

Summary
Regulation of nuclear receptor (NR) activity is driven by alterations in the conformational dynamics of the receptor upon ligand binding. Previously we demonstrated that hydrogen/deuterium exchange (HDX) can be applied to determine novel mechanism of action of PPARγ ligands and in predicting tissue specificity of selective estrogen receptor modulators. Here we applied HDX to probe the conformational dynamics of the ligand binding domain (LBD) of the vitamin D receptor (VDR) upon binding its natural ligand 1α,25-dihydroxyvitamin D3 (1,25D3), and two analogs, alfacalcidol and ED-71. Comparison of HDX profiles from ligands in complex with the LBD with full-length receptor bound to its cognate receptor retinoid X receptor (RXR) revealed unique receptor dynamics that could not be inferred from static crystal structures. These results demonstrate that ligands modulate the dynamics of the heterodimer interface as well as providing insight into the role of AF-2 dynamics in the action of VDR partial agonists.
Keywords: Vitamin D receptor, nuclear receptor, agonist, Hydrogen/deuterium exchange, protein dynamics, mass spectrometry
Introduction
The vitamin D receptor (VDR) is a ligand-dependent transcription factor and member of the nuclear receptor (NR) superfamily (Evans, 1988) that classically functions as a heterodimer with retinoid X receptor α (RXRα) (Forman et al., 1995) to control expression of genes traditionally associated with the regulation of mineral homeostasis. However, the scope of VDR biology has now been implicated in a wide range of physiological cellular responses including proliferation, differentiation and immunomodulation (Nagpal et al., 2005). The heterodimeric receptor complex is activated upon binding to 1α,25-dihydroxyvitamin D3 (1,25D3), the active form of vitamin D. Upon binding to vitamin D response elements (VDRE) on VDR target genes, liganded heterodimer recruits or dissociates coactivator/corepressor proteins that ultimately modulate the transcriptional output of the complex. Where molecular detail of VDR/RXR transactivation has been described, the chromatin environment, interacting protein partners, and temporal kinetics in and around the DNA-bound VDR/RXR complex can be highly diverse depending on the genomic target and cellular context in which such regulation is occurring (Meyer et al., 2007).
In addition to its well-described transcriptional regulation activities, VDR also displays the ability to rapidly initiate several key signal transduction pathways in a ligand-dependent manner that does not to require RXR (Mizwicki and Norman, 2009). These rapid signaling events appear to be initiated by ligand-bound VDR outside the nucleus perhaps within cytoplasmic signaling complexes at or near the plasma membrane. These non-genomic activities of VDR may occur via ligand-dependent recruitment of various co-interacting proteins in this cellular locale along with the appropriate ligand-induced VDR conformation (Mizwicki et al., 2004; Norman et al., 2004). Mizwicki et al have recently put forth a structural ensemble model to explain the mechanistic distinction between genomic vs. non-genomic activities (Mizwicki and Norman, 2009). However, this model remains to be empirically proven.
The VDR agonist 1,25D3 is used clinically for the treatment of renal osteodystrophy, osteoporosis, psoriasis, cancer, autoimmune diseases, and prevention of graft rejection. However, its therapeutic applications have been limited due to untoward effects associated with hypermineralization. As a result, drug discovery efforts have been focused on the development VDR ligands with improved side effect profile while maintaining maximal efficacy. One such ligand, the synthetic 1,25D3 analog alfacalcidol (1-hydroxycholecalciferol), was developed and it was determined that in vivo this analog is converted rapidly to 1,25D3 via 25-hydroxylase-mediated 25-hydroxylation primarily in liver and bone (Nishii, 2003). While it has been reported that alfacalcidol offers an improved therapeutic index, and as such, has been approved for treatment of osteoporosis in Japan and Europe (Cheskis et al., 2006), it is likely that alfacalcidol is a short-lived pro-drug rapidly converted to 1,25D3. Thus, clinical differentiation of 1,25D3 and alfacalcidol in vivo is likely due to differences in pharmacokinetic/pharmacodynamic (PK/PD) profiles of these two compounds.
Drug discovery efforts also lead to the development of ED-71, a VDR ligand which has been shown in vivo to have less hypercalcemia liability than 1,25D3 (Cheskis et al., 2006). While 1,25D3 and ED-71 have very similar chemical structures (Figure 1A) and comparable binding affinities for VDR, they afford distinct pharmacological profiles both in cellular and animal models. Ligand activation of NRs involves both ligand binding and ligand-induced conformational change of the receptor surface to facilitate cofactor protein interaction. This would suggest that the different pharmacology observed for ED-71 is related to the subtle changes in the chemical structure of the ligand leading to differences in protein-ligand contacts within the ligand binding pocket (LBP) imparting distinct conformations of the receptor-ED-71 complex. These distinct receptor-ligand complexes (VDR-1,25D3 and VDR-ED-71) would differentially interact with the milieu of cofactor proteins resulting in differences in transcriptional output following ligand treatment. As such, analysis of the molecular details of these conformational changes and subsequent interactions could provide an avenue for the development of VDR ligands with improved therapeutic index. The unique biological and pharmacological behavior of VDR ligands such as ED-71 has led to their classification as selective VDR modulators or VDRMs (Ma et al., 2006).
Figure 1.

VDR ligands and transcriptional responses. A) Chemical structures of the ligands. B) HEK293 T cells were transfected with the 4xVDRE reporter plasmid and VDR-FL or empty vector and treated with different VDR modulators (10 nM and 100 nM) or DMSO.
Hydrogen/deuterium exchange (HDX) particularly when coupled with mass spectrometry (MS) has emerged as a rapid and sensitive approach for characterization of protein folding, protein-protein interactions, protein-ligand interactions, and has been proven to provide complementary information to that gained via x-ray crystallography (Chandra et al., 2008; Hamuro et al., 2006; Hsu et al., 2009; Iacob et al., 2009). Our laboratory and others have successfully applied HDX to mechanistic analysis of NR activation (Bruning et al., 2007; Chalmers et al., 2007; Dai et al., 2009; Dai et al., 2008; Hamuro et al., 2006; Hamuro et al., 2003; Yan et al., 2004; Yan et al., 2007). In one example we described a novel mechanism of ligand-activation of PPARγ (peroxisome proliferator-activated receptor gamma) where different binding modes were detected for full agonists as compared to partial agonists and that partial agonists activate the receptor in a helix-12 independent fashion relying on stabilization of other regions of the LBP (Bruning et al., 2007). In another example we applied HDX to classify various selective ERα modulators (SERMs) based on their HDX signatures and these signatures were determined to be correlative to the pharmacology observed with these ligands in both pre-clinical and clinical settings (Dai et al., 2008). Most recently, we used HDX to reveal unique binding patterns of estrogens to ERα and ERβ (Dai et al., 2009). Although these studies were performed with isolated ligand binding domains (LBDs) of the specific NR, HDX analysis of intact PPARγ/RXRα heterodimer was published along with the crystal structure of the intact complex (Chandra et al., 2008). The HDX profile of PPARγ in the intact complex is in good agreement with that previously published for the LBD.
Based on these previous studies we postulated that HDX analysis would provide insight into the mechanism of ligand activation of the vitamin D receptor. To this end, we used HDX to probe the conformational dynamics of the LBD of VDR in complex with three ligands, 1,25D3, ED-71 and alfacalcidol. To examine the impact of the presence of the cognate co-receptor on ligand-induced dynamics of VDR, HDX profiles were also obtained with these ligands in complex with full length VDR/RXRα.
Comparison of the HDX profiles from ligands described above in complex with the VDR LBD to profiles obtained on full-length receptor heterodimerized with RXR revealed unique receptor dynamics that could not be inferred from static crystal structures. These comparisons demonstrate that VDR ligands modulate the dynamics of the VDR/RXR heterodimer interface as well as provide insight into the role of AF-2 dynamics in the action of VDR partial agonists. Furthermore, we demonstrate that these HDX profiles are predictive of the ligand-dependent functional behavior of VDR/RXRα in cofactor peptide interaction assays and cell-based transactivation activities.
Results
Characterization of VDR ligands
Cell-based reporter gene assays were used to assess the transcriptional potency and efficacy of 1,25D3, ED-71 and alfacalcidol ligands. Treatment of HEK293T cells transfected with a multimerized 4xVDRE luciferase reporter with 1,25D3 (10 nM) resulted in weak transactivation (~1.6 fold over DMSO only; Figure 1B) of luciferase. Under the same conditions, 10 nM ED-71 and alfacalcidol failed to stimulate expression of the reporter. The poor response of the reporter in this system likely reflects the low expression of the VDR in these cells. As expected, treatment of cells co-transfected with reporter and wild-type VDR with 10 nM 1,25D3 resulted in robust transactivation of luciferase (~10 fold over DMSO only; Figure 1B). Under the same conditions, treatment with ED-71 (10 nM) and alfacalcidol (10 nM) resulted in seven- and two-fold induction of the reporter respectively as compared to DMSO only (Figure 1B). Increasing the concentration of the ligands to 100 nM reveals that ED-71 reaches maximal efficacy matching that of 1,25D3 while the maximal induction of alfacalcidol is approximately 20% lower (Figure 1B).
We next examined the ability of these three ligands to modulate the ability of VDR to interact with an array of cofactor protein fragments using a biochemical assay. The cofactor peptide library, made up predominately of LXXLL-containing motifs, was designed from a full range of VDR interacting proteins including those of DRIP/TRAP and the p160 coactivator family. We utilized luminex-based xMAP technology to assess the global peptide interaction capacity while Alphascreen technology was used to quantify DRIP205-2 and SRC-1 NR2 peptide interactions independently. Importantly, both luminex and Alphascreen reactions were constructed such that signal was dependent on the formation of the heterodimer complex via Flag-hRXRα, as this was the source of signal generation in both settings. Controls were performed to verify that data generated was dependent of the presence of the heterodimer partner and could not be a function of homodimerization by either RXRα or VDR (data not shown).
Upon addition of saturating concentrations of each ligand (1μM) to the VDR and RXRα containing reaction, a highly significant but wide range of cofactor peptide interactions occurred across the peptide spectrum (Figure 2A). Most notably, the DRIP205, PGC-1, SRC, RIP140, and SHP/DAX peptide interactions were significantly driven by both ED-71 and 1,25D3. Interestingly, while the overall peptide interaction fingerprint for ED-71 vs. 1,25D3 was very similar, there were subtle differences in the magnitude of the various peptide interactions between these two ligands. However, alfacalcidol-bound VDR/RXRα clearly lacked capacity to drive significant peptide interactions across the entire peptide spectrum by comparison to ED-71 or 1,25D3 consistent with partial agonist activity. To quantify this ligand distinction, Alphascreen technology was used to measure individual cofactor peptide affinities for the ligand-bound VDR/RXRα complex. DRIP205-2 and SRC-1 NR2 cofactor peptides were chosen and a full concentration range of each ligand was tested (Figure 2B). Again, both ED-71 and 1,25D3 show robust but similar ligand and concentration-dependent capacities to interact with each cofactor peptide. Interestingly, there was a subtle but significant difference between ED-71 vs. 1,25D3 driven interaction with the SRC-1 NR2 peptide where a greater maximal interaction is achieved with 1,25D3 over ED-71 (~17% difference) while sharing similar potencies (Figure 2B). In agreement with luminex data (Figure 2A), alfacalcidol displays partial agonist activity with a reduced efficacy of interaction with the cofactor peptides (~57% and ~66% difference by comparison to 1,25D3). Alfacalcidol was also displayed significantly reduced potency relative to 1,25D3 and ED-71 (Figure 2B). The lack of the 25-hydroxyl group in alfacalcidol likely plays a critical role in the partial activity of this compound since the 25-hydroxyl group of 1,25D3 is known to play a role in positioning of H12 (Rochel et al., 2000). In general, the partial agonist activity of alfacalcidol was detected in both the biochemical and cell-based assays, but the partial agonist activity was somewhat less evident in the cell-based assay. Since HEK293 cells express vitamin D 25-hydroxylase (CYP2R1) (Cheng et al., 2004; Cheng et al., 2003) it is likely that alfacalcidol is 25-hydroxylated in the cell-based assay yielding a more efficacious compound than detected in the biochemical assays.
Figure 2.

Quantitative measurement of cofactor peptide interactions. A) Full length human His-VDR and Flag-RXR proteins were added to a multiplexed streptavidin-based bead-bound cassette of biotinylated NHR cofactor peptides. VDR ligands were added to this mixture at fully saturable concentrations (1μM) and incubated with shaking for 2hrs. Quantitation of VDR/ligand/RXR/bead-bound cofactor peptide complexes were measured using xMAP-based BioRad Luminex 120 instrument and Applied Cytometry Systems StarStation software. Cofactor peptide interactions with VDR/RXR are displayed as a heat map (fold interaction vs. the Apo DMSO state) using Spotfire DecisionSuite software. B) Using anti-Flag/streptavidin bead-based alphascreen technology, full length human His-VDR and Flag-RXR proteins were incubated with either biotinylated SRC-1 NR2 or DRIP205-2 33-mer cofactor peptides. VDR ligands were added to this mixture with increasing concentration (10 nM and 100 nM) and after 15min the formation of VDR/RXR/peptide complexes were quantified using a PerkinElmer Envision 2103 Multilabel Reader. Data analysis and curve-fitting was performed using GraphPad Prizm software.
HDX analysis of Apo VDR-LBD
The following studies were designed to provide optimal comparison between HDX and co-crystal structures. To that end the initial HDX experiments were carried out using the same domain context as was available in the PBD, which in the case of VDR is the isolated LBD. Prior to analyzing receptor-ligand complexes, HDX was used to characterize the conformational dynamics of the unliganded apo VDR LBD. As was elegantly shown by Englander and coworkers, the rate of amide hydrogen exchange with solvent deuterium is highly dependent on local fluctuations in protein structure (Englander and Kallenbach, 1983). While there is no published crystal structure of apo VDR LBD, HDX analysis provides information on the dynamics of functionally important regions of this domain such as H3 and AF2 (H3-H4 loop and H12) as well as relative dynamics of the LBP. HDX kinetics for 42 VDR peptic peptides covering ~90 percent of the construct used in this study (71 percent of the wt sequence of the LBD) were measured at specific time points. The average deuterium exchange (n=3) behavior of individual peptic peptides derived from apo VDR at each time point was overlaid onto the crystal structure of VDR in complex with 1,25D3 (PDB:1DB1 with ligand removed to avoid confusion) as shown in Figure 3. These data show that the entire C-terminal region including the H11-12 loop is very dynamic with over 80% of amide hydrogens exchange with 10 s exposure to heavy water. This is consistent with the fact that for most apo NRs the H11-H12 loop (residues 404-411 in VDR) is highly disordered (random-coil) and H12 (residues 412–419 in VDR), while structured, is assumed highly dynamic. Peptides derived from regions adjacent to the “insertion domain” (residues 219–224), a region of VDR that is poorly structured (Rochel et al., 2000), exhibited rapid exchange with solvent deuterium. The β sheet region (residues 280–308) was also found to be highly dynamic, which supports the distinct characteristics from crystallography of ligand-bound VDR LBD in that the tip of β sheet is shifted outwards to the surface, leading to a larger LBP (Rochel et al., 2000). Interestingly, most regions that form the LBP (H1, residues 133–150; H3, residues 225–233; H5, residues 273–279; H6-H7 and β sheet, residues 280–308; H11, residues 391–403;) had reached maximal on-exchange within 30 seconds. This property may correspond to the plasticity of the LBP needed to undergo a large local fluctuation to accommodate ligand insertion and binding (Yan et al., 2004). Not all of the LBD was found to be highly dynamic as slow exchanging regions were found mainly in the central core of the sandwich structure of the protein with the exception of H5 (residues 273–279) which exhibited fast exchange. The peptides derived from the region covering 352–365 were protected from exchange even after 5 min incubation (only <5% exchanged), revealing that this region is highly ordered. Regions remote of the central layer of the sandwich structure were also protected from exchange such as H10 (residues 384–391), where maximal exchange over the duration of the experiment was less than 15 percent. A movie illustrating the exchange behavior of apo VDR LBD versus time is provided in the Supplemental material.
Figure 3.

Dynamics of apo VDR-LBD. The percentage of deuterium levels for all peptides of apo VDR-LBD is mapped with color for each exchange point onto PDB:1DB1; the color code is explained at the bottom of the figure. Regions colored as white represent peptide are not detected. See also Movie S1.
Differential HDX analyses of VDR LBD/ligand complexes
To understand how ligands mediate the transcriptional activation of VDR, we performed comprehensive differential HDX analysis of the VDR LBD in the presence and absence of ligand. This approach provides a measure of the localized ligand-induced perturbation in the conformational dynamics of the receptor. HDX kinetics of peptic peptides derived from the VDR LBD were measured and the average difference in percentage of incorporated deuterium between apo VDR LBD and ligand-bound VDR LBD for the triplicate analysis of six on-exchange time points (see Methods) are presented in Table 1. A negative value represents an increase in protection to exchange (less dynamic) in that region of the LBD when bound to ligand as compared to apo whereas a positive value represents a decrease in protection to exchange (more dynamic) in that region of the LBD when bound to ligand as compared to apo. All of the underlying percent deuterium (D %) vs. time (log scale) plots for all peptides displayed in Table 1 are displayed in Supplemental Figure 2S. Plots that show statistically significant differential HDX dynamics following binding of 1,25D3, ED-71 and alfacalcidol are indicated as such.
Table 1.
Comparison of the conformational dynamics induced by different VDR modulators between VDR-LBD and full length VDR/RXR heterodimer. The values represent the average difference in percentage of deuterium incorporation of apo VDR-LBD and apo full length VDR/RXR in presence of difference VDR modulators across all H/D exchange time points (%). The regions with statistically significant differential deuteration level were colored. Statistical summary from a two-way ANOVA between apo and ligand bound data, P< 0.001. The value in parentheses represents the charge state of the peptide ion. Exchange kinetics for 41 different regions of the receptor LBD were measured and shown in Figure S2. See also Figure S1.
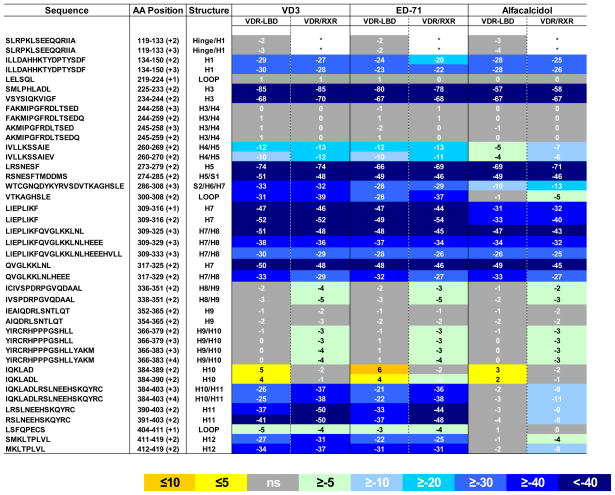 |
This exact peptide representing the hinge/H1 region (residues 119–133), the very N-terminus of VDR-LBD, was not identified in the HDX study of full length VDR/RXR heterodimer, as a different peptide is generated covering this region in the full length protein.
Upon close inspection the differential HDX kinetics induced by 1,25D3 binding to VDR correlates well with the co-crystal structure of VDR-LBD/1,25D3 (PDB:1DB1) (Rochel et al., 2000). As shown in Table 1 and Figure 4A, there are several regions within the VDR LBD that afforded dramatic protection to exchange following binding of 1,25D3. As can be inferred from PDB:1DB1 H12 (residues 412–419), a region known to be critical for coactivator binding and transcriptional activity of the receptor, interacts directly with 1,25D3 by van der Waals contacts with the methyl group of the ligand and internal interactions with other residues, some of which are directly interacting with the ligand (Rochel et al., 2000). Consistent with this structure, H12 showed significant protection to exchange upon binding 1,25D3. Regions 134–150 (H1), 225–244 (H3), 273–329 (H5-H7) and 391–403 (H11), comprising the ligand-binding pocket of VDR, showed decreased HDX kinetics, which can be attributed to hydrogen bonds formed between hydroxyl groups of 1,25D3 and residues within the LBP. As indicated by the active conformation of 1,25D3 in the LBP, the 1-OH group is hydrogen bonded to Ser-237 (H3) and Arg-274 (H5), the 3-OH group is hydrogen bonded to Ser-278 (H5) and Tyr-143 (H1), while the 25-OH group forms two hydrogen bonds with His-305 (loop H6-H7) and His-397 (H11) (Rochel et al., 2000). It would appear as the entire LBP is capable of forming a hydrogen bond network with 1,25D3 which would result in significant protection to exchange in these regions. In addition, it was observed that regions within the LBP that make Vander Wall contacts with ligand, such as L309, were also protected from exchange. Interestingly, regions remote of the LBP such as 317–325 (H7-H8) and 384–390 (H10), had altered HDX kinetics, a result that could not have been predicted from the VDR-LBD/1,25D3 co-crystal structure. Helix 10 is located within the dimerization interface of the VDR/RXR complex thus, we hypothesize that the ligand-dependent perturbation of this region impacts VDR interaction with RXR resulting in an overall stabilization of the VDR/RXR heterodimer. H10 is the only region of the LBD that is destabilized upon binding 1,25D3.
Figure 4.

Average differential HDX of each ligand mapped onto corresponding VDR-LBD/ligand crystal structures. A) 1,25D3 (PDB:1DB1), B) ED-71 (PDB:2HAR), C) Alfacalcidol (PDB:1DB1, since there is no VDR-LBD/alfacalcidol crystal structure available). The color legend shows the differential HDX between Apo VDR-LBD and ligand-bound ones. The regions in the crystal structure colored as white represent where are not covered in this study. See also Figures S1 and S3.
ED-71 is a 1,25D3 analog with a hydroxypropoxy group substituted at position 2 of the A ring (Figure 1A). ED-71 and 1,25D3 share similar HDX signatures (Table 1, Figure 4 and Figure 2S), which is consistent with the similarities observed by crystallography (Hourai et al., 2006). In the ED-71 VDR-LBD co-crystal structure, the position of H12 and the hydrogen bonds formed between 1-, 3-, 25- hydroxyl groups on the ligands and individual residuals from LBP are strictly maintained when compared to VDR/1,25D3 complex. The substitution of the 2-hydroxypropoxy group in ED-71 expels some water molecules from the LBP and thus disrupts the ability of Asp-144 in H1 to form a hydrogen bond with bound water. Disruption of this water-mediated hydrogen bond was reflected in the magnitude of HDX protection measured for H1 (Figure S1). Residues 134–150 exhibited a higher protection to exchange for 1,25D3 binding compared to that for ED-71 (29% and 23% respectively). Although the hydroxypropoxy group makes additional electrostatic and hydrophobic contacts with H1-loop-H2 and H3 (consistent with the observation that ED-71 has higher binding affinity than 1,25D3 (Hourai et al., 2006)), these contacts do not compensate for the loss of the hydrogen bond to H1 nor further stabilize H1-H3. The HDX data presented suggest that the only region of VDR LBD that undergoes differential conformational dynamics when comparing binding of 1,25D3 to ED-71 is H1. This observation is consistent with comparison of the two co-crystal structures.
Alfacalcidol is a precursor of 1,25D3 and lacks only a hydroxyl group at the C-25 position (Figure 1A). Interestingly, a very distinct HDX profile was obtained for the VDR/alfacalcidol complex when compared to VDR/1,25D3 and VDR/ED-71 complexes (Table 1 and Figure 4). Although there is no VDR-LBD/alfacalcidol X-ray structure available, we can predict the interactions between the protein and the ligand from HDX data. The regions 134–150 (H1), 234–244 (H3), 317–329 (H7-H8) present a similar reduction in deuterium incorporation induced by alfacalcidol compared to that induced by 1,25D3 and ED-71 (Table 1, Figure 4 and Figure 2S). This indicates that similar contacts are formed between the A, secoB, and C/D rings with the LBP of VDR. A feature of the alfacalcidol ligand compared to 1,25D3 and ED-71 is the notable absence of the 25-OH group. The crystal structure of VDR-agonists (Hourai et al., 2006; Rochel et al., 2000; Tocchini-Valentini et al., 2004) reveals that 25-OH forms hydrogen bonds with His-305 (loop H6–H7) and His-397 (H11). Therefore we expect that these two hydrogen bonds do not exist in VDR-alfacalcidol complex; a prediction that is supported by HDX analysis. HDX analysis clearly indicates that H11 (residues 391–403) and the loop of H6-H7 (residues 300–308) are not protected from exchange following alfacalcidol binding (Table 1, Figure 4C and Figure S2). In fact, regions of the protein containing His-397 and Tyr-401, which are involved in the internal-stabilization of H12, remain unchanged as compared to the apo receptor. Consequently, H12 remains in a highly dynamic state when bound to alfacalcidol. Moreover, the unprotected His-305 also increases the flexibility of the adjacent region (residues 309–316) which shows much higher protection following binding of 1,25D3 and ED-71. The region 225–233 (H3) also showed relatively lower protection, indicating the weaker interaction made in the alfacalcidol complex. These data suggest that although alfacalcidol binds to VDR, it is likely to display significant deficiency in AF-2 dependent transactivation and/or LXXLL-containing cofactor peptide interactions since it would be unable to stabilize and establish the proper cofactor interface. Hence, 25-hydroxylation is required to stabilize H12 and facilitate subsequent coactivator interaction at AF2.
Differential HDX of full length VDR in the context of VDR/RXR heterodimer
Having completed a thorough analysis of VDR LBD ligand complexes, we sought to compare these HDX profiles with that of full length VDR to determine if observations made using the isolated LBD could be translated to the full length receptor. Somewhat problematic, full length homodimeric VDR was found to be unstable in the absence of ligand making comprehensive HDX analysis impossible. As expected, addition of full length RXRα to full length VDR resulted in the formation of a stable complex even in the absence of ligand. Therefore, comprehensive HDX studies where performed on the heterodimeric complex in the absence and presence of ligands.
HDX data for VDR LBD/ligand and full length VDR/RXR/ligand complexes are shown in Table 1. Interestingly, the HDX profiles for the LBD and the full length complexes upon binding each of the three ligands are very similar. The magnitude of protection to exchange observed in most regions of the LBD was surprisingly similar with the some exceptions. For instance, subtle differences were observed in a region of the receptor that contains H8 and H9 (~5% difference in magnitude of protection). However, a larger difference in protection was observed for the region of the receptor that contains H10 and H11 region (~ 16% difference in magnitude of protection). It is important to note that these regions of the LBD are within or directly adjacent to the dimerization interface of VDR/RXR. These results suggest that the addition of ligand drives stronger interaction of VDR with RXR resulting in the formation of a more stable protein complex.
Discussion
To examine the mechanism of ligand activation of VDR we employed comprehensive differential HDX to study the conformational dynamics of VDR in the absence and presence of these three well characterized VDR ligands. Analysis of the HDX profile of 1,25D3 in complex with VDR reveals that a majority of the LBD protection is consistent with the co-crystal structure of VDR-LBD bound to 1,25D3. For instance, regions within the LBP of VDR that are in proximity and can make efficient hydrogen bonds to 1,25D3 were highly protected from exchange. In addition, it was observed that regions within the LBP that make Vander Wall contacts with ligand, such as L309, were also protected from exchange. Interestingly, regions remote of the LBP such as 317–325 (H7-H8) and 384–390 (H10), had altered HDX kinetics, a result that could not have been predicted from the VDR-LBD/1,25D3 co-crystal structure. Helix 10 is located within the dimerization interface of the VDR/RXR complex thus, we hypothesize that the ligand-dependent perturbation of this region impacts VDR interaction with RXR resulting in an overall stabilization of the VDR/RXR heterodimer.
Comparison of VDR LBD 1,25D3 and ED-71 complexes
The HDX profile of ED-71 was very similar to that of 1,25D3, with the exception of reduced protection in H1 (Table 1). It is possible that the small but statistically significant difference in HDX protection reflects the mechanistic differences of the two compounds that accounts for their different transcriptional activities and pharmacological properties. There have been many structural and biological studies addressing the important roles of H3, H5 and H12 in NR function (Danielian et al., 1992; Zhang et al., 1999; Zhang et al., 2005) and although H1 has not been addressed as intensively there have been reports indicating that H1 plays an important role in LBD function ((Pissios et al., 2001; Wu et al., 2002). Additionally, a recent study using HDX to characterize various PPARγ ligands showed differential protection in H1 of that receptor (Chalmers et al., 2007). Regardless, further studies such as mutagenesis will help to investigate the role of H1 in ligand-dependent transcriptional output of VDR.
Unique binding mode of Alfacalcidol
While there is no co-crystal available for VDR in complex with alfacalcidol, insight into its binding mode can be inferred from its HDX profile. It is evident that the lack of 25-hydroxylation of alfacalcidol results in a significantly different HDX profile for this ligand as compared to 1,25D3 and ED-71. Interestingly, in complex with alfacalcidol, H12 of VDR is not stabilized as compared to the apo receptor. It has been demonstrated that H12 is critical for cofactor protein interaction and ultimately the transcriptional output of the receptor. Furthermore, it is hypothesized that the position of H12 is the critical switch of the receptor between the inactive and active state. While HDX does not provide direct information on the location of H12, it does indicate its relative stability. Thus, the apo-like dynamics of H12 in both LBD and heterodimer HDX profiles with VDR/alfacalcidol suggests that alfacalcidol is deficient in its ability to stabilize H12 (Table 1, Figure 4C). This lack of stability would impair the receptors ability to stimulate the classical AF-2 dependent VDR transactivation activity. In agreement with the generated HDX profile we demonstrate that alfacalcidol, by comparison to 25-hydroxylated 1,25D3 and ED-71, clearly lacks full capacity to interact with LXXLL-containing cofactor peptides as well as displays significantly decreased efficacy consistent with partial agonist activity. Interestingly, biochemical-based cofactor interaction assays display a much greater loss in efficacy relative to 1,25D3 relative to the cell based assay. We postulate that this distinction may occur due to presence of 25-hydroxylase (CYP2R1) activity in HEK293 cells (Cheng et al., 2004; Cheng et al., 2003) resulting in the efficient conversion of alfacalcidol to 1,25D3 and therefore able to obtain maximal 1,25D3-like efficacy with increasing treatment dose of alfacalcidol.
In addition to H12, regions of the receptor contained within H7 (residues 309–316) were also less protected to exchange upon binding alfacalcidol as compared to 1,25D3 and ED-71. This is likely due to the absence of hydrogen bond between an adjacent region containing His 305 and the 25-OH of 1,25D3 and ED-71. Helix 7 is located at the interface of the dimer of VDR and RXR. Thus, the change in receptor dynamics of this region also suggest a more dynamic, less stable VDR/RXR heterodimer and is another unique feature exhibited by the partial agonist-bound VDR.
To better understand the interaction of alfacalcidol with VDR, docking studies were performed. As shown in Figure S3, docking suggests very good overlap of alfacalcidol with the active conformation of 1,25D3. However, the actual orientation of alfacalcidol in the LBP is predicted to be different from that of 1,25D3. Specifically, the three hydroxyl groups (1-OH, 3-OH, and 25-OH) of 1,25D3 and ED-71 should act as anchoring points to form hydrogen bonds with residues within the LBP (Figure 4A and Figure 4B). This would result in an elongated, curved ligand that comes in close contact with H3 (Rochel et al., 2000). HDX analysis would suggest that alfacalcidol is not elongated and curved within LBP, and consequently does not come in proximity to H3, as it only has two anchoring points (1-OH and 3-OH) on the same end of the molecule (ring A). This deviation might explain the reduced protection to exchange observed for H3 in the VDR/alfacalcidol complex as compared to 1,25D3 and ED-71.
Comparison of ligand-receptor dynamics of VDR LBD with full length VDR/RXR heterodimer
Initial experiments demonstrated that full length apo VDR, upon binding its cognate coreceptor RXR, forms a stable apo heterodimer complex that is amenable to detailed HDX analysis whereas the apo homodimer of VDR was not. Upon comparison, the HDX profiles for VDR LBD/ligand complexes were similar to those from analysis of full length VDR/RXR/ligand complexes. However, the subtle differences in the HDX profiles are intriguing. The VDR/RXR dimerization interface showed higher protection by the ligands in the full length receptor compared to the LBD alone, especially in H10-11 (Table 1). This enhanced protection in the full length receptor is attributed to the increased interactions between VDR and RXR, indicating that ligand binding influences heterodimer interactions in solution. This notion is more obvious when comparing the VDR LBD/alfacalcidol and the VDR/RXR/alfacalcidol complexes. In the VDR LBD complex, alfacalcidol does not impact the dynamics of H11 and H12 displaying apo-like HDX behavior due to the lack of 25-hydroxyl moiety on the ligand. Yet, in the VDR/RXR/alfacalcidol complex modest protection to exchange is observed likely arising from overall stabilization of the heterodimeric receptor complex. Thus, ligand binding indirectly impacts AF-2 dynamics via stabilization of the dimer interaction.
In summary, we have demonstrated that subtle differences in the chemical structure of VDR ligands can elicit distinct interactions within the receptor as determined by HDX. These observations are consistent with co-crystal structures and with biochemical and cellular assays. The perturbation in receptor dynamics induced by ligand binding to the isolated LBD as observed by HDX are well maintained in the full length heterodimer complex with the only deviations arising from within the VDR/RXR dimer interface. The HDX studies presented here provide novel insight into the molecular mechanisms of ligand activation of VDR and this new information may prove useful towards the design and development of VDR modulators with discriminate mechanistic behavior.
Experimental Procedures
Reagents
The luciferase reporter 4×VDRE containing a multimerized DR3 (AGGTCAtgaAGGTCA) is a kind gift from Dr. Ana Aranda. The full length VDR cDNA was cloned into an expressible vector pCMV-Sport 6 vector (Invitrogen). Vitamin D3 was purchased from Sigma (St. Louis, MO). ED-71 and alfacalcidol are from Lilly Research Laboratories (Indianapolis, IN).
Cell culture and transcriptional assays
Luciferase reporter assays were conducted using a multimerized VDRE luciferase reporter cotransfected with/or without a full length VDR construct into HEK293T cells (Kumar et al., 2010). Cells were seeded at 1~1.5 × 106 cells in a 6cm cell culture plate (Corning) in DMEM with 10 % heat inactivated FBS (Gibco BRL, Rockville, MD). Cells were transfected at a 3:1 ratio of fugene to DNA and 3 μg total amount of DNA was used for transfection. In details, day 1, 1.5 μg of reporter (4xVDRE-luc) was cotransfected with 1.5 μg of corresponding plasmid (VDR) or with an equivalent amount of empty vector. Day 2, transfected cells were transferred to a 384-well tissue culture plate and the medium replaced with DMEM+10% FBS. 4~5 hours later, cells were treated with compounds at 10 nM and 100 nM or DMSO. Day 3, luciferase activity was measured using Britelite™ (Perkin Elmer).
Luminex-based cofactor peptide interaction assays
A luminex-based biochemical assay (Berrodin et al., 2009) was used to assess the interaction of the receptor and cofactor drived by ligands. Thirty-three biotinylated cofactor peptides were synthesized by Anaspec, Inc (San Jose, CA). Amino acid peptide sequence design was based upon known LXXLL or LXXLL-like amphipathic helical cores from known NR cofactor proteins. Low capacity streptavidin beads were purchased from Radix Biosolutions (Georgetown, TX). To couple peptides to beads, 50μg/ml working concentrations of peptides were prepared in distilled H2O and used to couple to streptavidin beads overnight at 4°C. All bead/peptide conjugates were washed twice with PBS/BSA buffer (10mM NaH2PO4, 150mM NaCl, 0.1% (w/v) BSA, 2mM DTT, pH 7.4) and resuspended in 600μl of PBS/BSA buffer. All bead/peptide conjugates were mixed into a single homogenous bead mix prior to addition to the VDR/RXR/anti-Flag-Alexa532 complex. Alexa532 fluor-conjugated anti-Flag M2 antibody from Sigma (St. Louis, MO) was diluted to a final concentration of 0.8μg per ml in 1X luminex buffer (25mM Hepes, 100mM NaCl, 0.1% BSA, 2mM DTT, pH 7.4). For each 96-well luminex reaction, 10μl of anti-Flag M2-Alexa532 antibody was added to 10ul of 25X WT full length His-hVDR (60nM final) and Flag-hRXRα (40nM final). VDR/RXR/anti-Flag antibody complexing was allowed to occur for a minimum of 30 min prior to addition of 220μl cofactor peptide bead mix. 10μl of 25X VDR ligands (1μM final) were added to the appropriate wells. For each 250μl VDR/ligand/RXR/cofactor peptide luminex reaction, interactions were allowed to proceed for approximately 3 hours with shaking at room temperature. Quantification of cofactor peptide interaction was obtained by xMAP technology using the Bio-Plex 200 System and Suspension Array Platform (BioRad Laboratories, Hercules, CA).
Quantitative Alphascreen-based cofactor peptide interaction assays
Alphascreen assays (Rouleau et al., 2003) were performed in green-filtered light conditions in an aqueous assay buffer of PBS pH 7.2, 0.1% BSA and 2% DMSO in 384-well low volume white polystyrene assay plates (Greiner America #784705). Serial dilutions of 1α, 25 di-hydroxy vitamin D3, ED-71 or alfacalcidol were prepared in polypropylene plates and transferred to assay plates containing WT full length His-hVDR (30nM final). WT full length Flag-hRXRα (5nM final) was combined with 10ug/ml anti-flag acceptor and streptavidin donor Alphascreen beads (Perkin Elmer #6760613) and added to the assay plates. Finally, either biotinylated DRIP 205-2 peptide (20nM final) (DRIP205-2: Biotin- KGGTPPPVSSMAGNTKNHPMLMNLLKDNPAQDF) or biotinylated SRC1-NR2 peptide (30nM final) (SRC1-NR2: Biotin-KGGGGSCPSSHSSLTERHKILHRLLQEGSPSDI) was added and plates were incubated 15 minutes at room temperature before reading on an Envision 2103 multi-label reader (Perkin Elmer).
Protein and reagents for HDX analysis
His-hVDR LBD (residues 118–425, Δ[165–215]) was expressed in E. coli and purified via a three step purification Ni-NTA/Refolding/Q Sepharose FastFlow (QFF) chromatography. The final protein buffer is 10 mM Tris (pH8.0), 200mM NaCl, 2mM DTT. Full length WT His-hVDR and WT Flag-hRXRα were expressed in Baculovirus system and purified by Ni-NTASEC or Flag/SEC, respectively. The final protein buffer is 50mM Tris (pH8), 150mM NaCl, 10% glycerol, and 2mM DTT. The purity for each protein was >95% and identification of the protein was verified using SDS-PAGE and MALDI mass spectrometry. All purified proteins contained the epitope tag at the N-terminus. For HDX analysis, VDR LBD was incubated with compound at a ratio of 1:10 for 1 h on ice. The concentration of protein stock was 14 μM. For the full length HDX analysis, the heterodimer complex was formed by mixing VDR and RXRα at 1:1 molar ratio and the final concentration for each receptor was ~10 μM, respectively. VDR modulators were added at 10 fold molar excess to receptor.
Hydrogen/deuterium exchange mass spectrometry
Differential, solution phase HDX experiments were performed with a LEAP Technologies Twin HTS PAL liquid handling robot interfaced with an Orbitrap mass spectrometer (Exactive, ThermoFisher Scientific) (Chalmers et al., 2006). Each exchange reaction was initiated by incubating 4 μL of protein complex (with or without VDR modulators) with 16 μL of D2O protein buffer for a predetermined time (10s, 30s, 60s, 300s, 900s and 3600s) at 4 °C. The exchange reaction was quenched by mixing with 30 μL of 3 M Urea, 1% TFA at 1 °C. The mixture was passed across at an in-house packed pepsin column (2mm × 2cm) at 200 ul/min and digested peptides were captured onto a 2mm × 1cm C8 trap column (Agilent) and desalted (total time for digestion and desalting was 2.5 min). Peptides were then separated across a 2.1mm × 5cm C18 column (1.9μ Hypersil Gold, Thermo Scientific) with linear gradient of 4%–40% CH3CN, 0.3 % formic acid, over 5 min. Protein digestion and peptide separation were performed within a thermal chamber (Mécour) held at 1°C to reduce D/H back exchange. Mass spectrometric analyses were carried out with capillary temperature at 225 °C and data were acquired with a measured resolving power of 65,000 at m/z 400. Three replicates were performed for each on-exchange time point.
Peptide Identification and HDX data processing
MS/MS experiments were performed with a linear ion trap mass spectrometer (LTQ, ThermoFisher). Product ion spectra were acquired in a data-dependent mode and the five most abundant ions were selected for the product ion analysis. The MS/MS *.raw data files were converted to *.mgf files and then submitted to Mascot (Matrix Science, London, UK) for peptide identification. Peptides included in the peptide set used for HDX had a MASCOT score of 20 or greater. The MS/MS MASCOT search was also performed against a decoy (reverse) sequence and ambiguous identifications were ruled out. The MS/MS spectra of all of the peptide ions from the MASCOT search were further manually inspected and only those verifiable are used in the coverage. The intensity weighted average m/z value (centroid) of each peptide isotopic envelopes were calculated with a new version of our in-house developed software; HD Desktop (Pascal et al., 2009). The deuterium level was calculated as described previously(Zhang and Smith, 1993). Deuterium level (%)= {[m(P)-m(N)]/[m(F)-m(N)]}×100%, where m(P), m(N) and m(F) are the centroid value of partly deuterated peptide, non-deuterated peptide and fully deuterated peptide, respectively. The corrections for back-exchange were made based on an estimated 70% deuterium recovery and accounting for the known 80% deuterium content of the on-exchange buffer.
Highlights.
Dynamics of apo VDR are presented for which no crystal structure is available.
HDX of 1DB1 & 2HAR show perturbation of dimer interface upon ligand binding.
HDX demonstrates the role of 25-hydroxyl group for the activation of VDR.
HDX profiles of VDR LBD and intact RXR/VDR provide insight into dimer interface.
Supplementary Material
Acknowledgments
We are grateful for support from Mark Southern and Scooter Willis for software analyzing the HDX data, and John Bruning for docking studies. This work was supported in part by the Intramural Research Program of the National Institutes of Health National Institute of Mental Health [Grant U54-MH074404: H. Rosen PI] and by the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM084041].
Abbreviations
- NR
nuclear receptor
- HDX
hydrogen deuterium exchange
- VDR
vitamin D receptor
- RXR
retinoid X receptor
- 1,25D3
1a, 25-dihydroxyvitamin D3
- LBD
ligand binding domain
- LBP
ligand binding pocket
- VDRE
vitamin D response element
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berrodin TJ, Chang KC, Komm BS, Freedman LP, Nagpal S. Differential biochemical and cellular actions of Premarin estrogens: distinct pharmacology of bazedoxifene-conjugated estrogens combination. Mol Endocrinol. 2009;23:74–85. doi: 10.1210/me.2008-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JB, Chalmers MJ, Prasad S, Busby SA, Kamenecka TM, He Y, Nettles KW, Griffin PR. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure. 2007;15:1258–1271. doi: 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, He Y, Hendrickson CL, Marshall AG, Griffin PR. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2006;78:1005–1014. doi: 10.1021/ac051294f. [DOI] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, Southern MR, Griffin PR. A two-stage differential hydrogen deuterium exchange method for the rapid characterization of protein/ligand interactions. J Biomol Tech. 2007;18:194–204. [PMC free article] [PubMed] [Google Scholar]
- Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F. Structure of the intact PPAR-gamma-RXR-alpha nuclear receptor complex on DNA. Nature. 2008:350–356. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci U S A. 2004;101:7711–7715. doi: 10.1073/pnas.0402490101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JB, Motola DL, Mangelsdorf DJ, Russell DW. De-orphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxilase. J Biol Chem. 2003;278:38084–38093. doi: 10.1074/jbc.M307028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheskis BJ, Freedman LP, Nagpal S. Vitamin D receptor ligands for osteoporosis. Curr Opin Investig Drugs. 2006;7:906–911. [PubMed] [Google Scholar]
- Dai SY, Burris TP, Dodge JA, Montrose-Rafizadeh C, Wang Y, Pascal BD, Chalmers MJ, Griffin PR. Unique Ligand Binding Patterns between Estrogen Receptor alpha and beta Revealed by Hydrogen-Deuterium Exchange. Biochemistry. 2009 doi: 10.1021/bi901149t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai SY, Chalmers MJ, Bruning J, Bramlett KS, Osborne HE, Montrose-Rafizadeh C, Barr RJ, Wang Y, Wang M, Burris TP, et al. Prediction of the tissue-specificity of selective estrogen receptor modulators by using a single biochemical method. Proc Natl Acad Sci U S A. 2008;105:7171–7176. doi: 10.1073/pnas.0710802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian PS, White R, Lees JA, Parker MG. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 1992;11:1025–1033. doi: 10.1002/j.1460-2075.1992.tb05141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander SW, Kallenbach NR. Hydrogen-Exchange and Structural Dynamics of Proteins and Nucleic-Acids. Quarterly Reviews of Biophysics. 1983;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman BM, Umesono K, Chen J, Evans RM. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell. 1995;81:541–550. doi: 10.1016/0092-8674(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Hamuro Y, Coales SJ, Morrow JA, Molnar KS, Tuske SJ, Southern MR, Griffin PR. Hydrogen/deuterium-exchange (H/D-Ex) of PPARgamma LBD in the presence of various modulators. Protein Sci. 2006;15:1883–1892. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y, Coales SJ, Southern MR, Nemeth-Cawley JF, Stranz DD, Griffin PR. Rapid analysis of protein structure and dynamics by hydrogen/deuterium exchange mass spectrometry. J Biomol Tech. 2003;14:171–182. [PMC free article] [PubMed] [Google Scholar]
- Hourai S, Fujishima T, Kittaka A, Suhara Y, Takayama H, Rochel N, Moras D. Probing a water channel near the A-ring of receptor-bound 1 alpha,25-dihydroxyvitamin D3 with selected 2 alpha-substituted analogues. J Med Chem. 2006;49:5199–5205. doi: 10.1021/jm0604070. [DOI] [PubMed] [Google Scholar]
- Hsu YH, Burke JE, Li S, Woods VL, Jr, Dennis EA. Localizing the membrane binding region of Group VIA Ca2+-independent phospholipase A2 using peptide amide hydrogen/deuterium exchange mass spectrometry. J Biol Chem. 2009;284:23652–23661. doi: 10.1074/jbc.M109.021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacob RE, Pene-Dumitrescu T, Zhang J, Gray NS, Smithgall TE, Engen JR. Conformational disturbance in Abl kinase upon mutation and deregulation. Proc Natl Acad Sci U S A. 2009;106:1386–1391. doi: 10.1073/pnas.0811912106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N, Solt LA, Conkright JJ, Wang Y, Istrate MA, Busby SA, Garcia-Ordonez RD, Burris TP, Griffin PR. The benzenesulfoamide T0901317 [N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluorometh yl)ethyl]phenyl]-benzenesulfonamide] is a novel retinoic acid receptor-related orphan receptor-alpha/gamma inverse agonist. Mol Pharmacol. 2010;77:228–236. doi: 10.1124/mol.109.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Khalifa B, Yee YK, Lu J, Memezawa A, Savkur RS, Yamamoto Y, Chintalacharuvu SR, Yamaoka K, Stayrook KR, et al. Identification and characterization of noncalcemic, tissue-selective, nonsecosteroidal vitamin D receptor modulators. J Clin Invest. 2006;116:892–904. doi: 10.1172/JCI25901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MB, Zella LA, Nerenz RD, Pike JW. Characterizing early events associated with the activation of target genes by 1,25-dihydroxyvitamin D3 in mouse kidney and intestine in vivo. J Biol Chem. 2007;282:22344–22352. doi: 10.1074/jbc.M703475200. [DOI] [PubMed] [Google Scholar]
- Mizwicki MT, Keidel D, Bula CM, Bishop JE, Zanello LP, Wurtz JM, Moras D, Norman AW. Identification of an alternative ligand-binding pocket in the nuclear vitamin D receptor and its functional importance in 1alpha,25(OH)2-vitamin D3 signaling. Proc Natl Acad Sci U S A. 2004;101:12876–12881. doi: 10.1073/pnas.0403606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizwicki MT, Norman AW. The vitamin D sterol-vitamin D receptor ensemble model offers unique insights into both genomic and rapid-response signaling. Sci Signal. 2009;2:re4. doi: 10.1126/scisignal.275re4. [DOI] [PubMed] [Google Scholar]
- Nagpal S, Na S, Rathnachalam R. Noncalcemic actions of vitamin D receptor ligands. Endocr Rev. 2005;26:662–687. doi: 10.1210/er.2004-0002. [DOI] [PubMed] [Google Scholar]
- Nishii Y. Rationale for active vitamin D and analogs in the treatment of osteoporosis. J Cell Biochem. 2003;88:381–386. doi: 10.1002/jcb.10328. [DOI] [PubMed] [Google Scholar]
- Norman AW, Mizwicki MT, Norman DP. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov. 2004;3:27–41. doi: 10.1038/nrd1283. [DOI] [PubMed] [Google Scholar]
- Pascal BD, Chalmers MJ, Busby SA, Griffin PR. HD desktop: an integrated platform for the analysis and visualization of H/D exchange data. J Am Soc Mass Spectrom. 2009;20:601–610. doi: 10.1016/j.jasms.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pissios P, Tzameli I, Moore DD. New insights into receptor ligand binding domains from a novel assembly assay. J Steroid Biochem Mol Biol. 2001;76:3–7. doi: 10.1016/s0960-0760(00)00144-8. [DOI] [PubMed] [Google Scholar]
- Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5:173–179. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
- Rouleau N, Turcotte S, Mondou MH, Roby P, Bosse R. Development of a versatile platform for nuclear receptor screening using AlphaScreen. J Biomol Screen. 2003;8:191–197. doi: 10.1177/1087057103252605. [DOI] [PubMed] [Google Scholar]
- Tocchini-Valentini G, Rochel N, Wurtz JM, Moras D. Crystal structures of the vitamin D nuclear receptor liganded with the vitamin D side chain analogues calcipotriol and seocalcitol, receptor agonists of clinical importance. Insights into a structural basis for the switching of calcipotriol to a receptor antagonist by further side chain modification. J Med Chem. 2004;47:1956–1961. doi: 10.1021/jm0310582. [DOI] [PubMed] [Google Scholar]
- Wu Y, Delerive P, Chin WW, Burris TP. Requirement of helix 1 and the AF-2 domain of the thyroid hormone receptor for coactivation by PGC-1. J Biol Chem. 2002;277:8898–8905. doi: 10.1074/jbc.M110761200. [DOI] [PubMed] [Google Scholar]
- Yan X, Broderick D, Leid ME, Schimerlik MI, Deinzer ML. Dynamics and ligand-induced solvent accessibility changes in human retinoid X receptor homodimer determined by hydrogen deuterium exchange and mass spectrometry. Biochemistry. 2004;43:909–917. doi: 10.1021/bi030183c. [DOI] [PubMed] [Google Scholar]
- Yan X, Perez E, Leid M, Schimerlik MI, de Lera AR, Deinzer ML. Deuterium exchange and mass spectrometry reveal the interaction differences of two synthetic modulators of RXRalpha LBD. Protein Sci. 2007;16:2491–2501. doi: 10.1110/ps.073019707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hu X, Lazar MA. A novel role for helix 12 of retinoid X receptor in regulating repression. Mol Cell Biol. 1999;19:6448–6457. doi: 10.1128/mcb.19.9.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Simisky J, Tsai FT, Geller DS. A critical role of helix 3-helix 5 interaction in steroid hormone receptor function. Proc Natl Acad Sci U S A. 2005;102:2707–2712. doi: 10.1073/pnas.0409663102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.