 1993;75:1361–1370. doi: 10.1016/0092-8674(93)90622-w. [DOI] [PubMed] [Google Scholar]
- Katoh T, Suzuki T. Specific residues at every third position of siRNA shape its efficient RNAi activity. Nucleic Acids Res. 2007;35:e27. doi: 10.1093/nar/gkl1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DJ, Moore PB, Steitz TA. The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J Mol Biol. 2004;340:141–177. doi: 10.1016/j.jmb.2004.03.076. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. 29–32. [DOI] [PubMed] [Google Scholar]
- Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- Kuzmic P. A generalized numerical approach to rapid-equilibrium enzyme kinetics: application to 17beta-HSD. Mol Cell Endocrinol. 2006;248:172–181. doi: 10.1016/j.mce.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Legault P, Pardi A. In-Situ Probing of Adenine Protonation in Rna by C-13 Nmr. J Am Chem Soc. 1994;116:8390–8391. [Google Scholar]
- Lehmann KA, Bass BL. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39:12875–12884. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lei M, Samuel CE. Chimeric double-stranded RNA-specific adenosine deaminase ADAR1 proteins reveal functional selectivity of double-stranded RNA-binding domains from ADAR1 and protein kinase PKR. Proc Natl Acad Sci U S A. 2000;97:12541–12546. doi: 10.1073/pnas.97.23.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli H, Mosbacher J, Melcher T, Hoger T, Geiger JR, Kuner T, Monyer H, Higuchi M, Bach A, Seeburg PH. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science. 1994;266:1709–1713. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]
- Macbeth MR, Lingam AT, Bass BL. Evidence for auto-inhibition by the N terminus of hADAR2 and activation by dsRNA binding. RNA. 2004;10:1563–1571. doi: 10.1261/rna.7920904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan NA, Carpick BW, Hollis B, Toone WM, Zamanian-Daryoush M, Williams BR. Mutational analysis of the double-stranded RNA (dsRNA) binding domain of the dsRNA-activated protein kinase, PKR. J Biol Chem. 1995;270:2601–2606. doi: 10.1074/jbc.270.6.2601. [DOI] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature. 1996;379:460–464. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- Michlewski G, Guil S, Semple CA, Caceres JF. Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol Cell. 2008;32:383–393. doi: 10.1016/j.molcel.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA. 2008;14:1539–1549. doi: 10.1261/rna.1155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. Editor meets silencer: crosstalk between RNA editing and RNA interference. Nat Rev Mol Cell Biol. 2006;7:919–931. doi: 10.1038/nrm2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstrass FC, Lee A, Stefl R, Janis M, Chanfreau G, Allain FH. Shape-specific recognition in the structure of the Vts1p SAM domain with RNA. Nat Struct Mol Biol. 2006;13:160–167. doi: 10.1038/nsmb1038. [DOI] [PubMed] [Google Scholar]
- Ohman M, Kallman AM, Bass BL. In vitro analysis of the binding of ADAR2 to the pre-mRNA encoding the GluR-B R/G site. RNA. 2000;6:687–697. doi: 10.1017/s1355838200000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell. 2000;102:437–449. doi: 10.1016/s0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- Parker GS, Maity TS, Bass BL. dsRNA binding properties of RDE-4 and TRBP reflect their distinct roles in RNAi. J Mol Biol. 2008;384:967–979. doi: 10.1016/j.jmb.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson RD, Theimer CA, Wu H, Feigon J. New applications of 2D filtered/edited NOESY for assignment and structure elucidation of RNA and RNA-protein complexes. J Biomol NMR. 2004;28:59–67. doi: 10.1023/B:JNMR.0000012861.95939.05. [DOI] [PubMed] [Google Scholar]
- Ramos A, Grunert S, Adams J, Micklem DR, Proctor MR, Freund S, Bycroft M, St Johnston D, Varani G. RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J. 2000;19:997–1009. doi: 10.1093/emboj/19.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedmann EM, Schopoff S, Hartner JC, Jantsch MF. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA. 2008;14:1110–1118. doi: 10.1261/rna.923308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rould MA, Perona JJ, Soll D, Steitz TA. Structure of E. coli glutaminyl-tRNA synthetase complexed with tRNA(Gln) and ATP at 2.8 A resolution. Science. 1989;246:1135–1142. doi: 10.1126/science.2479982. [DOI] [PubMed] [Google Scholar]
- Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 1998;17:7505–7513. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansam CL, Wells KS, Emeson RB. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc Natl Acad Sci U S A. 2003;100:14018–14023. doi: 10.1073/pnas.2336131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Magn Reson Spectrosc. 1999;34:93–158. [Google Scholar]
- Stefl R, Allain FH. A novel RNA pentaloop fold involved in targeting ADAR2. RNA. 2005;11:592–597. doi: 10.1261/rna.7276805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefl R, Skrisovska L, Allain FH. RNA sequence- and shape-dependent recognition by proteins in the ribonucleoprotein particle. EMBO Rep. 2005a;6:33–38. doi: 10.1038/sj.embor.7400325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefl R, Skrisovska L, Xu M, Emeson RB, Allain FH. Resonance assignments of the double-stranded RNA-binding domains of adenosine deaminase acting on RNA 2 (ADAR2) J Biomol NMR. 2005b;31:71–72. doi: 10.1007/s10858-004-6058-x. [DOI] [PubMed] [Google Scholar]
- Stefl R, Xu M, Skrisovska L, Emeson RB, Allain FH. Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure. 2006;14:345–355. doi: 10.1016/j.str.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Tian B, Bevilacqua PC, Diegelman-Parente A, Mathews MB. The double-stranded-RNA-binding motif: interference and much more. Nat Rev Mol Cell Biol. 2004;5:1013–1023. doi: 10.1038/nrm1528. [DOI] [PubMed] [Google Scholar]
- Tonkin LA, Bass BL. Mutations in RNAi rescue aberrant chemotaxis of ADAR mutants. Science. 2003;302:1725. doi: 10.1126/science.1091340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature. 2009;459:1010–1014. doi: 10.1038/nature08025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui V, Zhu L, Huang TH, Wright PE, Case DA. Assessment of zinc finger orientations by residual dipolar coupling constants. J Biomol NMR. 2000;16:9–21. doi: 10.1023/a:1008302430561. [DOI] [PubMed] [Google Scholar]
- Valente L, Nishikura K. ADAR gene family and A-to-I RNA editing: diverse roles in posttranscriptional gene regulation. Prog Nucleic Acid Res Mol Biol. 2005;79:299–338. doi: 10.1016/S0079-6603(04)79006-6. [DOI] [PubMed] [Google Scholar]
- Valente L, Nishikura K. RNA binding-independent dimerization of adenosine deaminases acting on RNA and dominant negative effects of nonfunctional subunits on dimer functions. J Biol Chem. 2007;282:16054–16061. doi: 10.1074/jbc.M611392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiljeva L, Kim M, Mutschler H, Buratowski S, Meinhart A. The Nrd1-Nab3-Sen1 termination complex interacts with the Ser5-phosphorylated RNA polymerase II C-terminal domain. Nat Struct Mol Biol. 2008;15:795–804. doi: 10.1038/nsmb.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Henras A, Chanfreau G, Feigon J. Structural basis for recognition of the AGNN tetraloop RNA fold by the double-stranded RNA-binding domain of Rnt1p RNase III. Proc Natl Acad Sci U S A. 2004;101:8307–8312. doi: 10.1073/pnas.0402627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Wells KS, Emeson RB. Substrate-dependent contribution of double-stranded RNA-binding motifs to ADAR2 function. Mol Biol Cell. 2006;17:3211–3220. doi: 10.1091/mbc.E06-02-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwahlen C, Legault P, Vincent SJF, Greenblatt J, Konrat R, Kay LE. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage lambda N-peptide/boxB RNA complex. J Am Chem Soc. 1997;119:6711–6721. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.