

Abstract
The interferon-inducible, transmembrane protein BST-2 (CD317, tetherin) directly holds fully formed enveloped virus particles to the cells that produce them, inhibiting their spread. BST-2 inhibits members of the retrovirus, filovirus, arenavirus, and herpesvirus families. These viruses encode a variety of proteins to degrade BST-2 and/or direct it away from its site of action at the cell surface. Viral antagonism has subjected BST-2 to positive selection, leading to species-specific differences that presented a barrier to the transmission of simian immunodeficiency viruses (SIVs) to humans. This barrier was crossed by HIV-1 when its Vpu protein acquired activity as a BST-2 antagonist. Here, we review this new host-pathogen relationship and discuss its impact on the evolution of primate lentiviruses and the origins of the HIV pandemic.
BST-2/tetherin overview
BST-2, also known as CD317, HM1.24, or tetherin, is a newly identified component of the innate immune response to enveloped viruses1,2. This protein, whose original name stands for bone marrow stromal cell antigen 2, was cloned in 1995 from a rheumatoid-arthritis-derived synovial cell line3. However, its antiviral activity was not discovered until 2008, when BST-2 was identified as a host cell ‘restriction factor’ counteracted by the HIV-1 accessory protein Vpu1,2. Since then, BST-2′s spectrum of activity against enveloped viruses, its mechanism of action, and the ways in which viruses counteract it, have been intensely investigated. Here, we will introduce the concept of host cell restriction factors and viral countermeasures, present our current understanding of the BST-2 protein and how it limits the spread of enveloped viruses, describe how viruses have evolved to antagonize BST-2, and discuss the remarkable impact of this host-pathogen relationship on the evolution of primate lentiviruses and the pandemic spread of HIV-1.
Restriction factors are intrinsic host defense proteins that directly inhibit viral replication. In the case of HIV-1, the restriction factors identified so far include the APOBEC3 (A3) family of cytidine deaminases4, TRIM5α5, and BST-21,2, each of which are linked to the innate immune response by interferon-induction. As expected, HIV-1 has evolved countermeasures to each of these proteins. For example, the viral capsid protein, the target of restriction by Old World monkey TRIM5α, has evolved resistance to human TRIM5α5. However, neither A3 nor BST-2 target viral proteins, so HIV-1 has evolved entire accessory proteins to counteract them: Vif for the A3 proteins that damage viral reverse transcripts by deamination of cytosines6, and Vpu for BST-2, a transmembrane protein that directly traps virus particles (virions) on and within the cells that produce them1,2.
In humans, a single copy of the bst-2 gene is located on chromosome 193. The human gene so far appears to be relatively non-polymorphic. Orthologues of BST-2 exist in placental mammals, and substantial polymorphism exists in certain Old World monkeys7. BST-2 is expressed constitutively on a number of cell types including B cells, T cells, monocytes and macrophages, and plasmacytoid dendritic cells (PDCs)8,9; PDCs are major producers of type I interferon (IFN). The expression of BST-2 is induced in other cell types by type I IFN10. The protein is also constitutively expressed on several cancer cell lines, including cells of the B cell lineage in multiple myeloma11.
BST-2 is a type II transmembrane protein (the N-terminus is in the cytoplasm) whose lumenal C-terminus is modified with a glycosyl phosphatidyl inositol (GPI) membrane anchor (Figure 1)12. This unusual topology enables BST-2 to interact with the cell or virion lipid bilayer at each of its ends. BST-2 associates with cholesterol-enriched lipid rafts, presumably due to its modification with GPI12. BST-2 is found at the plasma membrane and within several endosomal membrane compartments, including the trans-Golgi network (TGN) as well as early and recycling endosomes13.
Figure 1. Features of the BST-2/tetherin monomer.
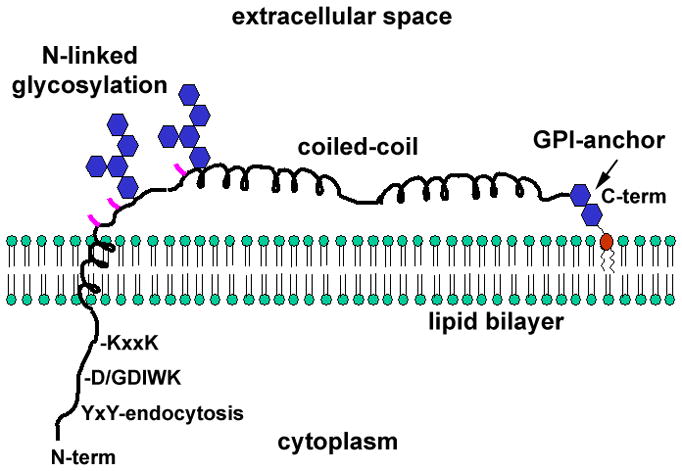
BST-2 binds the lipid bilayer via an N-terminal transmembrane α-helix and a C-terminal glycosylphophastidyl inositol (GPI) anchor. The cytoplasmic domain contains a YxY sequence that directs endocytosis by interacting with the clathrin adaptor AP-2. Non-human primate BST-2 contains an insertion relative to the human protein (D/GDIWK or a similar sequence), which is targeted by lentiviral Nef proteins. The KxxK sequence is ubiquitinated by the K5 protein of KSHV, leading to degradation. The transmembrane region is required for antagonism by HIV-1 Vpu. The ectodomain of the protein contains three cysteines (locations indicated in pink), which drive homo-dimerization. The ectodomain also contains an extended α-helical region that forms a parallel coiled-coil in BST-2 dimers. The transmembrane and GPI membrane anchors, the cysteines, and the integrity of the coiled-coil region are each required for the restriction of virion release.
The BST-2 protein has several key features (Figure 1). The short cytoplasmic domain contains a conserved YxY motif, which binds the clathrin adaptors AP-2 and AP-1 to direct endocytosis and retrieval to juxtanuclear endosomes14. The cytoplasmic domain of BST-2 also interacts indirectly with the subcortical actin network of the cell15, but whether this relates to its activity as a restriction factor is unknown. Conserved lysine residues in primate BST-2 proteins are targets for ubiquitination by the K5 protein of Kaposi’s sarcoma associated herpes virus (KSHV)16, and the D/GDIWK motif (or variations of this sequence) in the cytoplasmic domain of non-human primate orthologues of BST-2 confers susceptibility to antagonism by simian immunodeficiency virus (SIV) negative regulatory factor (Nef) proteins17,18. The transmembrane domain (TMD) of BST-2 is predicted to be a single α-helix that likely interacts with the α-helical TMD at the N-terminus of Vpu19. The ectodomain contains three cysteines that participate in intermolecular disulfide bonds to form parallel homodimers11. The ectodomain also contains two N-linked glycosylation sites that are substrates for heterogeneous glycosylation20.
Recently, a partial structure of the ectodomain of BST-2 has been solved by X-ray crystallography21. The ectodomain contains a parallel, dimeric, α-helical coiled-coil region 90 Å in length. Small angle X-ray scattering indicates that this structure is extended from the N-terminus of the coiled-coil to the lumenal end of the TMD at a slight angle (Figure 2)21. These data indicate that the ectodomain of BST-2 forms a rod-like structure with a maximum extended length of 150–170 Å. The coiled-coil region is partially splayed apart by structural irregularities along its length and is relatively unstable in the absence of disulfide bonds, suggesting potential conformational flexibility21.
Figure 2. Mechanism of restricted virion release by BST-2.

(a) Immuno-electron microscopic data indicate that BST-2 is appropriately positioned to directly tether mature virions to the plasma membrane and is incorporated into virions. The cell cytoplasm is at the left; the extracellular space is at the right and includes mature virions. Arrows indicate immuno-label for the BST-2 ectodomain. Reprinted with permission from Ref. [31]. (b) Model in which BST-2 dimers (green) embed one end in the plasma membrane and the other in the virion membrane, retaining virions on the cell surface. BST-2 dimers may also link virions to each other. An alternative model is discussed in the text. (c) Structure of the dimeric BST-2 ectodomain obtained by small angle X-ray scattering (molecular surface in gray) and X-ray crystallography (modified from Ref. [21]). The parallel, dimeric coiled-coil is colored orange. Abbreviations: Å, angstroms. (d) Crystallographic structure of the dimeric coiled-coil indicating residues important for the restriction of virion release (PDB accession code: 2x7a; structure drawn using PyMOL).
Restriction of virion release by BST-2/tetherin
BST-2 holds fully formed virions to the cell surface, preventing their release and spread (Figure 2). Retained virions may be internalized by endocytosis and subsequently degraded, or they may remain on the cell surface22. In either case, their spread as cell-free virions is restricted. An interaction between the cytoplasmic domain of BST-2 and the endocytic machinery contributes to the internalization and degradation of tethered virions: BCA2, a RING-type E3 ubiquitin ligase, associates with BST-2 and directs tethered virions to late endosomes23. Moreover, virions retained on the cell surface are aggregated by BST-2; this renders them poorly infectious even during direct cell-to-cell transmission24.
BST-2 has broad activity against diverse families of enveloped viruses, including retroviruses (HIV-1, HIV-2, SIV, HTLV-I, MLV, EIAV, spumaviruses, and XMRV)25–27, filoviruses (Ebola virus and Marburg virus)25,28, at least one arenavirus (Lassa virus)29, and at least one herpesvirus (KSHV)16. In principle, any lipid-enveloped virus could be restricted by BST-2. However, since the activity of BST-2 likely requires co-localization with budding virions13,30,31, its activity is probably limited to viruses that bud from cellular membranes that are enriched in BST-2.
The simplest model of restriction is that BST-2 directly holds completely budded, nascent virions to the plasma membrane of the cell (Figure 2). This ‘direct tethering’ model is supported by immuno-electron microscopy13,30–32; BST-2 is enriched at the sites of HIV-1 budding and is fully incorporated into virions (Figure 2). The notion that BST-2 acts autonomously, without obligate cellular cofactors, is supported by an elegant experiment: an ‘artificial tetherin’ constructed from parts of proteins with no primary sequence homology to BST-2 but containing a transmembrane domain, a dimeric coiled-coil ectodomain, and a GPI-anchoring sequence restricts HIV-1 release30. This result suggests that the general structural features and membrane-binding properties of BST-2 are sufficient for restrictive activity.
Exactly how does BST-2 retain virions on membranes? Currently, this question remains open. Perhaps the most intuitive topological model is one in which parallel homodimers of BST-2 insert one end in the host cell membrane and the other in the virion-membrane, forming a direct link between the virion and the cell (Figure 2). This ‘membrane spanning’ model takes advantage of the unusual ability of BST-2 to interact with lipid bilayers at each of its ends. However, a potential caveat regarding this model is that although virions retained on the cell surface by BST-2 should in principle be released by enzymatic hydrolysis of GPI anchors using phosphatidyl inositol specific phospholipase, they seem not to be31. Currently, the only method known to release retained virions is proteolysis of the cell and virion surface22.
For unclear reasons, the dimeric coiled-coil structure of the BST-2 ectodomain is required for restriction. Mutation of the cysteines that drive dimerization disrupts activity, as do selected mutations in the protein’s ectodomain, some of which destabilize the dimeric coiled-coil20,21,30. These data evoke an alternative to the membrane-spanning model: individual BST-2 molecules could embed both ends into either the host or virion membranes but interact with each other, potentially as the dimers described above or as tetramers or higher order multimers. This model can explain a requirement for dimerization, but it does not explain why BST-2 requires both of its membrane interacting domains for restrictive activity1,30. One approach to resolving the molecular topology of restriction is to assess the size of the gap between the juxtaposed membranes of retained virions and the host cell. This gap should be no less than 15–17 nm if the rod-like BST-2 ectodomain is extended to span the host and virion membranes, whereas it should be substantially less if the ectodomain is oriented along the lipid bilayers21. Regardless of orientation, BST-2 currently appears to form a covalent link between the virion and the cell. How BST-2 dimers (or higher order multimers) could maintain insertion into both the cell and virion membranes throughout the process of viral budding and membrane scission is an interesting question; one notion is that conformational flexibility of the ectodomain structure as noted above facilitates this21.
Viral antagonists and mechanisms of action
Many viruses have evolved specific countermeasures to overcome restriction by BST-2. Among the primate lentiviruses, three different viral gene products are known to antagonize BST-2 (Figure 3). While most SIVs use Nef to antagonize the BST-2 proteins of their respective non-human primate hosts, the Vpu protein of HIV-1 and the envelope glycoprotein of HIV-2 antagonize human BST-21,2,17,18,27,33. Additional viral antagonists of BST-2 include the KSHV K5 protein and the glycoproteins of SIV and Ebola virus16,28,34,35.
Figure 3. Viral antagonists of BST-2 and their potential mechanisms of action.

(a) Lentiviral antagonists. The HIV-1 Vpu protein associates with BST-2 through transmembrane domain interactions and recruits βTrCP, leading to the degradation of BST-2 within lysosomes and/or proteasomes. Alternatively, Vpu might simply trap BST-2 within the endosomal system. In either scenario, the concentration of BST-2 is decreased at the plasma membrane, its site of action as a tetherin. The SIV Nef protein interacts with the cytoplasmic domain of BST-2 and mediates downregulation from the cell surface, most likely via endocytosis. The HIV-2 Env protein downregulates and sequesters BST-2 within the trans-Golgi network (TGN) by a mechanism that depends on physical interactions between the two proteins and a conserved GYXXφ motif in the cytoplasmic tail of gp41. (b) Non-lentiviral antagonists. The Ebola GP physically associates with, and possibly sequesters, BST-2 within intracellular compartments. Alternatively, Ebola GP might directly interfere with the restrictive activity of BST-2 or it might direct viral assembly away from membrane domains containing BST-2. The KSHV K5 protein mediates the ubiquitination of BST-2, which induces degradation of the protein within proteasomes and/or lysosomes. The interactions between BST-2 and the lentiviral antagonist proteins Vpu, Nef, and Env are shown here as occurring at the plasma membrane, but they could occur alternatively or additionally within the endoplasmic reticulum (ER), the TGN, or recycling endosomes.
Vpu was initially proposed to antagonize BST-2 by removing it from the sites of virus assembly and release at the plasma membrane2. This model emerged from the observations that Vpu co-localizes with BST-2 in endosomes and decreases the levels of BST-2 on the cell surface1,2. Considerable progress has been made in elucidating the cellular pathways by which this occurs, although the ultimate fate of BST-2 remains controversial. Vpu was known to mediate the proteasomal degradation of CD4 through the recruitment of the β-transducin repeat-containing protein (βTrCP) subunit of the Skp1-Cullin1-F-box ubiquitin ligase complex36. βTrCP also contributes to the antagonism of BST-2, since disruption of the interaction between βTrCP and the cytoplasmic domain of Vpu reduces the capacity of Vpu to enhance virus release37–39. Moreover, Vpu and BST-2 appear to physically interact via their transmembrane domains40,41. Together, these observations suggest that Vpu acts as an adaptor for the recruitment of βTrCP to BST-2 and ultimately directs BST-2 along pathways that recognize ubiquitin as a trafficking signal37–39. Whereas initial studies indicated that proteasome inhibitors prevented Vpu-mediated degradation of BST-2, subsequent studies found that inhibitors of endosomal acidification also prevented this degradation and even prevented the Vpu-mediated removal of BST-2 from the cell surface in the apparent absence of degradation19,39,41,42. These observations led to the suggestion that Vpu induces the retention of BST-2 within the endolysosomal system, with concomitant partial lysosomal degradation (Figure 3). Indeed, the downregulation of BST-2 from the cell surface and its antagonism by Vpu do not strictly require degradation; endosomal sequestration appears sufficient to remove BST-2 from its site of action at the plasma membrane37,43–45. Such sequestration might occur along the biosynthetic pathway, trapping newly synthesized BST-2 within the TGN, or it could occur within recycling endosomes, preventing endocytosed BST-2 from returning to the cell surface37,44.
Prior to the identification of BST-2 as a restriction factor, the envelope glycoproteins of certain HIV-2 isolates were known to have ‘Vpu-like’ activity, since their expression could rescue the release of vpu-deleted HIV-1 from restrictive cells46. This activity of the HIV-2 envelope glycoprotein (Env) likely depends on a physical interaction between the ectodomain of the Env gp41 subunit and the ectodomain of BST-2; specific amino acid substitutions in the ectodomains of each protein abrogate Env-mediated antagonism of BST-247,48. Proteolytic cleavage of Env into the gp120 and gp41 glycoprotein subunits and a conserved tyrosine-based endocytosis motif (GYXXφ) in the cytoplasmic domain of gp41 are also required for activity27,49. Similar to Vpu, HIV-2 Env downregulates BST-2 from the cell surface, removing BST-2 from the sites of virus assembly at the plasma membrane (Figure 3)27,45. However, HIV-2 Env exclusively sequesters BST-2 within the TGN, with no concomitant degradation of BST-227,45. SIVagm Tan Env (isolated from tantalus monkeys) antagonizes BST-2 by a similar mechanism, requiring the same residue in the ectodomain of BST-2 that is necessary for antagonism by HIV-2 Env35. While the activity of SIVagm Tan Env seems at some odds with the role of Nef as the gene product of SIVagm that opposes African green monkey BST-217, the envelope glycoprotein of SIVagm Tan might have acquired the ability to antagonize BST-2 due to the passage of this particular isolate in human cells, in which Nef proteins are ineffective antagonists of BST-2 (see below).
Although the mechanism by which Nef counteracts restriction by BST-2 has not been determined, initial data provide a number of clues. The Nef proteins of diverse SIVs, including SIVsmm/mac (from sooty mangabeys and rhesus macaques), SIVagm (from African green monkeys), SIVblu (from the Blue monkey) and SIVcpz (from chimpanzees), enhance virus release from cells expressing the BST-2 proteins of their simian hosts, but not from cells expressing human BST-217,18,33. SIV Nef also downregulates macaque BST-2, but not human BST-2, from the cell surface17. This specificity maps to a five amino acid sequence in the cytoplasmic domain of simian BST-2 that is missing from the human protein17,18. Nef likely interacts with these residues to remove BST-2 from the plasma membrane (Figure 3). The residues in Nef that contribute to the putative interaction with BST-2 have not been defined, but mutations that eliminate the conserved myristoylation site at the N-terminus of Nef, or that disrupt CD4 downregulation, impair the ability of Nef to antagonize BST-217,18. These observations suggest that the antagonism of BST-2 requires the association of Nef with cellular membranes and involves the same pathway used by Nef to downregulate CD4: clathrin-mediated endocytosis50.
The K5 protein of KSHV was the first viral protein shown to reduce the levels of BST-2 at the plasma membrane, though at the time the function of BST-2 as a restriction factor was unknown 34. The K5 protein is homologous to a group of cellular transmembrane ubiquitin ligases, the RING-CH (MARCH) family, that promote the ubiquitination and degradation of transmembrane proteins. In addition to other roles in immune evasion including the degradation of class I major histocompatibility complex (MHC), K5 promotes the lysosomal degradation of BST-2 through the ubiquitination of a single lysine residues in the protein’s cytoplasmic domain; this targets BST-2 for transport to lysosomes via the ESCRT pathway16,51.
The Ebola virus glycoprotein (GP) antagonizes both human and murine BST-228. Similar to HIV-2 Env, the full-length form of the Ebola GP co-immunoprecipitates and co-localizes with BST-2, suggesting a physical interaction between the two proteins28. However, unlike HIV-2 Env, mutation of the furin cleavage site, which prevents the proteolytic processing of GP, does not affect the ability of GP to enhance virus release28. Moreover, the Ebola GP does not contain a tyrosine-based motif in its cytoplasmic tail. Since BST-2 preferentially associates with an incompletely glycosylated form of GP, and GP does not change the steady-state levels of BST-2 in cells, GP might intercept BST-2 during protein maturation and sequester it within intracellular compartments, away from the sites of virus assembly and release28. However, recent data indicate that Ebola GP does not remove BST-2 from the plasma membrane. Moreover, Ebola GP appears able to antagonize the ‘artificial tetherin’ described above, a protein with no primary sequence homology to BST-248. These results suggest that Ebola GP might directly interfere with the restriction mechanism of BST-2, or it might direct viral assembly away from membrane domains containing BST-2.
The role of BST-2/tetherin in shaping lentiviral evolution and the AIDS pandemic
Rapid evolution in response to the diversifying selective pressure imposed by viral pathogens is a hallmark of retroviral restriction factors. As a consequence of this ongoing evolutionary conflict, APOBEC3G and TRIM5α have acquired species-specific differences that are important host-range determinants of lentiviral infection52,53. This is also true for BST-2. Sequence comparisons of the bst-2 genes of different primate species suggest that BST-2 is rapidly evolving under positive selection7,19,54. Analysis of the ratio of nonsynonymous to synonymous nucleotide substitutions over the entire coding region revealed a concentration of nonsynonymous changes in regions of the gene encoding the cytoplasmic and transmembrane domains of the protein - regions expected to be evolving in response to the viral antagonists that target these domains.
These species-specific differences in BST-2 have had a significant impact on the evolution of the primate lentiviruses. While HIV-1 uses Vpu to antagonize BST-2, the majority of primate lentiviruses do not have a vpu gene. Only two major lineages of primate lentiviruses express Vpu: the SIVcpz/HIV-1 lineage and the SIVgsn lineage [a group of SIV isolates from Old World monkeys, including the greater spot-nosed monkey (SIVgsn), the mona monkey (SIVmon), the mustached monkey (SIVmus) and Dent’s mona monkey (SIVden)]55–58. In the absence of a vpu gene, SIV isolates from other species of Old World monkeys use Nef to counteract BST-2. The Nef proteins of SIV, SIVagm and SIVblu antagonize the BST-2 orthologues of their respective simian hosts, but are ineffective against human BST-217,18. This species-pecific activity maps to the five amino acid sequence (G/D14DIWK18) that is missing from the cytoplasmic domain of the human protein17,18.
The deletion of sequences recognized by Nef in human BST-2 could be the result of a previous encounter with a viral pathogen during human evolutionary history. Since the G/D14DIWK18motif is retained in the cytoplasmic domain of chimpanzee BST-2 (Figure 4)59, the absence of these residues from human BST-2 reflects a 15-nucleotide deletion that occurred since humans and chimpanzees last shared a common ancestor. Given that we are Old World primates, and that our ancestors might have lived in close proximity to primate species endemically infected with SIVs, a selective sweep by a lentiviral pathogen that used Nef (or a Nef-like protein) to antagonize BST-2 might explain the absence of the G/D14DIWK18 sequence in the human protein.
Figure 4. Adaptation of the primate lentiviruses for overcoming restriction by BST-2 in their respective hosts.

Chimpanzee SIV (SIVcpz) is thought to be a recombinant derived from Old World monkey SIVs similar to viruses presently found in the greater spot-nosed monkey (SIVgsn) and the red-capped mangabey (SIVrcm). HIV-1 and SIVgor are the result of the cross-species transmission of SIVcpz from chimpanzees to humans and from chimpanzees to gorillas. Likewise, HIV-2 and SIVmac reflect the cross-species transmission of SIVsmm from sooty mangabeys to humans and from sooty mangabeys to macaques. Arrows represent cross-species transmission events and the viral gene products that antagonize BST-2 in each species are indicated in red. The viral gene products that either retained, or lost (indicated by red X’s). Activity against BST-2 are indicated in black next to each arrow. Species-specific differences in BST-2 are depicted in the amino acid sequence alignment. Representative BST-2 sequences are shown for the rhesus macaque (rhBST2), sooty mangabey (smBST2), greater spot-nosed monkey (gsnBST2), chimpanzee (cpzBST2), gorilla (gorBST2) and humans (humBST2). Residues of the cytoplasmic domain that confer susceptibility to Nef are indicated by the red box, the transmembrane domain is highlighted in yellow, potential N-linked glycosylation sites are indicted in green and the site for cleavage and addition of the GPI anchor is indicated in blue. The five-residue deletion in the cytoplasmic domain of human BST-2 renders the protein resistant to antagonism by Nef, leading to the acquisition of BST-2 antagonist activity by HIV-1 Vpu and HIV-2 Env.
The absence of sequences in the cytoplasmic domain of human BST-2 that confer susceptibility to Nef has shaped the evolution of both HIV-1 and HIV-2. HIV-1 originated as the result of cross-species transmission of SIVcpz from chimpanzees to humans58,60. Likewise, SIVcpz is believed to have arisen from the cross-species transmission, and subsequent recombination, of the progenitors of SIVgsn and SIVrcm from Old World monkeys to chimpanzees61. While the Vpu proteins expressed by viruses of the SIVgsn lineage antagonize the BST-2 proteins of their respective hosts33, this is not the case for SIVcpz Vpu. Nef, rather than Vpu, is the gene product of SIVcpz that counteracts chimpanzee BST-233,62. HIV-1 Vpu apparently acquired the ability to antagonize BST-2 after the transmission of SIVcpz into humans. This adaptation was necessary since the sequences essential for recognition by Nef were missing from the cytoplasmic domain of human BST-2 (Figure 4). In contrast, Nef retained its role as an antagonist of BST-2 after the transmission of SIV into gorillas, since the sequences that confer susceptibility to Nef are present in the cytoplasmic domain of gorilla BST-2 (Figure 4)33.
The HIV-2/SIVsmm/mac lineage exhibits similar plasticity in adaptation to BST-2. Cross-species transmission of SIVsmm from sooty mangabeys gave rise to HIV-2 in humans and to SIVmac in macaques63–66. In the case of SIVmac, Nef retained the ability to antagonize BST-2 due to the presence of the G/D14DIWK18 sequence in rhesus macaque BST-2 (Figure 4)17,18. However, in the case of HIV-2, Nef was unable to antagonize human BST-2 due to the absence of these residues17,18. This, together with the lack of a vpu gene, explains the selective pressure for the envelope glycoprotein of HIV-2 to acquire activity against human BST-2 (Figure 4)27. The adaptation of the HIV-2 envelope glycoprotein for antagonism of human BST-2 might have facilitated the spread of HIV-2 in West Africa. However, the use of an essential viral structural protein to counteract BST-2 could have entailed a greater fitness cost to virus replication than the use of an accessory protein such as Vpu or Nef. We speculate that such a fitness cost might contribute to the lower level of virus replication, the slower rate of disease progression, and the more limited geographical distribution of HIV-2 in comparison to HIV-167.
The ability of HIV-1 Vpu to antagonize human BST-2 might have enhanced virus replication to a threshold that facilitated the global spread of HIV-1. Phylogenetic analyses indicate that at least three independent cross-species transmissions of SIVcpz gave rise to HIV-1 groups M, N and O65. Group M is responsible for the global HIV-1 pandemic, whereas groups N and O have not spread beyond West Africa. Comparison of the activities of Vpu proteins from each of these groups indicates that the Vpus of group M viruses efficiently antagonize human BST-2 (and mediate the degradation of CD4), while those of group N and O viruses are either weaker antagonists of BST-2 (and are unable to degrade CD4) or have no detectable activity against BST-233. Thus, the aggregate activities of the Vpu proteins of group M viruses could have contributed to the origin of pandemic HIV-1.
Potential roles of BST-2 in innate immunity beyond direct restriction
The intense recent investigation of BST-2 has focused on its role as a viral tetherin. In this role, BST-2 is a direct effector protein of the antiviral state induced by type I interferons, and it fulfills criteria as an intrinsic host restriction factor against which viruses have evolved counteracting activities.
However, two lines of evidence suggest that BST-2 might play roles in innate immunity beyond direct restriction. BST-2 is a ligand for the receptor ITL7, a member of the immunolglobulin-transcript like family found on human PDCs 68. Signaling through ILT7 suppresses the production of type I IFN by PDCs in response to signaling through the Toll-like receptors TLR7 and TLR9. BST-2 stimulates signaling by ILT7 and consequently provides negative feedback to the production of IFN by PDCs in response to viral infection68. How viral modulation of BST-2 might interact with this regulatory pathway to the pathogen’s advantage is an open question. One possibility is that the incorporation of BST-2 into virion membranes enables viruses to suppress the production of IFN by PDCs via the interaction of virion-associated BST-2 with ILT7. A similar IFN-suppression mechanism has been proposed to contribute to the tolerance of tumor cells that express BST-2 constitutively68.
Another observation that remains largely unexplored is the potential role of BST-2 itself as a signaling protein; BST-2 directly induces the expression of the transcription factor NF-κB69. This effect would nominally serve to activate transcriptional programs in a wide variety of immunologically active cells. In cells infected with viruses that encode BST-2 antagonist proteins, this effect of BST-2 on the activation of NF-κB could potentially be thwarted, but exactly how this would benefit the pathogen is an open question.
Conclusions and unanswered questions
The discovery of the antiviral activity of BST-2 opened a new field of inquiry into a novel mechanism of host defense. Many questions remain unanswered (Box 1). These include the exact molecular mechanism of restricted virion release, the breadth of activity of BST-2 against enveloped viruses, and the basis of BST-2 antagonism by viral proteins. Although BST-2 orthologues are found only in mammals, the minimal constraints on sequence and structure revealed by the activity of ‘artificial tetherin30’ allow the possibility of diverse host tetherins whose primary sequences are too distant from BST-2 for recognition.
Box 1. Outstanding questions.
What is the molecular topology and structural basis of virion-tethering by BST-2?
How exactly does each viral antagonist protein counteract BST-2?
What is the spectrum of antiviral activity of BST-2?
Do antiviral ‘tetherins’ exist in organisms other than mammals?
Can this host-pathogen relationship be exploited to develop new approaches to the prevention and therapy of infection with enveloped viruses?
The evolution of the primate lentiviruses, particularly HIV-1 and HIV-2, has been shaped by species-specific differences in BST-233. Human BST-2 is missing key residues that confer susceptibility to Nef17,18. Since the lentiviruses of non-human primates generally use Nef to antagonize this restriction factor, BST-2 likely presented a barrier to the initial transmission of primate lentiviruses to humans. In the case of HIV-1 group M, the Vpu protein solved this problem in a manner that might have facilitated the pandemic spread of the virus. Whether our new knowledge of BST-2 can be used to develop new approaches to the prevention and therapy of infections due to enveloped viruses clearly depends on the extent to which viral evasion of this innate host response facilitates the establishment of infection in the new host and enables the chronic and persistent replication of pathogens such as HIV.
While these new questions remain to be answered, the discoveries reviewed above exemplify how asking basic questions about the function of the so-called accessory proteins of viruses can reveal entirely new mechanisms of host defense.
Acknowledgments
This work was supported by Public Health Service grants AI087498, AI063993 and RR00168 to DTE and AI081668 to JCG. DTE is an Elizabeth Glaser Scientist supported by the Elizabeth Glaser Pediatric AIDS Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Neil SJ, et al. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 2.Van Damme N, et al. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishikawa J, et al. Molecular cloning and chromosomal mapping of a bone marrow stromal cell surface gene, BST2, that may be involved in pre-B-cell growth. Genomics. 1995;26:527–534. doi: 10.1016/0888-7543(95)80171-h. [DOI] [PubMed] [Google Scholar]
- 4.Sheehy AM, et al. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 5.Stremlau M, et al. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 6.Harris RS, et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 7.McNatt MW, et al. Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 2009;5:e1000300. doi: 10.1371/journal.ppat.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vidal-Laliena M, et al. Characterization of antibodies submitted to the B cell section of the 8th Human Leukocyte Differentiation Antigens Workshop by flow cytometry and immunohistochemistry. Cell Immunol. 2005;236:6–16. doi: 10.1016/j.cellimm.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Miyagi E, et al. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc Natl Acad Sci U S A. 2009;106:2868–2873. doi: 10.1073/pnas.0813223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blasius AL, et al. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol. 2006;177:3260–3265. doi: 10.4049/jimmunol.177.5.3260. [DOI] [PubMed] [Google Scholar]
- 11.Ohtomo T, et al. Molecular cloning and characterization of a surface antigen preferentially overexpressed on multiple myeloma cells. Biochem Biophys Res Commun. 1999;258:583–591. doi: 10.1006/bbrc.1999.0683. [DOI] [PubMed] [Google Scholar]
- 12.Kupzig S, et al. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic. 2003;4:694–709. doi: 10.1034/j.1600-0854.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- 13.Habermann A, et al. CD317/tetherin is enriched in the HIV-1 envelope and downregulated from the plasma membrane upon virus infection. J Virol. 2010;84:4646–4658. doi: 10.1128/JVI.02421-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rollason R, et al. Clathrin-mediated endocytosis of a lipid-raft-associated protein is mediated through a dual tyrosine motif. J Cell Sci. 2007;120:3850–3858. doi: 10.1242/jcs.003343. [DOI] [PubMed] [Google Scholar]
- 15.Rollason R, et al. A CD317/tetherin-RICH2 complex plays a critical role in the organization of the subapical actin cytoskeleton in polarized epithelial cells. J Cell Biol. 2009;184:721–736. doi: 10.1083/jcb.200804154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mansouri M, et al. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2009;83:9672–9681. doi: 10.1128/JVI.00597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia B, et al. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009;5:e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang F, et al. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe. 2009;6:54–67. doi: 10.1016/j.chom.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta RK, et al. Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 2009;5:e1000443. doi: 10.1371/journal.ppat.1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andrew AJ, et al. The formation of cysteine-linked dimers of BST-2/tetherin is important for inhibition of HIV-1 virus release but not for sensitivity to Vpu. Retrovirology. 2009;6:80. doi: 10.1186/1742-4690-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinz A, et al. Structural basis of HIV-1 tethering to membranes by the BST-2/tetherin ectodomain. Cell Host Microbe. 2010;7:314–323. doi: 10.1016/j.chom.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neil SJ, et al. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006;2:e39. doi: 10.1371/journal.ppat.0020039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyakawa K, et al. BCA2/Rabring7 promotes tetherin-dependent HIV-1 restriction. PLoS Pathog. 2009;5:e1000700. doi: 10.1371/journal.ppat.1000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casartelli N, et al. Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog. 2010;6:e1000955. doi: 10.1371/journal.ppat.1000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jouvenet N, et al. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. 2009;83:1837–1844. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Groom HC, et al. Susceptibility of xenotropic murine leukemia virus-related virus (XMRV) to retroviral restriction factors. Proc Natl Acad Sci U S A. 2010;107:5166–5171. doi: 10.1073/pnas.0913650107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le Tortorec A, Neil SJ. Antagonism and intracellular sequestration of human tetherin by the HIV-2 envelope glycoprotein. J Virol. 2009;83:11966–11978. doi: 10.1128/JVI.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaletsky RL, et al. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A. 2009;106:2886–2891. doi: 10.1073/pnas.0811014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakuma T, et al. Inhibition of Lassa and Marburg virus production by tetherin. J Virol. 2008;83:2382–2385. doi: 10.1128/JVI.01607-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perez-Caballero D, et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 2009;139:499–511. doi: 10.1016/j.cell.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fitzpatrick K, et al. Direct restriction of virus release and incorporation of the interferon-induced protein BST-2 into HIV-1 particles. PLoS Pathog. 2010;6:e1000701. doi: 10.1371/journal.ppat.1000701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammonds J, et al. Immunoelectron microscopic evidence for Tetherin/BST2 as the physical bridge between HIV-1 virions and the plasma membrane. PLoS Pathog. 2010;6:e1000749. doi: 10.1371/journal.ppat.1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sauter D, et al. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe. 2009;6:409–421. doi: 10.1016/j.chom.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartee E, et al. Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog. 2006;2:e107. doi: 10.1371/journal.ppat.0020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta RK, et al. Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST-2/CD317 by intracellular sequestration. Proc Natl Acad Sci U S A. 2009;106:20889–20894. doi: 10.1073/pnas.0907075106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Margottin F, et al. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998;1:565–574. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 37.Mitchell RS, et al. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009;5:e1000450. doi: 10.1371/journal.ppat.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mangeat B, et al. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 2009;5:e1000574. doi: 10.1371/journal.ppat.1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Douglas JL, et al. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a βTrCP-dependent mechanism. J Virol. 2009;83:7931–7947. doi: 10.1128/JVI.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rong L, et al. The transmembrane domain of BST-2 determines its sensitivity to down-modulation by human immunodeficiency virus type 1 Vpu. J Virol. 2009;83:7536–7546. doi: 10.1128/JVI.00620-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iwabu Y, et al. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J Biol Chem. 2009;284:35060–72. doi: 10.1074/jbc.M109.058305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goffinet C, et al. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe. 2009;5:285–297. doi: 10.1016/j.chom.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 43.Goffinet C, et al. Antagonism of CD317 restriction of human immunodeficiency virus type 1 (HIV-1) particle release and depletion of CD317 are separable activities of HIV-1 Vpu. J Virol. 2010;84:4089–4094. doi: 10.1128/JVI.01549-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dube M, et al. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010;6:e1000856. doi: 10.1371/journal.ppat.1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hauser H, et al. HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology. 2010;7:51. doi: 10.1186/1742-4690-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bour S, et al. The envelope glycoprotein of human immunodeficiency virus type 2 enhances viral particle release: a Vpu-like factor? J Virol. 1996;70:820–829. doi: 10.1128/jvi.70.2.820-829.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bour S, et al. Naturally occurring amino acid substitutions in the HIV-2 ROD envelope glycoprotein regulate its ability to augment viral particle release. Virology. 2003;309:85–98. doi: 10.1016/s0042-6822(02)00128-9. [DOI] [PubMed] [Google Scholar]
- 48.Lopez LA, et al. Ebola virus glycoprotein counteracts BST-2/Tetherin restriction in a sequence-independent manner that does not require tetherin surface removal. J Virol. 2010;84:7243–7255. doi: 10.1128/JVI.02636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abada P, et al. Functional domains within the human immunodeficiency virus type 2 envelope protein required to enhance virus production. J Virol. 2005;79:3627–3638. doi: 10.1128/JVI.79.6.3627-3638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenberg ME, et al. Co-localization of HIV-1 Nef with the AP-2 adaptor protein complex correlates with Nef-induced CD4 down-regulation. EMBO J. 1997;16:6964–6976. doi: 10.1093/emboj/16.23.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pardieu C, et al. The RING-CH ligase K5 antagonizes restriction of KSHV and HIV-1 particle release by mediating ubiquitin-dependent endosomal degradation of tetherin. PLoS Pathog. 2010;6:e1000843. doi: 10.1371/journal.ppat.1000843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sawyer SL, et al. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004;2:E275. doi: 10.1371/journal.pbio.0020275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sawyer SL, et al. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci U S A. 2005;102:2832–2837. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ortiz M, et al. Evolutionary trajectories of primate genes involved in HIV pathogenesis. Mol Biol Evol. 2009;26:2865–2875. doi: 10.1093/molbev/msp197. [DOI] [PubMed] [Google Scholar]
- 55.Dazza MC, et al. Characterization of a novel vpu-harboring simian immunodeficiency virus from a Dent’s Mona monkey (Cercopithecus mona denti) J Virol. 2005;79:8560–8571. doi: 10.1128/JVI.79.13.8560-8571.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Courgnaud V, et al. Characterization of a novel simian immunodeficiency virus with a vpu gene from greater spot-nosed monkeys (Cercopithecus nictitans) provides new insights into simian/human immunodeficiency virus phylogeny. J Virol. 2002;76:8298–8309. doi: 10.1128/JVI.76.16.8298-8309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Courgnaud V, et al. Identification of a new simian immunodeficiency virus lineage with a vpu gene present among different cercopithecus monkeys (C. mona, C. cephus, and C. nictitans) from Cameroon. J Virol. 2003;77:12523–12534. doi: 10.1128/JVI.77.23.12523-12534.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao F, et al. Origins of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature. 1999;397:436–441. doi: 10.1038/17130. [DOI] [PubMed] [Google Scholar]
- 59.Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 60.Keele BF, et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science. 2006;313:523–526. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bailes E, et al. Hybrid origin of SIV in chimpanzees. Science. 2003;300:1713. doi: 10.1126/science.1080657. [DOI] [PubMed] [Google Scholar]
- 62.Yang SJ, et al. Anti-tetherin activities in Vpu-expressing primate lentiviruses. Retrovirology. 2010;7:13. doi: 10.1186/1742-4690-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mansfield KG, et al. Origins of simian immunodeficiency virus infection in macaques at the New England Regional Primate Research Center. J Med Primatol. 1995;24:116–122. doi: 10.1111/j.1600-0684.1995.tb00156.x. [DOI] [PubMed] [Google Scholar]
- 64.Murphey-Corb M, et al. Isolation of an HTLV-III-related retrovirus from macaques with simian AIDS and its possible origin in asymptomatic mangabeys. Nature. 1986;321:435–437. doi: 10.1038/321435a0. [DOI] [PubMed] [Google Scholar]
- 65.Hahn BH, et al. AIDS as a zoonosis: scientific and public health implications. Science. 2000;287:607–614. doi: 10.1126/science.287.5453.607. [DOI] [PubMed] [Google Scholar]
- 66.Gao F, et al. Human infection by genetically diverse SIVsm-related HIV-2 in West Africa. Nature. 1992;358:495–499. doi: 10.1038/358495a0. [DOI] [PubMed] [Google Scholar]
- 67.de Silva TI, et al. HIV-2: the forgotten AIDS virus. Trends Microbiol. 2008;16:588–595. doi: 10.1016/j.tim.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 68.Cao W, et al. Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J Exp Med. 2009;206:1603–1614. doi: 10.1084/jem.20090547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuda A, et al. Large-scale identification and characterization of human genes that activate NF-kappaB and MAPK signaling pathways. Oncogene. 2003;22:3307–3318. doi: 10.1038/sj.onc.1206406. [DOI] [PubMed] [Google Scholar]