

Abstract
In our efforts to discover neuronal isoform selective nitric oxide synthase (NOS) inhibitors we have developed a series of compounds containing a pyrrolidine ring with two stereogenic centers. The enantiomerically pure compounds, (S,S) vs. (R,R), exhibited two different binding orientations, with (R,R) inhibitors showing much better potency and selectivity. To improve the bioavailability of these inhibitors we have introduced a CF2 moiety geminal to an amino group in the long tail of one of these inhibitors, which reduced its basicity, resulting in compounds with monocationic character under physiological pH conditions. Biological evaluations have led to a nNOS inhibitor with a Ki of 36 nM and high selectivity for nNOS over eNOS (3800-fold) and iNOS (1400-fold). MM-PBSA calculations indicated that the low pKa NH is, at least, partially protonated when bound to the active site. A comparison of rat oral bioavailability of the difluorinated compound to the parent molecule shows 22% for the difluorinated compound versus essentially no oral bioavailability for the parent compound. This indicates that the goal of this research to make compounds with only one protonated nitrogen atom at physiological pH to allow for membrane permeability, but which can become protonated when bound to NOS, has been accomplished.
Introduction
Since its first discovery as the endothelium-derived relaxing factor (EDRF) in 1987,1 nitric oxide (NO) has emerged as an important biological messenger involved in a wide variety of physiological functions as well as pathophysiological states. In living organisms, NO is produced by nitric oxide synthase (NOS), a homodimeric flavohemoprotein, through the oxidation of L-arginine to L-citrulline with NADPH and O2 as cosubstrates.2–4
To date, three distinct isoforms of mammalian NOS, namely neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS), have been identified with high homology (>55%).5 nNOS catalyzes the oxidation of L-arginine in the central nervous system, generating NO as a critical neurotransmitter.6–7 Although normal activity of nNOS is important for neurotransmission, NO overproduction by nNOS has been shown to be associated with chronic neurodegenerative pathologies such as Parkinson’s,8 Alzheimer’s,9 and Huntington’s diseases,10 also with chronic headaches,11 as well as neuronal damage in stroke.12 iNOS, the isozyme of NOS that produces cytotoxic NO, is responsible for maintaining normal function of the immune system.13 eNOS, on the other hand, regulates blood pressure. It has been shown experimentally that the reduced NO production by eNOS causes hypertension and atherosclerosis.14 Therefore, selective inhibition of nNOS activity over its closely related isoforms, eNOS and iNOS, represents an exciting drug approach for the development of new therapeutic agents to treat neurodegenerative diseases.5,15–16
In our continuing efforts to develop nNOS selective inhibitors, we have discovered a pyrrolidine-based compound (1, Figure 1), which showed great potency (Ki = 15 nM) and excellent selectivity for nNOS over eNOS (2100-fold) and iNOS (630-fold).17 However, further application of this compound as a drug candidate for neurodegenerative diseases is limited by its poor bioavailability.18 The two positive charges of 1 at physiological pH, derived from the two basic amino (NH) groups, dramatically impair the ability of 1 to penetrate the blood brain barrier (BBB) by passive diffusion.18 In addition, the benzylic position of the m-fluorophenylethyl substituent is susceptible to metabolic oxidation.19
Figure 1.

Chemical structures of 1 and 2
To circumvent these problems, different strategies have been employed to improve the bioavailability of inhibitors by limiting the number of basic functional groups in 1.18,20,21 For example, by incorporating an electron-withdrawing group adjacent to the amino group in the hydrophobic tail, a series of low pKa inhibitors were synthesized with monocationic or pseudo-monocationic characters at physiological pH.20 Evaluation of these inhibitors led to the discovery of inhibitor 2, with two fluorines substituted at the benzylic position of the m-fluorophenylethyl tail of 1. The racemic mixture of 2 indicated good potency (Ki = 80 nM) and high selectivity for nNOS over both eNOS (1500-fold) and iNOS (650-fold). More importantly, it showed improved cell-membrane permeability compared to the parent compound 1.20 The CF2 group is widely used in drug development because of its unique pharmacological properties.22 For inhibitor 2, the additional CF2 group not only decreases the basicity of the adjacent amino group dramatically (pKa ≈ 5.5), but also effectively blocks the potential metabolic oxidation at the benzylic position of the m-fluorophenyl ring.19 We report here the structure-based design, synthesis, and biological evaluation of a series of enantiomerically pure inhibitors (3a–f) derived from inhibitor 2, in an attempt to further improve the pharmacodynamic and pharmacokinetic properties of these inhibitors.
Results and Discussion
Chemistry
An improved synthesis of enantiomerically pure pyrrolidine core (4a–b) is shown in Scheme 1. First, racemic trans-alcohol 5 underwent a Mitsunobu reaction with (S)-(−)-camphanic acid as the nucleophile to produce two separable diastereomers (6a and 6b) in excellent yields. Next, the ester in 6a and 6b was hydrolyzed in aqueous Na2CO3 to generate 4a and 4b in high yields.
Scheme 1.

Synthesis of 4a–ba
a Reagents and conditions: (a) (S)-(−)-camphanic acid, PPh3, DIAD, rt, 16 h, 95%; (d) Na2CO3, rt, 4 h, 95%.
As shown in Scheme 2, single enantiomer 4a or 4b was treated with NaH, and the resulting anion was allowed to react with allyl bromide to generate 7a and 7b in excellent yields. Ozonolysis of 7a and 7b using Zn dust as the reducing reagent yielded 8a and 8b in good yields. Aldehydes 8a and 8b were subjected to reductive amination reactions with different ethanamines in the presence of NaHB(OAc)3 to generate secondary amines, which were further protected by another Boc-protecting group to produce fully protected inhibitors 9a–d in good yields. Next, the benzyl-protecting group was removed by catalytic hydrogenation using Pd(OH)2 at 60 °C to provide 10a–d in modest yields. Finally, the three Boc-protecting groups were removed at the same time in HCl to generate the final inhibitors (3a–d) in high yields.
Scheme 2.

Synthesis of 3a–da
a Reagents and conditions: (a) (i) NaH, (ii) allyl bromide, rt, 1 h, 96%; (b) O3, −78 ° C, (ii) Zn, −78 °C to rt, 2 h, 81%; (c) (i) ethanamines, THF, r.t., 5 min, (ii) NaHB(OAc)3, r.t., 3 h; (d) (Boc)2O, Et3N, MeOH, r.t., 3 h, 48–60% for two steps; (e) Pd(OH)2/C, H2, EtOH, 60 ° C, 30 h, 45–60%; (f) 6 N HCl/MeOH (2:1), r.t., 16 h, 95–100%.
The synthesis of inhibitor 3e began with 9a (Scheme 3). Catalytic hydrogenation of 9a using Pd(OH)2 at elevated temperature removed the benzyl-protecting group; when run for 48 h, the pyridinyl ring also was reduced to generate 10e in modest yields. Then, the three Boc-protecting groups were removed in HCl to generate final inhibitor 3e in high yields.
Scheme 3.

Synthesis of 3ea
a Reagents and conditions: (a) Pd(OH)2/C, H2, EtOH, 60 ° C, 48 h, 55%; (d) 6 N HCl/MeOH (2:1), r.t., 16 h, 96%.
The synthesis of inhibitor 3f is shown in Scheme 4. Reductive amination between aldehyde 8b and 2,2-difluoro-2-(pyridin-2-yl)ethanamine generated a secondary amine, which was further protected by another Boc-protecting group to provide 9f in good yields. Next, catalytic hydrogenation of 9f removed the benzyl protecting group and also reduced the pyridinyl group adjacent to the CF2 group to generate 10f in modest yields. Finally, the three Boc-protecting groups were removed at the same time in HCl to generate inhibitor 3f in high yields.
Scheme 4.

Synthesis of 3fa
a Reagents and conditions: (a) (i) 2,2-difluoro-2-(pyridin-2-yl)ethanamine, THF, r.t., 5 min, (ii) NaHB(OAc)3, r.t., 3 h; (b) (Boc)2O, Et3N, MeOH, r.t., 3 h, 55% for two steps; (c) Pd(OH)2/C, H2, EtOH, 60 ° C, 30 h, 60%; (d) 2 N HCl/MeOH (1:1), r.t., 16 h, 100%.
Crystal Structures of nNOS with 3a–3f Bound
Consistent with the binding preference of enantiomerically pure parental compound 1,23 the binding mode of these difluorinated derivatives is also dependent on the configuration around the two chiral centers, the 3′ and 4′ positions, of the pyrrolidine ring. While (S,S) inhibitor 3a was found to bind with its aminopyridine moiety hydrogen bonded to the side chain of Glu592 in nNOS (Fig. 2A), (R,R) inhibitor 3b adopted a 180° flipped binding mode with its aminopyridine making bifurcated hydrogen bonds with the heme propionate of pyrrole ring D (Fig. 2B). To make room for these new hydrogen bonds, the Tyr706 side chain rotates away and π-stacks against the aminopyridine ring of the inhibitor. The pyrrolidine nitrogen in 3b also makes favorable hydrogen bonds with propionate A as well as the O4 atom of H4B. In comparison, the pyrrolidine nitrogen in 3a is only loosely hydrogen bonded to Glu592. The long, flexible linker extending from the pyrrolidine allows the fluorophenyl group in 3b to reach the vicinity of Glu592 and to π-stack with the heme. The amino group vicinal to the CF2 moiety points toward the Glu592 side chain, resulting in an alternate conformation of the carboxylate enabling a hydrogen bond to form between the amino nitrogen and the carboxylate oxygen (Fig. 2B). This alternate conformation of Glu592 was not observed in the structure of nNOS bound with 3a because the tight bifurcated hydrogen bonds from the inhibitor aminopyridine to the carboxylate of Glu592 made an excellent match to the original conformation of the Glu side chain. The fit of the fluorophenyl group in 3b near Glu592 is not ideal, and to avoid close van der Waals contacts with the inhibitor, Glu592 adopts the alternate conformation. In addition, the fluorophenyl tail of 3b is disordered, as indicated by the weak electron density for this group. When only one conformation of the tail was modeled with the two fluorine atoms pointing away from the heme plane, strong negative difference density clustered around the fluorine atoms. Also, the electron density of the fluorophenyl ring could not be accounted for with only one ring orientation. The tail portion of 3b was, therefore, modeled with two different conformations (0.6 and 0.4 occupancy) as shown in Fig. 2B. There also is a partially occupied site for a water molecule bridging between the heme propionate and the amino group in the tail portion of 3b in the minor conformation. In contrast, the fluorophenyl ring in 3a fits into a pocket formed by Met336, Leu337, and Tyr706 (Fig. 2A), but the density for 3a is clear only up to the position of the two fluorine atoms thus precluding determination of the fluorophenyl ring orientation.
Figure 2.

The nNOS active site with inhibitor 3a (A) or 3b (B) bound. The sigmaA weighted 2Fo − Fc density for inhibitor is also shown at contour level of 1 σ. Hydrogen bonds are depicted with dashed lines. The atomic color schemes are: N, blue; O, red; S, yellow; F, light cyan. Alternate conformations for 3b (yellow and green) and E592 were observed. Figures 2, 3, and 4 were prepared with PyMol (http://www.pymol.org)
Inhibitor 3c has its fluorine position in the phenyl ring changed from meta in 3b to para in 3c. Because the C-F bond is 1.34 Å, 3c is at least 1.3 Å longer than 3b. As a result, the para-F in 3c makes a H-bond directly to the protein backbone amide (Gly586), while the meta-F of 3b makes only a weak van der Waals contact with the protein (Fig. 3A). Because of the tighter contact of 3c, the CF2 moiety is forced into a downward conformation. The major conformation in 3b, however, has the CF2 moiety pointing upward. The downward conformation causes clashes between the CF2 and heme propionate A, so the upward CF2 conformation of 3b represents a more relaxed fit in the active site, leading to higher potency.
Figure 3.
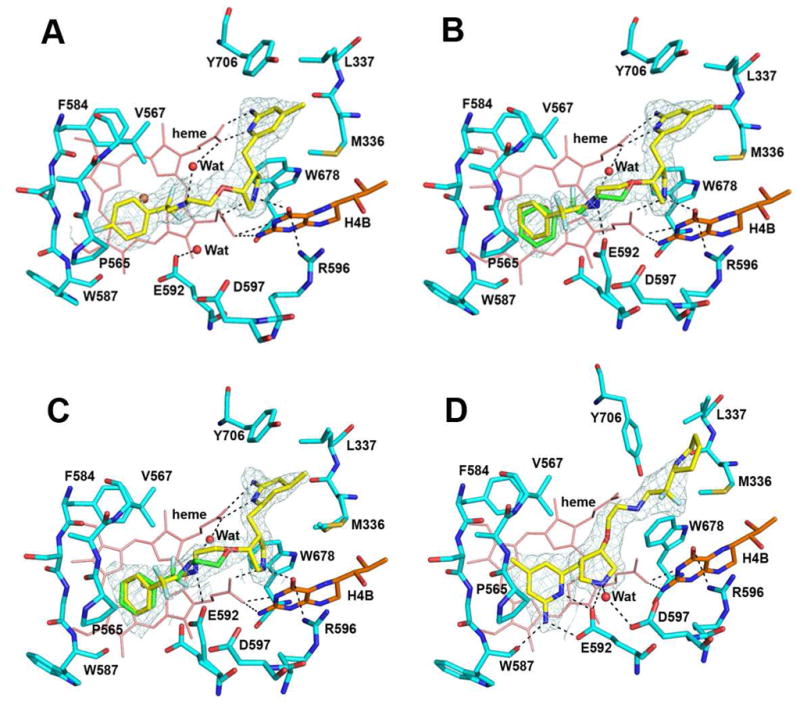
The nNOS active site with 3c (A), 3d (B), 3e (C), or 3f (D) bound. Around each inhibitor is the sigmaA weighted 2Fo − Fc density contoured at 1 σ. Major hydrogen bonds are depicted with dashed lines. The same atomic color schemes as in Figure 1 are used. Note the alternate conformations for E592 occur when the inhibitor shows multiple conformations in the nNOS-3d or nNOS-3e structure.
Inhibitor 3d is a derivative of 3b and differs only by the absence of the fluorine on the phenyl ring. Its binding mode to nNOS is, therefore, almost identical to that of 3b (Fig. 3B). Without the meta-fluorine the phenyl ring makes a looser contact with the hydrophobic pocket defined by Pro565, Val567, and Phe584. The phenyl tail portion in 3d exhibits two alternate conformations similar to what is observed for 3b.
Inhibitor 3e has its aminopyridine ring partially reduced from that in 3d, which has a negligible impact on the binding mode of the inhibitor compared to 3d (Fig. 3C). The amino and the ring nitrogens remain planar and are still tightly hydrogen bonded to the heme propionate of pyrrole D, and the pyrrolidine nitrogen is hydrogen bonded to propionate A and H4B. As in 3b and 3d, the phenyl tail in 3e shows two conformations above the heme plane, and the Glu592 side chain also has two corresponding conformations.
Another (S,S) inhibitor, 3f, is similar to 3a except the fluorophenyl tail has been replaced by a piperidine ring. The binding of 3f is similar to 3a with its aminopyridine hydrogen bonded to the Glu592 side chain while the piperidine ring fits into the pocket of Met336, Leu337, and Tyr706 (Fig. 3D). The density for the tail portion is good only up to the CF2 moiety with the piperidine partially disordered. Therefore, the orientation of the ring (position of nitrogen) is not well defined.
Crystal Structures of eNOS with 3b and 3f Bound
The eNOS structure bound with (R,R) inhibitor 3b (Fig 4A) shows that it has adopted a ‘flipped’ binding mode, the same orientation as in nNOS. The Tyr477 side chain rotates farther out than in nNOS and, therefore, does not experience optimized π-stacking interactions with the aminopyridine of the inhibitor, as was observed in nNOS. Similar to what was seen in nNOS, the aminopyridine in 3b makes bifurcated hydrogen bonds with the heme propionate of pyrrole ring D, while the pyrrolidine nitrogen makes favorable hydrogen bonds with propionate A as well as the O4 atom of H4B (Fig. 4A). A hydrogen bond between the inhibitor amino group with alternate conformations for Glu363 also is observed. With the available density at the limited resolution only one conformation of the fluorophenyl tail portion can be modeled.
Figure 4.

The eNOS active site with 3b (A) and 3f (B) bound. Around each inhibitor is the sigmaA weighted 2Fo − Fc density contoured at 1 σ. Major hydrogen bonds are depicted with dashed lines. The same atomic color schemes as that in Figure 2 are used. As observed with nNOS, alternate conformations for Glu363 occur in the eNOS-3b structure.
The binding of the (S,S) inhibitor 3f in eNOS is similar to that in nNOS, with its aminopyridine hydrogen bonded to the Glu363 side chain (Fig. 4B) in the normal binding mode. The pyrrolidine nitrogen also hydrogen bonds with the conserved glutamate residue, but the density for the tail portion is good only up to the CF2 moiety with the piperidine partially disordered. Therefore, the orientation of the ring (position of nitrogen) and the exact configuration of the puckered ring are not clear.
Inhibitory Assays and Structure-Based Evaluation
Inhibitors 3a–f were evaluated for in vitro inhibition activities against three isozymes of NOSs including rat nNOS, bovine eNOS, and murine iNOS,24 as summarized in Table 1. Compared to racemic lead compound 2, (S,S) enantiomer 3a is a weak inhibitor of nNOS with a Ki value of 390 nM, which is five-fold less potent than 2. In addition, the selectivity of this inhibitor for nNOS over eNOS and iNOS also decreases by 5-fold and 2-fold, respectively. The (R,R) enantiomer (3b), however, shows excellent potency for nNOS (Ki = 36 nM) and remarkable selectivity over eNOS (3800-fold) and iNOS (1400-fold). These results indicate that the chirality around the cis-chiral pyrrolidine core plays a key role in determining the potency and selectivity of inhibitors, as we have observed for another series of trans- or cis-chiral pyrrolidine inhibitors.23 The potency and selectivity shown with racemic compound 2 can be attributed mainly to (R,R)-component 3b. We now know that a large difference in binding affinity to nNOS between 3a and 3b originates from two different binding modes.23 The flipped binding mode of 3b relative to 3a allows both the aminopyridine and pyrrolidine nitrogen atoms to make extensive hydrogen bonds with the heme and H4B (Fig. 2B). In addition, as we have argued elsewhere,23 the conformation of 3a when bound to NOS places the pyrrolidine nitrogen atom very close to the aminopyridine, and because of electrostatic repulsion, this aminopyridine is only partially protonated. However, in the 3b flipped orientation the aminopyridine is farther from the pyrrolidine nitrogen atom and remains fully protonated. Thus, the 3b conformation provides greater electrostatic stabilization than the 3a conformation, which accounts for the 10-fold lower Ki for 3b than 3a.
Table 1.
Kia values of inhibitors for rat nNOS, bovine eNOS, and murine iNOS.
| Compound | nNOS (μM) | eNOS (μM) | iNOS (μM) | selectivityb |
|
|---|---|---|---|---|---|
| n/e | n/i | ||||
| 2 | 0.080 | 120 | 52 | 1500 | 650 |
| 3a | 0.390 | 110 | 130 | 280 | 330 |
| 3b | 0.036 | 140 | 51 | 3800 | 1400 |
| 3c | 0.160 | 31 | 190 | 190 | 1200 |
| 3d | 0.085 | 130 | 85 | 1500 | 1000 |
| 3e | 0.170 | 130 | 26 | 770 | 150 |
| 3f | 2.70 | 64 | 450 | 24 | 170 |
The Ki values were calculated based on the directly measured IC50 values, which represent at least duplicate measurements with standard deviations of ±10%.
The ratio of Ki (eNOS or iNOS) to Ki (nNOS).
As a p-fluorophenyl derivative of 3b, inhibitor 3c shows a significant drop in potency for nNOS (Ki = 160 nM), which is 4.5-fold less potent than 3b. More interestingly, 3c loses 20-fold of selectivity over eNOS observed with 3b by moving one single F atom from the m-position to the p-position of the phenyl tail. The new fluorine position leads to a ‘difluoromethylene-down’ binding mode of the inhibitor’s phenyl tail. As a result, the hydrogen bond between the Glu592 side chain and the amino nitrogen in the tail seen in 3b has been eliminated, which may explain the weaker potency and selectivity of 3c.
Inhibitor 3d, with the F atom removed from the phenyl tail, has restored good potency for nNOS (Ki = 85 nM) and high isozyme-selectivity (1500-fold over eNOS and 1000-fold over iNOS). This is because 3d has retained the binding mode of 3b. The only difference is that without a fluorine atom on the phenyl ring the van der Waals contacts to the protein (Pro565, Val567, and Phe584) are less optimal compared to that for 3b. This results in a bit less potency and selectivity for 3d than 3b.
The amidino inhibitor 3e, with the aromatic system of the aminopyridine fragment partially reduced, exhibits only a 2-fold drop in potency against nNOS compared to 3d. Although π-π stacking of 3e with Tyr706 should be greatly diminished relative to that with 3d, the increased basicity of the amidine of 3e over the aminopyridine of 3d may compensate by producing a tighter electrostatic interaction with the heme propionate, resulting in only a factor of two difference for the Ki values of the these inhibitors. The removal of the aromatic system of the aminopyridine ring from inhibitor 3d allows 3e to bind more than 3-fold better to iNOS, thus exhibiting a much lower selectivity.
Finally, inhibitor 3f, with an (S,S) configuration of the pyrrolidine core and a piperidinyl tail, showed poor inhibition activity and isozyme-selectivity. Both 3a and 3f have similar binding orientations and rather poor potency among the inhibitors reported in this work. This result emphasizes again that the chirality of the pyrrolidine core is the key to higher inhibitory activity with (R,R) inhibitors. In addition, a less polar aromatic ring such as the fluorophenyl group in 3a seems to fit better into the pocket surrounded by Met336, Leu337, and Tyr706 in nNOS than does the polar piperidinyl ring in 3f, and the π-stacking with the heme is lost in the case of 3f. However, both 3a and 3f bind to eNOS with similar potency, possibly because of the smaller Val106 side chain in the pocket in eNOS compared to Met336 in nNOS.
The isoform selection shown for this series of inhibitors in Table 1 is not that straightforward to interpret based only on the structures presented here. For any one particular inhibitor among those reported in Table 1 the binding mode in eNOS is no different from that seen in nNOS. Similar to what we have discussed for another series of chiral pyrrolidine containing inhibitors,23 one of the potential reasons for the isoform selectivity shown by 3b is the difference in π-stacking interaction between the aminopyridine and Tyr706 in nNOS (or Tyr 477 in eNOS). We have consistently observed tighter interactions between this Tyr and the aminopyridine with nNOS complexed with inhibitors that adopt the 3b flipped orientation.23
Calculations to Determine the Protonation State of Bound Inhibitors
The principal aim of this research was to lower the pKa of one of the NH groups in the inhibitors, so that, at physiological pH, there would be effectively only one positive charge on the molecules, which should be acceptable for CNS penetration.25 However, on the basis of past results, two positive charges are required for potency and isozyme selectivity. The aim was to find the appropriate pKa so that once bound to NOS, the low pKa NH would become protonated and could undergo the appropriate electrostatic interaction with a carboxylate.
In our initial work on flipped inhibitors,23 we employed the Molecular Mechanics Poisson–Boltzmann/Surface Area (MM-PBSA)26 computational method to calculate the free energy of binding of a series of aminopyridine inhibitors to nNOS and eNOS. We employed the empirical equation derived from Fig. 5, ΔGcalc = (PB-5.2874)/6.029, to obtain computed free energies. In our earlier work23 we found that the best fits with ΔGexp resulted when the orientation of the inhibitor with the aminopyridine stacked over the heme (normal orientation) was assumed to be 50% protonated, while in the flipped orientation in which the aminopyridine interacts with the heme propionate, a +1 charge was assigned to the aminopyridine. The way partial charges were handled was to carry out the MM-PBSA calculations with a 0 or +1 charge and average the results. We carried out the same types of calculations with the difluorinated inhibitors used in this study, but the goal was to probe the state of protonation of the NH group whose pKa has been lowered to ≈5.5 as a result of the fluorine atoms. The results of these calculations are shown in Table 2. With inhibitor 3b, additional calculations were carried out because we observed two orientations in the nNOS active site. The best fit to ΔGexp results from assigning a full +1 charge to the aminopyridine in the “flipped” orientation and a +0.5 charge in the “normal” orientation and a +0.5 charge for the NH group in all the inhibitors independent of orientation. Our strategy for lowering the pKa of the NH group was to decrease the total charge of the inhibitors in solution for better bioavailability but with the hope that they would become at least partially protonated once bound in the active site, where the nearby heme propionates and Glu592 might promote protonation. The calculations support this scenario and indicate that the NH group is partially protonated. This is reasonable because the NH group in these inhibitors, no matter which binding orientation, always H-bonds with one carboxylate and is within 4Å from another carboxylate (either the Glu592 side chain or the heme propionate).
Figure 5.

Plot of the computed free energy (PB) vs experimental free energy (ΔGexp) obtained from Ki values.
Table 2.
Calculated free energies for three inhibitors used in this study assuming different charges. For 3b two different conformations of the inhibitor were observed in the crystal structure referred to here as conformations A and B.
| inhibitor | charge on pyrrolidine | charge on NH | charge on amino pyridine | PB | ΔGcalc | ΔGexp | ΔΔG |
|---|---|---|---|---|---|---|---|
| 3a | 1 | 1 | 1 | −77.3 | −13.70 | −8.8 | 4.90 |
| 3a | 1 | 1 | 0 | −47.66 | −8.78 | −8.8 | 0.018 |
| 3a | 1 | 0 | 0 | −18.75 | −3.99 | −8.8 | 4.81 |
| average | −8.82 | −8.8 | 0.022 | ||||
| 3c | 1 | 0 | 1 | −24.02 | −4.86 | −7.92 | 3.06 |
| 3c | 1 | 1 | 1 | −55.9 | −10.15 | −7.92 | 2.23 |
| average | −7.50 | −7.92 | 0.42 | ||||
| 3b A | 1 | 1 | 1 | −66.03 | −11.83 | −10.2 | 1.63 |
| 3b B | 1 | 1 | 1 | −82.7 | −14.59 | −10.2 | 4.39 |
| 3b A | 1 | 0 | 1 | −45.02 | −8.34 | −10.2 | 1.86 |
| 3b B | 1 | 0 | 1 | −36.25 | −6.89 | −10.2 | 3.31 |
| average | −10.41 | −10.2 | 0.21 |
Pharmacokinetic Data for 3b and (R,R)−1
The goal for insertion of two gem-fluorines into 1 was to lower the pKa of one of the two basic nitrogen atoms to enhance metabolic stability and oral bioavailability but allow for potential reprotonation of that nitrogen atom once in the active site of nNOS. Pharmacokinetic properties of compound 3b (the (R,R) isomer of the gem-difluorinated analogue corresponding to (R,R)−1) were compared to those of (R,R)−1 to determine the effect of the difluorine substitution on pharmacokinetics. The in vivo compound half-life (t1/2) for i.v. dosing at 1 mg/kg in rats (average of 3 rats) was 0.33 h for (R,R)−1 and 7.5 h for 3b; the t1/2 for oral dosing at 5 mg/kg was too low to measure for (R,R)−1 and 3.7 h for 3b; oral bioavailability for (R,R)−1 was essentially zero and 22.2% for 3b. It is apparent that the addition of the two fluorines has a remarkable effect on in vivo stability and oral bioavailability.
In summary, we have designed and synthesized a new series of selective nNOS inhibitors (3a–f) with monocationic character, and, therefore, potentially possessing better bioavailability. Biological evaluation of these new inhibitors based on crystal structures led to the discovery of inhibitor 3b, which not only retains most of the inhibitory activity of lead compound 1, but also shows remarkable selectivity for nNOS over both eNOS and iNOS. MM-PBSA calculations demonstrate that despite the pKa of one of the nitrogen atoms in 3b being 5.5, when bound to the active site, it becomes partially protonated for good binding. Whereas the in vivo half-life (t1/2) and oral bioavailability in rats for the parent compound [(R,R)−1] are nil, when the two fluorines are added (3b), the t1/2 rises to 10 h and oral bioavailability increases to 22%. This work represents a significant step forward toward the goal of developing drugs with therapeutic potential in the treatment of diseases caused by unregulated NO generation from nNOS.
Experimental Section
1. General Methods
All experiments were conducted under anhydrous conditions in an atmosphere of argon, using flame-dried apparatus and employing standard techniques in handling air-sensitive materials. All solvents were distilled and stored under an argon or nitrogen atmosphere before use. All reagents were used as received. Aqueous solutions of sodium bicarbonate, sodium chloride (brine), and ammonium chloride were saturated. Analytical thin layer chromatography was visualized by ultraviolet, ninhydrin, or phosphomolybdic acid (PMA). Flash column chromatography was carried out under a positive pressure of nitrogen. 1H NMR spectra were recorded on 500 MHz spectrometers. Data are presented as follows: chemical shift (in ppm on the δ scale relative to δ = 0.00 ppm for the protons in TMS), integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constant (J/Hz). Coupling constants were taken directly from the spectra and are uncorrected. 13C NMR spectra were recorded at 125 MHz, and all chemical shift values are reported in ppm on the δ scale, with an internal reference of δ 77.0 or 49.0 for CDCl3 or MeOD, respectively. High-resolution mass spectra were measured on liquid chromatography/time-of-flight mass spectrometry (LC-TOF).
2. Preparation and Characterization of New Compounds
General Method A: Reductive Amination
To a solution of aldehyde 8a or 8b (0.1 mmol) in THF (3 mL) was added the ethanamine (0.2 mmol), followed by NaHB(OAc)3 (0.12 mmol). The mixture was stirred at room temperature for an additional 3 h and then concentrated. The crude product was purified by flash column chromatography (EtOAc/hexanes, 2:1–4:1) to yield the corresponding secondary amines as colorless oils, which were used without further purification.
General Method B: Boc-protection
To a solution of the secondary amine (0.5 mmol) in MeOH (10 mL) was added (Boc)2O (164 mg, 0.75 mmol) and TEA (140 μL, 1.0 mmol). The reaction mixture was allowed to stir at room temperature for 30 min. The solvent was removed by rotary evaporation, and the resulting material was purified by flash column chromatography (EtOAc/hexanes, 1:4-1:2) to yield 9a–f as colorless oils.
General Method C: Catalytic hydrogenation
To a solution of 9a–f (0.2 mmol) in EtOH (20 mL) was added Pd(OH)2/C (100 mg). The reaction vessel was charged with H2, heated at 60 °C for 24–48 h, then cooled to room temperature. The catalyst was removed by filtration, and the resulting solution was concentrated by rotary evaporation. The crude material was purified by flash column chromatography (EtOAc/hexanes, 1:4-1:2) to yield 10a–f as white foamy solids.
General Method D: Boc-deprotection
To a solution of 10a–f (50 μmol) in MeOH (0.5 mL) was added 6 N HCl (1.0 mL). The reaction mixture was allowed to stand at room temperature for 12 h. The solvent was removed by rotary evaporation. The crude product was recrystallized with cold diethyl ether to provide 3a–f as pale yellow solids.
(9a)
9a was synthesized by general methods A and B using aldehyde 8b as the starting material (60%): 1H NMR (500 MHz, CDCl3) δ 1.35–1.55 (m, 27H), 2.20–2.40 (s, 3H), 2.50–2.80 (m, 2H), 2.80–2.95 (m, 1H), 3.00–3.21 (m, 2H), 3.30–3.79 (m, 6H), 3.80–4.00 (m, 2H), 5.10–5.25 (s, 2H), 6.60–6.70 (br s, 1H), 7.00–7.30 (m, 8H), 7.31–7.60 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 14.4, 21.2, 21.3, 27.8, 28.1, 28.4, 28.5, 28.7, 29.9, 34.8, 42.3, 42.9, 47.5, 47.7, 49.0, 49.4, 50.1, 50.4, 50.9, 60.6, 67.9, 76.9, 78.9, 79.3, 79.7, 79.8, 80.6, 81.4, 113.0, 113.1, 117.2, 117.5, 120.1, 121.4, 126.7, 126.8, 126.9, 127.0, 127.1, 127.2, 127.5, 127.6, 128.3, 128.5, 128.6, 130.4, 140.0 148.8, 154.1, 154.5, 154.9, 157.8, 161.5, 163.4; LCQ-MS (M+H+) calcd for C43H58F3N4O7 799, found 799; LC-TOF (M+H+) calcd for C43H58F3N4O7 799.4252, found 799.4258.
(9b)
9b was synthesized by general methods A and B using aldehyde 8a as the starting material (58%): 1H NMR (500 MHz, CDCl3) δ 1.35–1.55 (m, 27H), 2.20–2.40 (s, 3H), 2.50–2.80 (m, 2H), 2.80–2.95 (m, 1H), 3.00–3.21 (m, 2H), 3.30–3.79 (m, 6H), 3.80–4.00 (m, 2H), 5.10–5.25 (s, 2H), 6.60–6.70 (br s, 1H), 7.00–7.30 (m, 8H), 7.31–7.60 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 14.4, 21.2, 21.3, 27.8, 28.1, 28.4, 28.5, 28.7, 29.9, 34.8, 42.3, 42.9, 47.5, 47.7, 49.0, 49.4, 50.1, 50.4, 50.9, 60.6, 67.9, 76.9, 78.9, 79.3, 79.7, 79.8, 80.6, 81.4, 113.0, 113.1, 117.2, 117.5, 120.1, 121.4, 126.7, 126.8, 126.9, 127.0, 127.1, 127.2, 127.5, 127.6, 128.3, 128.5, 128.6, 130.4, 140.0 148.8, 154.1, 154.5, 154.9, 157.8, 161.5, 163.4; LCQ-MS (M+H+) calcd for C43H58F3N4O7 799, found 799; LC-TOF (M+H+) calcd for C43H58F3N4O7 799.4252, found 799.4248.
(9c)
9c was synthesized by general methods A and B using aldehyde 8a as the starting material (48%): 1H NMR (500 MHz, CDCl3) δ 1.20–1.50 (m, 27H), 2.25–2.35 (m, 3H), 2.45–2.65 (m, 1H), 2.66–2.70 (m, 1H), 2.80–2.95 (m, 1H), 3.00–3.10 (m, 1H), 3.10–3.20 (m, 1H), 3.25–3.70 (m, 7H), 3.80–4.00 (m, 2H), 5.10–5.20 (m, 2H), 6.60–6.70 (m, 1H), 7.00–7.15 (m, 2H), 7.16–7.20 (m, 1H), 7.21–7.27 (m, 4H), 7.35–7.60 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 13.7, 14.2, 19.1, 21.0, 21.1, 24.7, 27.91, 27.96, 28.01, 28.2, 28.3, 28.5, 30.6, 34.4, 34.5, 34.6, 42.0, 42.1, 42.6, 42.7, 47.4, 47.5, 47.6, 47.8, 48.8, 49.3, 49.9, 50.1, 50.3, 50.8, 53.4, 53.8, 54.0, 60.4, 64.4, 67.7, 67.8, 68.0, 78.7, 78.8, 79.1, 79.2, 79.3, 79.6, 80.3, 81.17, 81.22, 115.2, 115.4, 115.6, 115.7, 117.0, 117.1, 119.9, 126.5, 126.6, 126.9, 127.0, 127.5, 128.1, 131.5, 139.7, 139.8, 148.6, 153.9, 154.3, 154.4, 154.5, 154.6, 154.7, 154.8, 155.1, 157.4, 157.5, 157.6, 162.7, 164.6, 171.2; LC-TOF (M+H+) calcd for C43H58F3N4O7 799.4258, found 799.4237.
(9d)
9d was synthesized by general methods A and B using aldehyde 8a as the starting material (55%): 1H NMR (500 MHz, CDCl3) δ 1.20–1.50 (m, 27H), 2.25–2.35 (m, 3H), 2.45–2.65 (m, 1H), 2.66–2.71 (m, 1H), 2.80–2.95 (m, 1H), 3.00–3.10 (m, 1H), 3.10–3.20 (m, 1H), 3.25–3.70 (m, 7H), 3.80–4.00 (m, 2H), 5.10–5.20 (m, 2H), 6.60–6.70 (m, 1H), 7.00–7.60 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 13.7, 14.2, 19.1, 21.0, 21.1, 24.7, 27.91, 27.96, 28.01, 28.2, 28.3, 28.5, 30.6, 34.4, 34.5, 34.6, 42.0, 42.1, 42.6, 42.7, 47.4, 47.5, 47.6, 47.8, 48.8, 49.3, 49.9, 50.1, 50.3, 50.8, 53.4, 53.8, 54.0, 60.4, 64.4, 67.7, 67.8, 68.0, 78.7, 78.8, 79.1, 79.2, 79.3, 79.6, 80.3, 81.17, 81.22, 115.2, 115.4, 115.6, 115.7, 117.0, 117.1, 119.9, 126.5, 126.6, 126.9, 127.0, 127.5, 128.1, 131.5, 139.7, 139.8, 148.6, 153.9, 154.3, 154.4, 154.5, 154.6, 154.7, 154.8, 155.1, 157.4, 157.5, 157.6, 162.7, 164.6, 171.2; LC-TOF (M+H+) calcd for C43H59F2N4O7 781.4352, found 781.4366.
(9f)
9f was synthesized by general methods A and B using aldehyde 8b as the starting material (55%): 1H NMR (500 MHz, CDCl3) δ 1.40–1.55 (m, 27H), 2.27–2.29 (m, 3H), 2.45–2.67 (m, 1H), 2.68–2.75 (m, 1H), 2.85–2.95 (m, 1H), 3.00–3.11 (m, 1H), 3.12–3.20 (m, 1H), 3.30–3.45 (m, 3H), 3.46–3.65 (m, 3H), 4.05–4.20 (m, 2H), 5.16 (s, 2H), 6.67 (s, 1H), 7.17–7.20 (m, 1H), 7.21–7.26 (m, 4H), 7.30–7.45 (m, 2H), 7.50–7.70 (m, 1H), 7.75–7.85 (m, 1H), 8.60–8.71 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 14.2, 21.11, 21.13, 24.7, 28.0, 28.1, 28.2, 28.3, 28.5, 29.7, 34.4, 34.5, 36.6, 42.2, 42.6, 42.7, 47.6, 47.7, 48.0, 48.8, 49.2, 49.89, 49.92, 50.1, 50.2, 50.8, 60.4, 67.5, 67.6, 67.7, 78.7, 79.1, 79.6, 80.2, 81.1, 81.2, 117.0, 117.1, 120.0, 120.4, 120.5, 124.76, 124.84, 126.4, 126.5, 126.6, 126.9, 127.0, 128.06, 128.11, 136.9, 137.0, 139.8, 139.9, 148.5, 149.3, 149.5, 153.8, 154.3, 154.4, 154.5, 154.7, 155.0, 155.4, 157.7; LC-TOF (M+H+) calcd for C42H58F2N5O7 782.4304, found 782.4299.
(3a)
3a was synthesized by general methods C and D using 9a as the starting material (95%): 1H NMR (500 MHz, D2O) δ 2.29 (s, 3H), 2.78–2.81 (m, 2H), 2.95–3.05 (dd, J = 8.0, 15.0 Hz, 1H), 3.15–3.20 (t, J = 6.0, 1H), 3.31–3.35 (dd, J = 3.0, 13.0 Hz, 1H), 3.40–3.55 (m, 3H), 3.63–3.66 (d, J = 13.0 Hz, 1H), 3.71–3.79 (m, 1H), 3.87–3.95 (m, 3H), 4.24–4.26 (t, J = 3.0 Hz, 1H), 6.55 (s, 1H), 6.64 (s, 1H), 7.25–7.29 (dt, J = 2.5, 8.5 Hz, 1H), 7.34–7.36 (dd, J = 2.5, 14.0 Hz, 1H), 7.38–7.40 (dd, J = 2.5, 8.0 Hz, 1H), 7.49–7.52 (dd, J = 6.0, 8.0 Hz, 1H); 13C NMR (125 MHz, D2O) δ 21.0, 29.1, 41.3, 47.0, 47.5, 49.2, 51.5, 51.7, 51.9, 63.6, 78.3, 110.4, 112.3, 112.5, 114.0, 118.2, 118.4, 118.6, 121.0, 131.2, 131.3, 134.2, 145.5, 153.9, 158.1, 161.4, 163.3; LC-TOF (M+H+) calcd for C21H28F3N4O 409.2215, found 409.2226.
(3b)
3b was synthesized by general methods C and D using 9b as the starting material (100%): 1H NMR (500 MHz, D2O) δ 2.29 (s, 3H), 2.78–2.81 (m, 2H), 2.95–3.05 (dd, J = 8.0, 15.0 Hz, 1H), 3.15–3.20 (t, J = 6.0, 1H), 3.31–3.35 (dd, J = 3.0, 13.0 Hz, 1H), 3.40–3.55 (m, 3H), 3.63–3.66 (d, J = 13.0 Hz, 1H), 3.71–3.79 (m, 1H), 3.87–3.95 (m, 3H), 4.24–4.26 (t, J = 3.0 Hz, 1H), 6.55 (s, 1H), 6.64 (s, 1H), 7.25–7.29 (dt, J = 2.5, 8.5 Hz, 1H), 7.34–7.36 (dd, J = 2.5, 14.0 Hz, 1H), 7.38–7.40 (dd, J = 2.5, 8.0 Hz, 1H), 7.49–7.52 (dd, J = 6.0, 8.0 Hz, 1H); 13C NMR (125 MHz, D2O) δ 21.0, 29.1, 41.3, 47.0, 47.5, 49.2, 51.5, 51.7, 51.9, 63.6, 78.3, 110.4, 112.3, 112.5, 114.0, 118.2, 118.4, 118.6, 121.0, 131.2, 131.3, 134.2, 145.5, 153.9, 158.1, 161.4, 163.3; LC-TOF (M+H+) calcd for C21H28F3N4O 409.2215, found 409.2223.
(3c)
3c was synthesized by general methods C and D using 9c as the starting material (95%): 1H NMR (500 MHz, D2O) δ 2.30 (s, 3H), 2.78–2.81 (m, 2H), 2.95–3.05 (dd, J = 8.0, 15.0 Hz, 1H), 3.15–3.20 (t, J = 6.0, 1H), 3.31–3.35 (dd, J = 3.0, 13.0 Hz, 1H), 3.40–3.55 (m, 3H), 3.63–3.66 (d, J = 13.0 Hz, 1H), 3.71–3.79 (m, 1H), 3.87–3.95 (m, 3H), 4.24–4.26 (t, J = 3.0 Hz, 1H), 6.55 (s, 1H), 6.64 (s, 1H), 7.21–7.25 (dd, J = 8.5, 8.5 Hz, 2H), 7.59–7.62 (dd, J = 5.0, 8.5 Hz, 2H); 13C NMR (125 MHz, D2O) δ 21.0, 29.0, 41.3, 47.0, 47.4, 49.2, 51.7, 51.9, 51.9, 63.6, 78.3, 110.4, 113.9, 116.0, 116.1, 118.7, 127.42, 127.47, 127.55, 127.59, 145.5, 153.9, 158.1; LC-TOF (M+H+) calcd for C21H28F3N4O 409.2215, found 409.2230.
(3d)
3d was synthesized by general methods C and D using 9d as the starting material (95%): 1H NMR (500 MHz, D2O) δ 2.30 (s, 3H), 2.73–2.84 (m, 2H), 2.95–3.05 (dd, J = 8.5, 15.0 Hz, 1H), 3.10–3.20 (t, J = 6.0, 1H), 3.31–3.35 (dd, J = 3.0, 13.5 Hz, 1H), 3.40–3.55 (m, 3H), 3.63–3.66 (d, J = 13.5 Hz, 1H), 3.71–3.79 (m, 1H), 3.87–3.95 (m, 3H), 4.24–4.26 (t, J = 3.0 Hz, 1H), 6.55 (s, 1H), 6.64 (s, 1H), 7.45–7.65 (m, 5H); 13C NMR (125 MHz, D2O) δ 21.0, 29.0, 41.3, 47.0, 47.4, 49.2, 51.6, 51.8, 52.0, 63.6, 78.2, 110.4, 113.9, 119.0, 120.9, 124.81, 124.86, 124.91, 129.1, 131.6, 131.9, 132.1, 145.5, 153.9, 158.1;; LC-TOF (M+H+) calcd for C21H29F2N4O 391.2309, found 391.2337.
(3e)
3e was synthesized by general methods C and D using 9e as the starting material (96%): 1H NMR (500 MHz, D2O) δ 0.85–0.95 (d, J = 2.5, 3H), 1.45–1.55 (m, 1H), 1.61–1.70 (m, 1H), 1.71–1.97 (m, 3H), 2.00–2.10 (m, 1H), 2.35–2.45 (m, 1H), 2.46–2.57 (m, 1H), 2.90–3.00 (t, J = 6.0, 1H), 3.16–3.21 (m, 1H), 3.30–3.50 (m, 4H), 3.51–3.60 (d, J = 13.5 Hz, 1H), 3.61–3.70 (m, 1H), 3.75–3.90 (m, 3H), 4.14s-4.16 (t, J = 3.0 Hz, 1H), 7.45–7.65 (m, 5H); 13C NMR (125 MHz, D2O) δ 17.5, 17.6, 18.2, 26.8, 27.2, 27.3, 32.6, 32.9, 40.0, 40.2, 48.3, 48.9, 49.1, 49.6, 50.6, 52.2, 58.8, 65.2, 79.9, 80.0, 115.4, 115.6, 116.8, 117.0, 126.0, 132.1, 132.2, 140.2, 163.1, 165.0, 168.4; LC-TOF (M+H+) calcd for C21H33F2N4O 395.2622, found 395.2633.
(3f)
3f was synthesized by general methods C and D using 9f as the starting material (100%): 1H NMR (500 MHz, D2O) δ 1.40–1.50 (m, 1H), 1.51–1.60 (m, 2H), 1.79–1.90 (m, 2H), 1.91–1.98 (d, J = 13.5 Hz, 1H), 2.20 (s, 3H), 2.65–2.75 (m, 1H), 2.79–2.85 (m, 1H), 2.94–2.96 (d, J = 7.5 Hz, 1H), 2.97–3.00 (m, 1H), 3.07–3.12 (dd, J = 11.5, 11.5 Hz, 1H), 3.20–3.26 (dd, J = 0.5, 13.0 Hz, 1H), 3.31–3.47 (m, 4H), 3.52–3.55 (d, J = 13.5 Hz, 1H), 3.61–3.64 (m, 1H), 3.74–3.82 (m, 5H), 4.11 (s, 1H), 6.52 (s, 1H), 6.57 (s, 1H); 13C NMR (125 MHz, D2O) δ 20.4, 21.05, 21.06, 22.4, 28.95, 29.01, 41.48, 41.51, 45.1, 47.0, 48.08, 48.14, 49.4, 57.7, 63.7, 63.9, 78.3, 78.4, 110.4, 114.09, 114.12, 145.7, 153.9, 158.1; LC-TOF (M+H+) calcd for C20H34F2N5O 398.2742, found 398.2726.
Enzyme Assays
The three isozymes, murine macrophage iNOS, rat nNOS, and bovine eNOS, were recombinant enzymes, overexpressed (in E. coli) and isolated as reported.27 IC50 values for inhibitors 3a–f were measured for the three different isoforms of NOS using L-arginine as a substrate. The formation of nitric oxide was measured using a hemoglobin capture assay described previously.24 All NOS isozymes were assayed at room temperature in a 100 mM Hepes buffer (pH 7.4) containing 10 μM L-arginine, 1.6 mM CaCl2, 11.6 μg/mL calmodulin, 100 μM DTT, 100 μM NADPH, 6.5 μM H4B, 3.0 μM oxyhemoglobin (for iNOS assays, no Ca2+ and calmodulin were added). The assay was initiated by the addition of enzyme, and the initial rates of the enzymatic reactions were determined by monitoring the formation of NO-hemoglobin complex at 401 nm for 60 sec. The corresponding Ki values of inhibitors were calculated from the IC50 values using equation 1 with known Km values (rat nNOS, 1.3 μM; iNOS, 8.3 μM; eNOS, 1.7 μM).
| (1) |
Inhibitor Complex Crystal Preparation
The nNOS or eNOS heme domain protein used for crystallographic studies were produced by limited trypsin digest from the corresponding full length enzymes and further purified through a Superdex 200 gel filtration column (GE Healthcare) as described previously.28 The enzyme-inhibitor complex crystals were obtained by soaking rather than co-crystallization as reported in the earlier structural work.29 The nNOS heme domain at 7–9 mg/mL containing 20 mM histidine or the eNOS heme domain at 20 mg/mL with 2 mM imidazole were used for the sitting drop vapor diffusion crystallization setup under the conditions reported before.28,30 Fresh crystals (1–2 day old) were first passed stepwise through cryo-protectant solutions described28,30 and then soaked with 10 mM inhibitor for 4–6 h at 4 °C before being mounted on nylon loops and flash cooled by plunging into liquid nitrogen. Crystals were stored in liquid nitrogen until data collection.
X-ray Diffraction Data Collection, Processing, and Structure Refinement
The cryogenic (100K) X-ray diffraction data were collected remotely at various beamlines at Stanford Synchrotron Radiation Lightsource through the data collection control software Blu-Ice31 and the crystal mounting robot. Raw data frames were indexed, integrated, and scaled using HKL2000.32 Typically, each data set consisted of 90 to 100 degree of data with a 0.5 degree frame width for both nNOS and eNOS crystals because of their identical orthorhombic P212121 space group symmetry.
The binding of inhibitors was detected by the initial difference Fourier maps calculated with REFMAC.33 The inhibitor molecules were then modeled in O34 or COOT35 and refined using REFMAC. Water molecules were added in REFMAC and checked by COOT. The TLS36 protocol was implemented in the final stage of refinements with each subunit as one TLS group. The refined structures were validated in COOT before deposition to RCSB protein data bank. The crystallographic data collection and structure refinement statistics are summarized in Table 3 with PDB accession codes included.
Table 3.
Crystallographic data collection and refinement statistics
| Data set1 | nNOS-3a | nNOS-3b | nNOS-3c | nNOS-3d |
|---|---|---|---|---|
| Data collection | ||||
| PDB code | 3NLV | 3NLX | 3NLY | 3NLZ |
| Space group | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | ||||
| a, b, c (Å) | 52.3, 111.7, 164.4 | 52.3, 111.5, 164.4 | 51.8, 111.2, 164.1 | 51.9, 110.4, 164.0 |
| Resolution (Å) | 2.10 (2.14-2.10) | 1.87 (1.90-1.87) | 2.00 (2.03-2.00) | 1.92 (1.95-1.92) |
| Rsym or Rmerge | 0.080 (0.59) | 0.053 (0.36) | 0.078 (0.40) | 0.050 (0.32) |
| I/σI | 9.1 (2.2) | 13.5 (3.9) | 7.6 (1.8) | 11.5 (2.6) |
| No. unique reflections | 56,857 | 79,054 | 63,439 | 73,017 |
| Completeness (%) | 99.4 (99.5) | 98.2 (96.5) | 96.7 (80.9) | 99.4 (89.8) |
| Redundancy | 5.6 (2.9) | 4.1 (4.1) | 3.9 (3.1) | 4.1 (3.6) |
| Refinement | ||||
| Resolution (Å) | 2.10 | 1.87 | 2.00 | 1.92 |
| No. reflections used | 54,011 | 75,069 | 60,245 | 69,320 |
| Rwork/Rfree | 0.175/0.211 | 0.178/0.209 | 0.201/0.249 | 0.173/0.210 |
| No. atoms | ||||
| Protein | 6,671 | 6,689 | 6,676 | 6,707 |
| Ligand/ion | 187 | 223 | 190 | 217 |
| Water | 434 | 479 | 190 | 417 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.013 | 0.012 | 0.015 | 0.013 |
| Bond angles (°) | 1.325 | 1.390 | 1.554 | 1.456 |
| Data set1 | nNOS-3e | nNOS-3f | eNOS-3b | eNOS-3f |
|---|---|---|---|---|
| Data collection | ||||
| PDB code | 3NM0 | 3NLW | 3NLU | 3NLT |
| Space group | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | ||||
| a, b, c (Å) | 52.1, 110.9, 164.2 | 52.2, 111.4, 164.7 | 58.0, 107.0, 156.9 | 57.9, 106.9, 157.0 |
| Resolution (Å) | 1.81 (1.84-1.81) | 2.10 (2.14-2.10) | 2.65 (2.70-2.65) | 2.75 (2.80-2.75) |
| Rsym or Rmerge | 0.048 (0.38) | 0.069 (0.55) | 0.121(0.58) | 0.103 (0.65) |
| I/σI | 12.1 (2.7) | 9.2 (2.4) | 9.7 (1.9) | 12.4 (1.9) |
| No. unique reflections | 86,908 | 56,364 | 28,366 | 25,670 |
| Completeness (%) | 99.0 (92.5) | 99.2 (100.0) | 97.3 (99.1) | 95.6 (94.4) |
| Redundancy | 4.0 (3.8) | 4.1 (4.1) | 3.7 (3.7) | 3.9 (4.0) |
| Refinement | ||||
| Resolution (Å) | 1.81 | 2.10 | 2.65 | 2.74 |
| No. reflections used | 82,532 | 53,523 | 26,957 | 24,379 |
| Rwork/Rfree2 | 0.178/0.209 | 0.171/0.207 | 0.185/0.254 | 0.186/0.262 |
| No. atoms | ||||
| Protein | 6,716 | 6,676 | 6,451 | 6,419 |
| Ligand/ion | 217 | 185 | 197 | 195 |
| Water | 459 | 319 | 111 | 60 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.013 | 0.013 | 0.014 | 0.014 |
| Bond angles (°) | 1.360 | 1.347 | 1.502 | 1.519 |
Rfree was calculated with the 5% of reflections set aside throughout the refinement. For each NOS isoform the set of reflections for the Rfree calculation were kept the same for all data sets according to those used in the data of the starting model.
Molecular Mechanics Poisson–Boltzmann/SurfaceA (MM-PBSA) Method for Free Energy of Binding of Inhibitors
The MM-PBSA method26 as implemented in Amber 9.0 was used to compute the free energy of binding of inhibitors to NOS and was previously described in detail.23 To briefly summarize, the free energies of 10 different aminopyridine inhibitors-NOS complexes were calculated from a single energy minimized structure starting from the known crystal structures described in our previous work.23 A plot of the resulting computed free energy (PB) vs ΔGexp, obtained from experimental Ki values. The empirical equation derived from Fig. 5, ΔGcalc = (PB-5.2874)/6.029, was used to calculate free energies of binding for the inhibitors used in the present study.
Pharmacokinetic Data
All pharmacokinetic results were obtained by LC-MS-MS analysis and calculation of PK parameters using WinNonlin software at BioDuro, Inc., Shanghai, China.
Supplementary Material
Acknowledgments
The authors are grateful for financial support from the National Institutes of Health (GM49725 to RBS and GM57353 to TLP). We thank Dr. Bettie Sue Siler Masters (NIH grant GM52419, with whose laboratory P.M. and L.J.R. are affiliated). B.S.S.M. also is grateful to the Welch Foundation for a Robert A. Welch Distinguished Professorship in Chemistry (AQ0012). P.M. is supported by grants 0021620806 and 1M0520 from MSMT of the Czech Republic. We also thank the staff at SSRL for their assistance during the remote X-ray diffraction data collections.
Footnotes
Supporting Information Available: Copies of complete spectroscopic data of compounds 9a–d, 9f, and 3a–f are available.
References
- 1.Palmer RMJ, Ferrige AG, Moncada S. Nature. 1987;327:524. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 2.Marletta MA. J Biol Chem. 1993;268:12231. [PubMed] [Google Scholar]
- 3.Marletta MA. Cell. 1994;78:927. doi: 10.1016/0092-8674(94)90268-2. [DOI] [PubMed] [Google Scholar]
- 4.Griffith OW, Stuehr DJ. Annu Rev Physiol. 1995;57:707. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 5.Alderton WK, Cooper CE, Knowles RG. Biochem J. 2001;357:593. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall AV, Antoniou H, Wang Y, Cheung AH, Arbus AM, Olson SL, Lu WC, Kau CL, Marsden PA. J Biol Chem. 1994;269:33082. [PubMed] [Google Scholar]
- 7.Wang Y, Newton DC, Marsden PA. Crit Rev Neurobiol. 1999;13:21. doi: 10.1615/critrevneurobiol.v13.i1.20. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Dawson VL, Dawson TM. Pharmacol Ther. 2006;109:33. doi: 10.1016/j.pharmthera.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Dorheim MA, Tracey WR, Pollock JS, Grammas P. Biochem Biophys Res Commun. 1994;205:659. doi: 10.1006/bbrc.1994.2716. [DOI] [PubMed] [Google Scholar]
- 10.Norris PJ, Waldvogel HJ, Faull RLM, Love DR, Emson PC. Neuronsicence. 1996;72:1037. doi: 10.1016/0306-4522(95)00596-x. [DOI] [PubMed] [Google Scholar]
- 11.Ashina M. Exp Opin Pharmacother. 2002;3:395. doi: 10.1517/14656566.3.4.395. [DOI] [PubMed] [Google Scholar]
- 12.Sims NR, Anderson MF. Neurochem Int. 2002;40:511. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 13.Hobbs AJ, Higgs A, Moncada S. Annu Rev Pharmacol. 1999;39:191. doi: 10.1146/annurev.pharmtox.39.1.191. [DOI] [PubMed] [Google Scholar]
- 14.Dawson VL, Dawson TM, London ED, Bredt DS, Snyner SH. Proc Natl Acad Sci. 1991;88:6368. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Southan GJ, Szabo C. Biochem Pharmacol. 1996;51:383. doi: 10.1016/0006-2952(95)02099-3. [DOI] [PubMed] [Google Scholar]
- 16.Babu BR, Griffith OW. Curr Opin Chem Biol. 1998;2:491. doi: 10.1016/s1367-5931(98)80125-7. [DOI] [PubMed] [Google Scholar]
- 17.Ji H, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. J Med Chem. 2009;52(3):779. doi: 10.1021/jm801220a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawton GR, Ranaivo HR, Wing LK, Ji H, Xue F, Martesek P, Roman LJ, Watterson DM, Silverman RB. Bioorg Med Chem. 2009;17:2371. doi: 10.1016/j.bmc.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silverman RB. The Organic Chemistry of Drug Design and Drug Action. 2. Elsevier; 2004. [Google Scholar]
- 20.Xue F, Fang J, Lewis WW, Martásek P, Roman LJ, Silverman RB. Bioorg Med Chem Lett. 2010;20:554. doi: 10.1016/j.bmcl.2009.11.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xue F, Huang J, Ji H, Fang J, Li H, Martásek P, Roman LJ, Poulos TP, Silverman RB. Bioorg Med Chem. 2010;18:6526. doi: 10.1016/j.bmc.2010.06.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smart BE. J Fluorine Chem. 2001;109:3. [Google Scholar]
- 23.Delker DL, Ji H, Li H, Jamal J, Fang J, Xue X, Silverman RB, Poulos TL. J Am Chem Soc. 2010;132:5437. doi: 10.1021/ja910228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hevel JM, Marletta MA. Method Enzymol. 1994;233:250. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 25.Kerns EH, Di L. Drug-like Properties: Concepts, Structure Design and Methods. Academic Press; Amsterdam: 2008. p. 130. [Google Scholar]
- 26.Massova I, Kollman PA. J Am Chem Soc. 1999:8133. [Google Scholar]
- 27.(a) Hevel JM, White KA, Marletta M. J Biol Chem. 1991;266(34):22789. [PubMed] [Google Scholar]; (b) Roman LJ, Sheta EA, Martásek P, Gross SS, Liu Q, Masters BSS. Proc Natl Acad Sci USA. 1995;92(18):8428. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Martasek P, Liu Q, Roman LJ, Gross SS, Sessa WC, Masters BSS. Biochem Biophys Res Commun. 1996;219(2):359. doi: 10.1006/bbrc.1996.0238. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. Biochemistry. 2002;41:13868. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 29.Flinspach ML, Li H, Jamal J, Yang W, Huang H, Hah JM, Gomez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Nat Struct Mol Biol. 2004;11:54. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- 30.Raman CS, Li H, Martasek P, Kral V, Masters BS, Poulos TL. Cell. 1998;95:939. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 31.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. J Synchrotron Radiat. 2002;9:401. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Murshudov GN, Vagin AA, Dodson EJ. Acta Cryst. 1997;D53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 34.Jones TA, Zou JY, Cowan SW, Kjeldgaarrd M. Acta cryst. 1991;A47:110. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. Acta Cryst. 2004;D60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Winn MD, Isupov MN, Murshudov GN. Acta Cryst. 2001;D57:122. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.