

Abstract
Purpose
The aim of the study was to resolve the genetic etiology in families having inherited cataracts.
Methods
Families afflicted with congenital/childhood cataracts were registered in Chennai and Orissa (India). Blood samples were collected from the probands and available family members. Selected functional candidate genes were amplified by polymerase chain reaction (PCR) and characterized by direct sequencing. Putative mutations were confirmed in healthy controls.
Results
We observed interesting new polymorphisms of ethnic specificity, some of frequent nature, such as a 3-bp deletion in intron 3 of CRYBB2 (encoding βB2-crystallin) and IVS1+9 c>t variation in HSF4 (encoding heat-shock factor 4). Some rare single nucleotide polymorphisms (SNPs) co-segregate with the respective phenotype such as IVS3+120c>a of CRYBB2, while M44V of CRYGD (encoding γD-crystallin), although found in association with blue dot opacity was seen in a few healthy controls too. We identified two new mutations co-segregating along with the respective cataract phenotype within the families that were not seen in healthy controls from India or Germany. These include two missense mutations; one in GJA3 (encoding gap junction protein α3, which is also referred to as connexin 46); the mutation affects codon 19 (T19M), and the corresponding phenotype is a posterior-polar cataract. The other missense mutation affects CRYBB2 (W59C; total cataract). Additionally, a cDNA variation (G54A) identified in a zonular cataract affects a highly conserved splice site of CRYBB2. This mutation, however, showed reduced penetrance in the family, which might be explained by different molecular consequences in the affected family members: nonsense-mediated decay of the mutated mRNA might have no clinical phenotype in heterozygotes, whereas the translation of the mutated mRNA is predicted to lead to a small hybrid protein (consisting of 16 amino acids of the βB2-crystallin and 18 new amino-acids), which might have a dominant-negative function in the lens.
Conclusions
This report identifies in families with childhood cataract some new alleles, which may be considered as causative for cataracts. Furthermore, we report some geographically restricted rare polymorphic sites, whose significance might be considered in some context as modifiers or alleles in sensitizing ocular lens toward cataractogenesis.
Introduction
Childhood cataracts are fairly common, and timely clinical intervention is important for a good visual prognosis. Globally, about 20 million children suffer from blindness; over 50% of them indicate a genetic basis, and one third is familial [1]. In India, cataract accounts for ~12% of childhood blindness [2]. When isolated, it exhibits Mendelian inheritance with autosomal dominance as the commonest mode. Molecular analysis revealed ~60 loci to be associated with several phenoypes of childhood cataracts (for recent reviews see [3,4]). Candidate genes include those coding for αA- and αB-crystallins (CRYAA; CRYAB), for βA1- or βA4-crystallin (CRYBA1, CRYBA4), for βB1 - βB3-crystallins (CRYBB1; CRYBB2; CRYBB3), for γC-, γD-, or γS-crystallins (CRYGC, CRYGD, CRYGS), for the beaded filament structural proteins (BFSP1; BFSP2), for connexin 46 (GJA3) or connexin 50 (GJA8), for the lens membrane intrinsic protein 2 (LIM2) and for the major intrinsic protein of lens fibers (MIP). Another group of candidate genes codes for transcription factors (heat shock factor 4, HSF4; paired-like homeodomain transcription factor 3, PITX3; paired-box gene 6, PAX6; eyes-absent 1, EYA1, and for a homolog to the musculoaponeurotic fibrosarcoma oncogene, MAF). Finally, also genes encoding enzymes were tested (glucoseaminyl(N-acetyl)transferase-2, GCNT2; chromatin modifying protein-4B, CHMP4). Among them, the crystallin-encoding genes are those having the highest probability to be involved in cataract formation; however, some of them are also associated with other diseases besides cataract [5]. Other known causes of congenital, hereditary cataracts are mutations affecting enzymes of sugar metabolism (e.g. galactokinase 1, GALK1 [6]) or other metabolic disorders like hyperferritinemia (FTL) [7]. Clinical as well as genetic heterogeneity of childhood cataracts has been well established. It is suggested that additional genes or environmental factors might act as modifiers [8]. The identified genes and their mutations give us some insight into the process of maintenance of lens transparency and may increase our understanding of the complexity in age related cataracts.
The present study is an attempt to screen five families (CBE21, DJC1, JEE13, JPM1, and SEC18) with autosomal dominant congenital or childhood cataract for possible mutations in selected candidate genes like CRYAA; CRYBB2; CRYGC and CRYGD; GJA3; GJA8,PITX3, and HSF4. We report the identification of three novel mutations - one in CRYBB2 (W59C) in a family (JPM1) with total cataract, and another in GJA3 (T19M) in a family (SEC18) with posterior polar cataract and the third one with reduced penetrance in CRYBB2 (G54A) in a proband (DJC1) with zonular cataract. The probable pathogenicity of these mutations as disease causing molecular lesions is discussed in the light of earlier reports.
Methods
As part of our ongoing study at the Regional Institute of Ophthalmology (RIO), Government Eye Hospital, Chennai, India, several families having visual impairment caused by hereditary cataracts were registered. One familial case (JPM1) was registered from the Rotary Eye Hospital at Cuttack, Orissa, India. The probands and their relatives were examined by senior pediatric ophthalmologists (P.S. and N.G.) with slit-lamp microscope (Zeiss, Oberkochen, Germany). Family history was recorded according to the information given by available family members. The study adopted the tenets of the Declaration of Helsinki, as family members were enlightened about the study, its outcome and their role in regional language before seeking informed consent as per standard norms. The study was also approved by the Institutional Ethical Committee of Dr. ALM - Post Graduate Institute of Basic Medical Sciences, University of Madras, India.
Blood samples (5–10 ml) were collected from available affected and unaffected family members. Genomic DNA was isolated from peripheral blood using salt extraction to precipitate contaminating proteins as described previously [9]. Genomic DNA was amplified by PCR for the exons (and their flanking regions) of the CRYAA [10], CRYBB2 [11], CRYGC, CRYGD [12], GJA8, GJA3 [13], HSF4 [14]), and PITX3 (Table 1) genes. PCR products were checked in 1.5% agarose gels and purified through Nucleospin columns (Macherey and Nagel, Düren, Germany). Sequencing was done commercially (GATC Biotech AG, Konstanz, Germany; or SequiServe, Vaterstetten, Germany) according to standard procedures.
Table 1. Primers for amplification of PITX3 from genomic DNA.
| Primer | Sequence | Tm (°C) | Size (bp) |
|---|---|---|---|
| PITX3-Ex1-L1 |
CTGCCATAAAGTGAATGGGCGC |
49–60 |
278 |
| PITX3-Ex1-R1 |
TCCAAGGTCCAGCAATAGCTCCTC |
|
|
| PITX3-Ex2-L1 |
CCATTACCCTGGTCTGTGTCTTCTCTC |
56–60 |
221 |
| PITX3-Ex2-R1 |
CCCGCACTGGGGATGAAGC |
|
|
| PITX3-Ex3-L1 |
TGGGCGCTCTGTGACCTGC |
56–60 |
273 |
| PITX3-Ex3-R1 |
CGCGGGTGCGAGTCGC |
|
|
| PITX3-Ex4-L1 |
CCTTCAGCCGCTGGGACC |
63–67 |
1000 |
| PITX3-Ex4-R1 | TCAAGCGCAACTTTGAATCATCAC |
The mutations were confirmed by the presence/absence of the cleavage site for restriction enzymes or by direct sequencing, if no informative restriction site was available. As controls, 30–100 ethnically matched healthy individuals were used. Additionally, we took 96 randomly chosen, healthy individuals of the KORA Survey 4 (Cooperative Health Research in the Region of Augsburg, Germany), which studied a population-based sample of 4,261 subjects aged 25–74 years over the period 1999–2001 [15].
Copy-number variation analysis was done using PCR, the cloning and counting approach as described previously [16]. Briefly, the following primers were used to amplify the region around exon 2 of CRYBB2 (UCSC Genome Browser [17]: human genome hg18, chr22:23,947,149–23,947,711): 5′-GAC CTC GTT TTT CCC TCC TC-3′ and 5′-GTG GCA AAA CAG GTA AGG GA-3′. Amplicons were cloned and around 90 clones per sample were sequenced. Copy number was determined by calculating the frequency/ratios of alleles at variations rs16979774, rs7291633, G54A, rs56191632, and rs739315. Obtained allele ratios were searched for statistical significant deviations (Fisher’s exact test) from 1:1 which is the typical situation for two copies per diploid genome (no copy number variability). A copy number, e.g., of three, would result in a ratio of 2:1 / 1:2 or frequency of 0.66 / 0.33.
To predict consequences of mutations or polymorphic sites, the Bioinformatics tool of the ExPASy proteomics server was adopted. For secondary structure predictions, we applied the GOR4 method [18]. To predict splice sites the NetGene2 World Wide Web Server from the Center for Biologic Sequence Analysis was employed (Technical University of Denmark [19]; ). Prediction of regulatory RNA motifs was performed using RegRNA, an integrated web server for identifying the homologs of regulatory RNA motifs and elements against an input mRNA sequence.
Results
Family CBE21
In family CBE21, the proband was referred from the Institute of Child Health, Chennai, India, as a case of congenital Rubella syndrome to the RIO Egmore Eye Hospital, Chennai. The child was 11 months at the time of case registration, had developmental delay, microcephaly, congenital heart disease, microcornea, nystagmus, and had dense nuclear opacity with peripheral cortical opacity of the lens. The child died at the age of 5 years. No DNA sample was collected from the child. The mother, when examined through slit lamp, was found to have blue dot opacities. One of the mother’s sib (II.6) at the age of 12 years is said to have undergone surgery for cataract. Follow up visit of this family revealed the family to have a second child who is said to be healthy. The pedigree of this family is given in Figure 1, and the case history of the family in Table 2.
Figure 1.
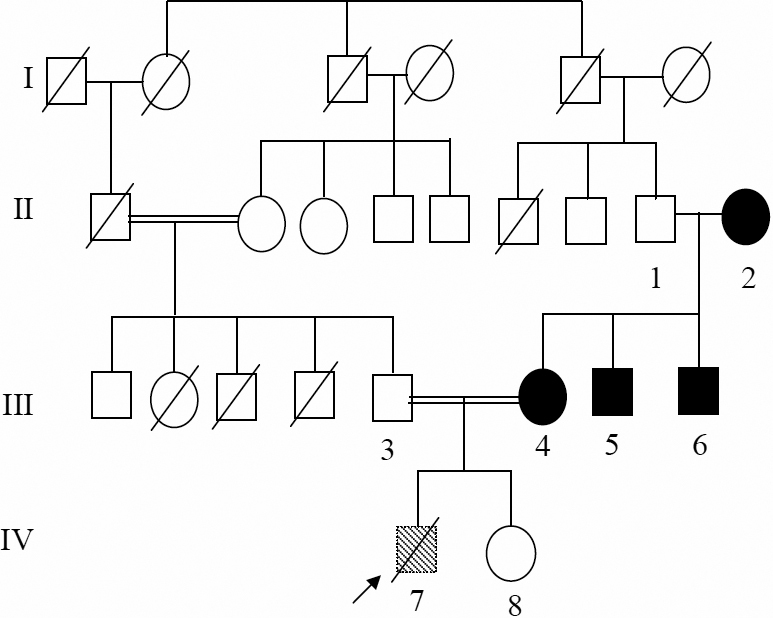
Cataract phenotype in family CBE21. Pedigree of the family CBE21 indicates consanguinity after one generation. The blue dot opacity appeared in the second generation and affects four members of the family. The deceased proband (IV-7) is indicated by an arrow. The numbers below the symbol indicate the laboratory number of the samples.
Table 2. Case history of family CBE21.
| Subject code | Relationship | Age (Years) | VA (L/R) | VA (L/R) | SLE |
|---|---|---|---|---|---|
| II.2 |
Proband’s grandmother |
55 |
6/60;6/60 |
6/6;6/9 |
Fine blue dot cataract opacity |
| III.5 |
Proband’s elder uncle |
21 |
6/60;6/60 |
6/6;6/9 |
Fine scattered blue dot opacities |
| III.6 | Proband’s younger uncle | 17 | 6/6;- | -;- | Fine blue dot opacities minimal and scattered |
The proband IV.7 was deceased at the age of 5 years. Subject III.6 was operated on at the age of 13 years for removal of a traumatic cataract in his right eye due to a stick injury at the age of 5 years.
Molecular genetics of Family CBE21
The mother’s genomic DNA was screened for all exons and their flanking sequences of some functional candidate genes (CRYAA, CRYGC, CRYGD, and PITX3). This functional candidate gene approach detected a mutation in exon 2 of CRYGD. Comparison of the wild-type sequence with that of the proband’s mother demonstrated a heterozygous C→T mutation at position 130 (C130T; Figure 2A), which leads to an amino acid exchange from Met to Val at pos. 44 (M44V; Figure 2B). Further, the mutation creates a new Alw21I restriction site (Figure 2B). Restriction digest showed co-segregation of the genotype with only one other affected member of the family (Figure 2C; III.6). The third affected member was PGM (II.2; 60 years), who also had blue dot opacities, but the restriction site for Alw21l was absent. This restriction site is absent in the proband’s father (III.3) and in unaffected relatives of other cataract probands (n=5; data not shown), it was also not detected in 60 healthy controls from the general population of India but was observed in 6 of the 96 healthy controls (KORA) from Germany. Therefore, it could not be considered as a causative mutation, but may be interpreted as a polymorphism with an allele frequency of 6.2% in the German population.
Figure 2.

CRYGD mutation in the family CBE21. A: Sequence analysis of exon 2 of CRYGD indicates heterozygosity (arrow) for the mother of the proband (III.4). B: Comparison of the wild-type sequence (WT) with the proband’s mother’s sequence (CBE21) demonstrates that the C→T mutation at cDNA position 130 leads to an amino acid exchange from Met to Val at pos. 44 (M44V); moreover, it shows the creation of a new Alw21I restriction site (underlined) in two (III.4 and III.6) of the affected members of the family checked, while the same was absent in the proband’s grand mother (II.2). One of the proband’s uncle (III.5) could not be checked (in the cDNA sequence, the A of the ATG start codon is counted as #1; in the amino-acid sequence, the first Met is counted as #1). C: Restriction analysis with (+) or without (-) the enzyme Alw21I in the members of the core family leads to an additional fragment of 370 bp in the mutant DNA (red); it demonstrates the presence of the mutation in the affected mother (III.4) and the affected brother of the proband’s mother (III.6). The asterisks mark the additional band of 370 bp indicating the mutation; the band of 533 bp indicates the undigested DNA. WT represents an independent control from the laboratory; M=marker.
Family JEE13
The proband, a female child of 6 years (as on 2005) of family JEE13 had a triangular central cataract surrounded by cataractous dots and fibers in the lens cortex (Figure 3A). The other eye (LE) was operated earlier on. The proband’s mother (23 years) was examined to have lamellar with peripheral coronary cataract at both eyes and her visual acuity being 3/60. The eyes of the proband’s sib (3 years) however are healthy. One of the mother’s sibs was also affected who has three healthy children (not clinically examined). The pedigree (Figure 3B) demonstrated an autosomal dominant mode of inheritance as the cataract appeared in all three generations affecting both sexes.
Figure 3.

Cataract phenotype in family JEE13. A: A lamellar cataract was observed in the 6-year-old proband. B: The pedigree of a 3-generation family demonstrates that the cataract appeared in all three generations affecting both sexes; it indicates an autosomal dominant mode of inheritance. The proband is indicated by an arrow.
Molecular genetics of Family JEE13
Molecular analysis of some functional candidate genes like CRYAA, CRYBB2, CRYGC, CRYGD, GJA3, GJA8, HSF4, and PITX3 revealed a C→T mutation at the beginning of intron 1 of HSF4 (position #9; Figure 4A); this heterozygous mutation was also present in the affected mother. The mutation leads to a loss of a HaeIII restriction site (Figure 4B) and segregates perfectly in the family (Figure 4C), and was not detected in 96 unrelated controls of the German population. However, among the 35 unrelated and healthy controls from India, 6 of them showed the same restriction pattern as observed in the cataract patients. Therefore, this SNP might be specific for the Indian ethnicity (~17%), and is not associated with cataract formation. This inference is further being supported by the fact that the mutation occurs outside of the conserved recognition sites for splicing; several splice site prediction programs failed to show any influence on splicing.
Figure 4.

HSF4 mutation in family JEE13. A: Sequence analysis of HSF4 genomic DNA indicates heterozygosity at position +9 of the first intron (red arrow) of the proband’s HSF4 gene. B: Comparison of the wild-type sequence (AB029347) with the proband’s sequence demonstrates a C→T exchange at position 9 of intron 1. The mutation leads to a loss of a HaeIII restriction site (underlined). C: Restriction analysis with (+) or without (-) the enzyme HaeIII in members of the family leads to an additional fragment of 98 bp in the mutant DNA (red); it demonstrates the presence of the mutation in the affected mother (II.3) and in the proband (III.4). The asterisks mark the additional band of 98 bp indicating the mutation; the band of 262 bp indicates the undigested DNA. C40 and WT represent independent controls from our laboratories; M=marker.
Family DJC1
The pedigree of the large family DJC1 is shown in Figure 5A. The phenotype “congenital zonular cataract” seems to appear spontaneously in the youngest daughter as proband (1.5 years as on 2005) whose visual acuity (by ivory ball testing) was found to be >6/60 at a distance of 0.5–1.0 m. Both eyes of the proband were operated on, with no other systemic involvement. Her parents were related as first degree cousins. Both the parents and her three sibs underwent complete ophthalmologic check-up and were declared to have regular vision.
Figure 5.

CRYBB2 mutation in family DJC1. A: The pedigree of family DJC1 indicates that a zonular cataract appeared in the youngest daughter of healthy, but consanguineous parents. There is no other report of any type of cataract over three generations. B: Sequence analysis of CRYBB2 genomic DNA indicates heterozygosity for the proband at cDNA pos. 54 (red arrow). C: Comparison of the wild-type sequence (Z99916) with the proband’s sequence demonstrates that the G→A exchange at cDNA-position 54 leads to an altered splice site; it is predicted that the mutated mRNA contains at least part of intron 2 (the A of the ATG start codon is counted as #1; in the amino-acid sequence, the first Met is counted as #1). The mutation leads to a loss of an Eco130I restriction site. D: Restriction analysis using the enzyme Eco130I creates a larger fragment of 216 bp in the mutant DNA (red); it demonstrates the presence of the mutation in the proband (III.4), but surprisingly also in the healthy mother (II.6) and one healthy sister (III.3) of the proband; the asterisks mark the additional band of 216 bp indicating the mutation. Undigested PCR fragments of 393 bp are indicated by “-” or “+” are digested PCR products. The small fragment of 51 bp in the wild-type DNA is not visible; the two bands of 165 and 177 in the wild type are not separated under the conditions used here. C1 and C2 represent independent controls from the laboratory; M=marker.
Molecular genetics of Family DJC1
Molecular analysis of functional candidate genes CRYAA, CRYBB2, CRYGC, CRGYD, GJA8, and PITX3 revealed a single heterozygous base-pair exchange at the last base of exon 2 of the CRYBB2 gene (cDNA-G54A; Figure 5B). The mutation does not affect the amino acid in the corresponding codon 18 (it remains Leu; Figure 5C), however, the A instead of the G destroys the common donor splice site, and a splice-site prediction program suggests that the corresponding splice site is completely lost. The most likely next donor splice site might be 338 bp downstream (calculated probability is 79%). If this would be true, the corresponding intronic sequences will be present in the mRNA and translated resulting in 16 novel amino acids; translation will then be terminated by a premature stop codon.
As the mutation leads to a loss of an Eco130I restriction site, its presence or absence was then tested in the core family (father II.5, mother II.6, and the siblings III.1–3) as well as in controls of German origin (n=96) and from unrelated controls of India (n=35). We did not observe the mutation in the German controls, also it was absent in controls from India. Surprisingly, the healthy mother (II.6) and one of the healthy sisters (III.3) showed the same digestion pattern as the proband. Therefore, we sequenced also the corresponding CRYBB2 exon in the parents (II.5 and II.6) and siblings (III.1–3) of the proband. The mother and the sister III.3 had a small, but significant fluorescence A peak suggestive for copy-number variability of the locus. Therefore, we quantified the allele frequencies of the cDNA-G54A mutation and surrounding SNPs, but the result indicated a 1:1 ratio of the respective alleles typical for a heterozygous single-copy locus. Therefore, this mutation has to be evidently described as having reduced penetrance. Since no operated lens material is available it remains speculative whether the 34-amino acid peptide is actually present in the cataractous lenses or if the mutated mRNA is subjected to nonsense-mediated decay in the clinically healthy family members.
Family JPM1
The proband, a female child of age 2.5 years at the time of registration, was diagnosed to have bilateral congenital cataract (Figure 6A) along with amblyopia and nystagmus in the left eye. There was no other associated ocular or systemic anomaly. She was operated for removal of total cataract in both eyes by lens aspiration, primary posterior capsulotomy and anterior vitrectomy. The mother had no history of any systemic ailments during pregnancy. The child’s developmental milestones were reported to be normal. The child was affected since birth. The father and a male sib were also affected; three of father‘s sibs were said to be affected with cataract. The entire pedigree is shown in Figure 6B.
Figure 6.

Cataract phenotype in family JPM1. A: The eye picture shows a central opacity of both lenses of the female proband (III.2; 2.5 years). B: The central lens opacity appeared in 4 out of 7 siblings of seemingly healthy parents (generation I); one of the brothers (generation II) transmitted the cataract to both his offspring (generation III) indicating an autosomal dominant mode of inheritance. The mutation might have originated in the germ cells of the healthy grandparents (generation I).
Molecular genetics of Family JPM1
Molecular analysis of some functional candidate genes like CRYAA, CRYBB2, CRYGC, CRYGD, GJA8, and PITX3 revealed a G→C mutation at the beginning of exon 4 of CRYBB2 at position 177 leading to an exchange of Trp by Cys (W59C; Figure 7A); the mutation is predicted not to affect the formation of 2nd Greek key motif. Rough computer-assisted prediction of biophysical properties suggests that the hydrophobicity of the area around W59 slightly increases (from 1.4 up to 1.7); although the isoelectric point of the protein is not altered. The mutation destroys a BseY1 restriction site (Figure 7B) which is seen only in affected family members (Figure 7C); it is not present in unrelated controls from India (n=100) or from Germany (n=96). Therefore, this mutation is most likely to be the causative genetic lesion for the cataract in this particular family.
Figure 7.
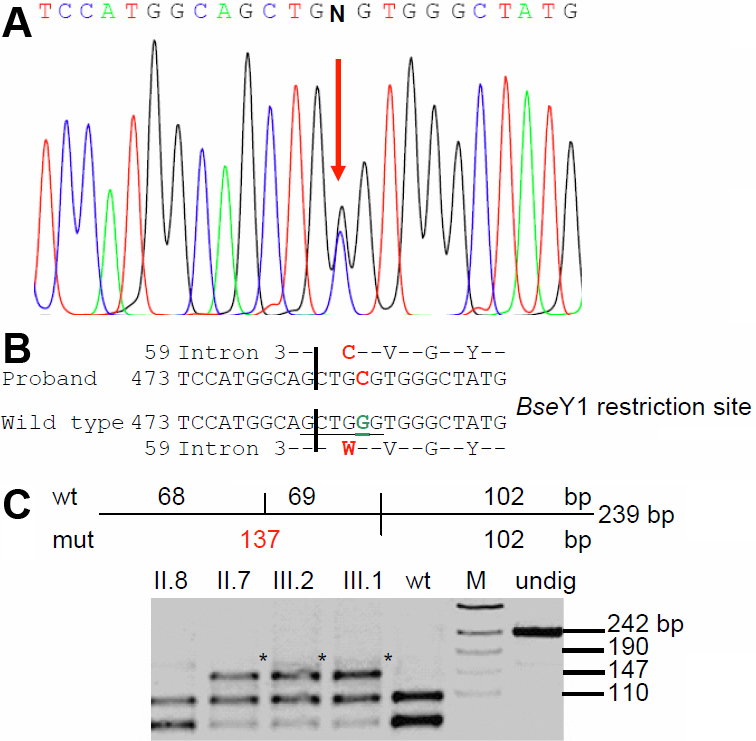
CRYBB2 mutation in family JPM1. A: Sequence analysis of CRYBB2 genomic DNA indicates heterozygosity in the proband’s father (II.7) at cDNA pos. 177 (red arrow). B: Comparison of the wild-type sequence (Z99916) with the proband’s father sequence demonstrates that the G→C exchange at cDNA-position 177 leads to an exchange of Trp by Cys (W59C). The BseY1 restriction site in the wild-type sequence is underlined. C: Restriction analysis using the enzyme BseYI leads to an additional fragment of 137 bp in the mutant DNA (red); it demonstrates the presence of the mutation in the affected father (II.7) and the affected brother (III.1) of the proband (III.2). The asterisks mark the additional band of 137 bp indicating the mutation; “undig” is an undigested PCR fragment of 239 bp. WT represents an independent control from the laboratory; M=marker.
Family SEC18
The proband (III.2) is a male child of 9 years at the time of case registration at RIO in 2007, with a complaint of posterior polar cataract in his left and was pseudoaphakic in his right eye. He underwent cataract surgery at the age of 4y. Presently, his visual acuity is 6/12p with PH 6/12. Subsequently, he had Yag capsulotomy done on his left eye at the age of 9 years (Figure 8A). His elder brother (III.1) is also affected. The pedigree (Figure 8B) demonstrates that the cataract developed spontaneously in the father (II.2) who was operated earlier at the age of 22 years for his right eye. He was 25 years old at the time of his second surgery at left eye for removal of posterior polar cataract. His visual acuity was 6/36 with PH 6/18 at left eye. His pupil is said to be irregular with no other ocular anomalies. Fundus examination revealed his retina with no pathological findings.
Figure 8.

Cataract phenotype in family SEC18. A: The posterior polar cataract in the proband (9 years) is demonstrated in the left eye; the right eye is pseudoaphakic. B: Pedigree of family SEC18 indicates the occurrence of the cataract in the second generation of two unrelated families; it is fully transmitted to both sons in the third generation.
Molecular genetics of family SEC18
Molecular analysis of some functional candidate genes (CRYAA, CRYGC, CRYGD, GJA3, GJA8, and PITX3) revealed a C→T mutation of GJA3 (cDNA position # 56; Figure 9A,B); at the amino-acid level, it exchanges the Thr at codon 19 by a Met (T19M). The mutation forms a new restriction site for NcoI. The mutation segregated within the family (Figure 9C) among the affected members (n=3) and was not observed in unrelated, healthy controls of either Indian (n=100) or German origin (n=96).
Figure 9.
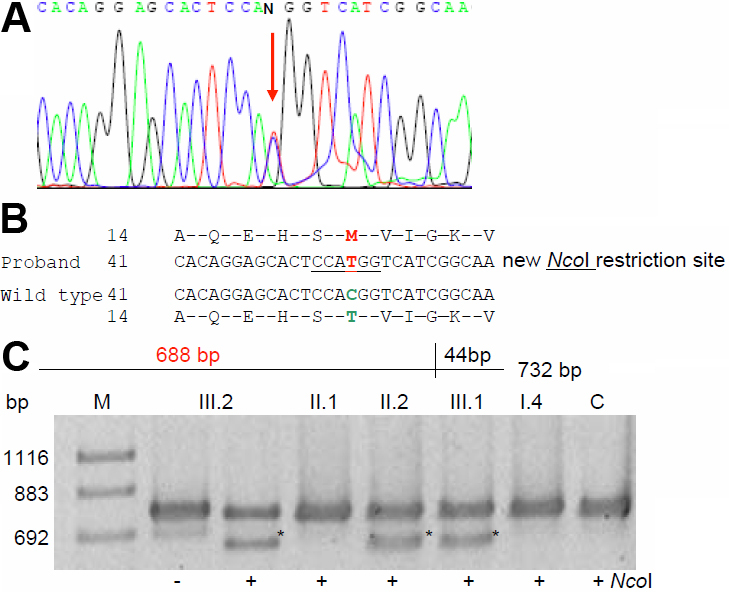
GJA3 mutation in family SEC18. A: Sequence analysis of GJA3 genomic DNA indicates heterozygosity (red arrow) in the proband. B: Comparison of the wild-type sequence with the proband’s sequence demonstrates a C→T exchange at position 56 leading to an amino acid alteration at codon 19 (T19M). C: Restriction analysis with (+) or without (-) the enzyme NcoI in members of the family leads to an additional fragment of 688 bp in the mutant DNA (red); it demonstrates the presence of the mutation in the affected father (II.2), in the proband (III.2) and in his brother (III.1). The asterisks mark the additional band of 688 bp indicating the mutation; the band of 732 bp indicates the undigested DNA. WT represents independent control from the laboratory; M=marker.
The mutation affects the last amino acid of the NH2-terminal cytoplasmic domain and is predicted not to alter the first transmembrane domain. However, secondary structure prediction program (GOR4) suggested that the extended-strand and random-coil structure between amino acids 17 and 26 was then changed to an α-helical structure, which runs in the mutant protein from amino acid 5 to 24. It enhances the fraction of the α-helical structure from 42.5% in the wild-type protein to 44.6% in the mutant connexin46.
Discussion
Here we report a screen of five families with members having congenital or childhood cataracts (CBE21, DJC1, JEE13, JPM1, and SEC18). We identified three mutations being most likely causative for congenital or childhood cataracts in these families from India – one affects the GJA3 gene encoding connexin 46, the other two affect CRYBB2 encoding β-crystallin. As the size of the families was rather small we resorted to apply a functional candidate gene approach.
While investigating, two new rare polymorphic sites were documented in some of the candidate genes. The nonsynonymous change M44V in the CRYGD gene was identified in two (of four) affected members of a family (CBE21) who suffer from blue dot opacities. This variation was not observed in 60 of the unrelated population controls from India while the same was observed in ~6% of the German controls. In family JEE13, the SNP, IVS1+9 c>t in HSF4 was observed rather frequently (~17%) in controls of Indian origin. These results suggest ethnic specificity of certain polymorphisms; since these SNPs do not explain the Mendelian type of inheritance, they might act as susceptibility alleles for cataracts in the respective populations. In our screening, several common SNPs were also documented in some of the candidate genes (Table 3) representing coding as well as non-coding regions. SNPs in non-coding regions may influence promoter activity, splicing (in introns), translational efficiency (by different codon usage) or mRNA stability (in 3′-UTR). Therefore, it is evident that SNPs may alter gene function and lead to phenotypic modifications.
Table 3. SNPs documented in cataract probands/affected relatives of Indian origin.
| Genes | Non coding | Family code | Coding | Family code |
|---|---|---|---|---|
|
CRYAA |
IVS2+27 g>c |
CBE21;DJC1 |
rs60485881 |
CBE21 |
|
CRYBB2 |
65843t>a;(IVS1+31t>a) |
JEE13 |
54 g>a; K18K |
DJC1 |
| |
65894a>g;(IVS1+84a>g) |
JEE13 |
rs17842553 |
SEC18 |
| |
71307c>a;IVS3+120c>a) |
DJC1; JPM1 |
|
|
| |
73644a>g;(IVS3–360a>g) |
JEE13;DJC1 |
|
|
| |
75738 g>a;(IVS5+9g>a) |
JEE13;DJC1 |
77788 g>a;G161G |
JEE13;DJC1 |
|
CRYGC |
none |
none |
rs3189020 |
Sec18 |
|
CRYGD |
517t>c; IVS2+30t>c |
CBE21;DJC1;JPM1; |
286t>c; Y16Y |
JPM1;DJC1; |
| |
|
JEE13 |
365a>g;M43V |
CBE21 |
| |
570c>t; IVS2+83t>c |
CBE21;DJC1;JPM1; |
285a>g;R94R |
CBE21 |
| |
|
JEE13 |
|
JEE13 |
| |
553c>t-3′UTR |
JEE13 |
|
|
|
GJA3 |
rs968566 |
SEC18 |
none |
none |
|
GJA8 |
none |
none |
none |
none |
|
PITX3 |
none |
none |
rs17858134 |
SEC18;CBE21 |
| HSF4 | IVS1+9 c>t | JEE13 | none | none |
In the present study, two new mutations in CRYBB2 have been identified. In family DJC1; a splice-site prediction program suggests that the G54A mutation of CRYBB2 disrupts the conserved donor splice site. Unfortunately, this could not be validated further, because experiments on splicing aspects need surgical lens exudates from the proband. This mutation was not observed in the general population of neither Indian (n=35) nor of German (n=96) origin, but it showed reduced penetrance in the family, since two members (the mother and one sister) carry the mutation without clinical findings. Given this ambiguity, it would be interesting to have an animal model for this particular mutation to further understand its mode of action: If the splice site is destroyed, a premature stop codon is present after 16 codons resulting in a short peptide of 34 amino acids. If this peptide is formed, a dominant-negative phenotype might be expected; in case of nonsense-mediated decay of the mutated mRNA, a loss-of-function phenotype without clinical relevance in heterozygotes might be expected. Individual variations among the affected family members might explain the observed reduced penetrance, since the corresponding Crybb2 knockout mutation in the mouse indicates a less severe phenotype in the homozygotes as compared to the heterozygous dominant point-mutations [20] (the phenotype of heterozygous knockout mutants is not reported and might be considered to be indistinguishable from the wild type).
The second novel mutation, W59C in CRYBB2, has been identified to segregate in all affected members of the JPM1 family afflicted with total cataract; no other systemic ailments were obvious. Human βB2-crystallin, has five Trp residues, of which three are in the 2nd Greek key motif at amino acid positions (59, 81, and 84), one in the 4th Greek key motif (151), and the last one in the COOH-terminal extension (194). At position 59, Trp (single letter code W) seems to be a buried residue conserved both in acidic as well as in basic β-crystallins [21] depicting phylogenetic significance. The sequence variation W59C disrupts the conserved Trp residue by substitution of Cys. In human βB2-crystallin, two Cys residues are located at amino acid positions 48 (1st Greek key motif) and 67 (2nd Greek key motif). The putative mutation W59C introduces a third Cys residue in the mutant βB2-crystallin. Theoretically, this might increase the scope for more Cys-mediated crosslinks through di-sulphide bridges which might disrupt the folding and change the topography of the amino acids in the vicinity. As per an earlier report, formation of a disulphide bond involving Cys-37 and Cys-67 of βB2-crystallin has been observed in a human nuclear cataract [22]. Moreover, the microenvironment surrounding the W59 might change in consequence to the mutation, but this hypothesis needs to be verified by appropriate biophysical methods. Interestingly, both the variations W59C (JPM1) and IVS2+1g>a (DJC1) are also in association with an SNP viz., IVS3+120c>a (71307c>a) as was observed earlier in a central nuclear cataract with the mutation W151C of βB2-crystallin [11]. This SNP could therefore act as a molecular tag denoting a marker/modifier for putative mutations in CRYBB2. Putting together the above points, the mutation W59C could well be considered as the molecular lesion for the dominant cataract in the family JPM1.
The appropriate association of β-crystallins into higher-order complexes is critical to the maintenance of lens transparency and a high refractive index [23]. Besides the lens, β-crystallins are expressed in other tissues such as retina, brain, and testis [24,25] implying its role in other biologic functions as well, like elongation of axons during regeneration of retinal ganglion cells [26].
Mutations in human CRYBB2 have been reported to be associated with variable dominant cataract phenotypes [5,8] like Cerulean cataract [27], Coppock-like cataract [28], Cerulean and sutural cataract [29], a variable phenotype in a Chinese family [30], a dominant congenital cataract in a Chilean family [31], and progressive polymorphic congenital coronary cataract [32]. The mutation Q155X depicted a decrease in protein–protein interaction and decreased ordered structure and stability. However, the partially unfolded protein was found to retain some dimer structure [33]. It is understandable that such heterogeneity in dominant cataracts may be attributed either to ethnic background, or environmental or other modifiers.
Moreover, three other mutations have been identified in CRYBB2: W151C in a dominant central nuclear cataract [11] in an Indian family, D128V, affecting exon 5 of the human CRYBB2 gene in a German family depicting nuclear cataract with an additional ring like cortical opacity [34], and third the mutation S31W co-segregated in a Chinese family with inherited coronary cataract [35].
In mice, three mutant lines have been reported to affect βB2–crystallins, the Philly mouse, AEY2, and O377. In the Philly mouse, the progressive opacity involves the nucleus and the anterior sutures [36], while in Aey2 a progressive opacification of the whole lens sets in by the age of 11 weeks [37], and in O377 the phenotype is a progressive cataract with a small lens [25]. Further studies of Crybb2 mutations in animal models might throw light on potential regions of β-crystallin molecule trivial for protein association.
Connexins-46 and −50 are important for proper lens development and differentiation of their fiber cells; they enable contact not only to the anterior epithelial cells, but also among the lens fiber cells [38-42]. Several mutations documented in GJA3 and GJA8 substantiate this statement [8]. Here we report a new mutation as T19M in GJA3, which was observed in a 9-year-old proband having a posterior polar cataract; the same phenotype and the same mutation (T19M) was confirmed in his elder brother and father, but it was not observed in any of his unaffected family members checked (SEC18). The putative mutation T19M occurs at a highly conserved site both across the family of connexins as well as across species. As per secondary structure prediction program (GOR4), it enhances the fraction of the α-helical structure in the mutant connexin-46.
Altered function and/or expression of specific connexin-encoding genes have been linked to several diseases, including genetic deafness, skin disease, peripheral neuropathies, and cataracts [39]. The cataract-causing mutations in GJA3 include Asp3Tyr [43], Leu11Ser [44], Val28Met [45], Phe32Leu [46], Arg33Leu [47], Pro59Leu [48], Asn63Ser [49], Arg76Gly [45], Thr87Met [50], Pro187Leu [51], Asn188Thr [52], and c.1137insC causing a frameshift at codon 380 [49]. It is obvious from this list that most mutations identified in human autosomal dominant congenital cataracts (ADCC) so far affect the first half of the protein and the majority of murine cataracts occur in E1 domain [4]. Except the COOH-terminal frame-shift mutation viz., S380fs, all others are missense mutations spread over the NH2-terminus (2nos.), M1 (3nos), E1 (3nos.), at boundary of E1/M2 (2nos.), M2 (1no) and at E2 domains (2nos) of Connexin-46. In human, most mutations seem to be associated with zonular pulverulent opacities featuring nuclear or perinuclear regions. However, other variable phenotypes such as anterior capsular with cortical opacities [45], nuclear [53] and total cataracts [45] have also been encountered. Interestingly, the mutation T19M in the present study and the one in an earlier report (T87M) are missense mutations substituting a polar amino acid (Thr) for a non-polar one (Met); they are phenotypically different in a way that might reflect their domain location.
In rats, a non-conservative missense mutation was reported (Glu42Lys [54]), and a knockout mutation of Gja3−/− in mouse causes nuclear opacity [55]. Gja3 knock outs in mice present various degrees of cataractous opacity depending on the genetic background [40] as was also seen with the intra familial variation in the Chinese family. Murine Gja8 dominant as well as null mutations also display different types of cataracts [56,57]. On the contrary to the mutations mapped in Gja8, all missense mutations in Gja3 are dominant as discussed earlier [13]. Therefore, it is well documented that genetic background does influence the severity of cataract phenotypes with connexin mutations [58]. Moreover, electrophysiological studies of the Gja8-G22R mutation at the NH2-terminal region have confirmed that mutant subunits alter the gating and conductance of gap junction channels in vitro [59]. Similar experiments on the new T19M GJA3 mutant protein might help to understand the biophysical properties of the mutant connexin-46 molecule in conjunction with the wild type protein.
In conclusion, we present here ample evidence for new cataract-causing mutations in CRYBB2 and in GJA3. The data presented focuses on the complexity of genetic and phenotypic diversity of congenital or childhood cataracts in humans.
Acknowledgments
We thank the family members for their kind cooperation as well as Erika Bürkle and Monika Stadler (Helmholtz Center Munich, Neuherberg) for their expert technical assistance. This work was supported at least in part by the German Federal Ministry of Research and Technology, Grant BMBF/DLR IND 03/001 (J.G.) and the Department of Biotechnology, Govt. of India, Proj. No. BT/IN/FRG/JRS/2003-’04, India (S.T.S. and P.M.G.).
References
- 1.Francis PJ, Moore AT. Genetics of childhood cataract. Curr Opin Ophthalmol. 2004;15:10–5. doi: 10.1097/00055735-200402000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Dandona L, Williams JD, Williams BC, Rao GN. Population-based assessment of childhood blindness in Southern India. Arch Ophthalmol. 1998;116:545–6. [PubMed] [Google Scholar]
- 3.Graw J. The genetic and molecular basis of congenital eye defects. Nat Rev Genet. 2003;4:876–88. doi: 10.1038/nrg1202. [DOI] [PubMed] [Google Scholar]
- 4.Graw J. Mouse Models for Cataracts. J Genet. 2009;88:469–86. doi: 10.1007/s12041-009-0066-2. [DOI] [PubMed] [Google Scholar]
- 5.Graw J. Genetics of crystallins: Cataract and beyond. Exp Eye Res. 2009;88:173–89. doi: 10.1016/j.exer.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 6.Sangiuolo F, Magnani M, Stambolian D, Novelli G. Biochemical characterization of two GALK1 mutations in patients with galactokinase deficiency. Hum Mutat. 2004;23:396. doi: 10.1002/humu.9223. [DOI] [PubMed] [Google Scholar]
- 7.Aguilar-Martinez P, Schved JF, Brissot P. The evaluation of hyperferritinemia: an updated strategy based on advances in detecting genetic abnormalities. Am J Gastroenterol. 2005;100:1185–94. doi: 10.1111/j.1572-0241.2005.40998.x. [DOI] [PubMed] [Google Scholar]
- 8.Hejtmancik JF. Congenital cataracts and their molecular genetics. Semin Cell Dev Biol. 2008;19:134–49. doi: 10.1016/j.semcdb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graw J, Klopp N, Illig T, Preising MN, Lorenz B. Congenital cataract and macular hypoplasia in humans associated with a de novo mutation in CRYAA and compound heterozygous mutations in P. Graefes Arch Clin Exp Ophthalmol. 2006;244:912–9. doi: 10.1007/s00417-005-0234-x. [DOI] [PubMed] [Google Scholar]
- 11.Santhiya ST, Manisastry SM, Rawlley D, Malathi R, Anishetty S, Gopinath PM, Vijayalakshmi P, Namperumalsamy P, Adamski J, Graw J. Mutation analysis of congenital cataracts in Indian families: identification of SNPS and a new causative allele in CRYBB2 gene. Invest Ophthalmol Vis Sci. 2004;45:3599–607. doi: 10.1167/iovs.04-0207. [DOI] [PubMed] [Google Scholar]
- 12.Santhiya ST, Shyam MM, Rawlley D, Vijayalakshmi P, Namperumalsamy P, Gopinath PM, Löster J, Graw J. Novel mutations in the gamma-crystallin genes cause autosomal dominant congenital cataracts. J Med Genet. 2002;39:352–8. doi: 10.1136/jmg.39.5.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt W, Klopp N, Illig T, Graw J. A novel GJA8 mutation causing a recessive triangular cataract. Mol Vis. 2008;14:851–6. [PMC free article] [PubMed] [Google Scholar]
- 14.Forshew T, Johnson CA, Khaliq S, Pasha S, Williis C, Abbasi R, Tee L, Smith U, Trembath RC, Mehdi SQ, Moore AT, Maher ER. Locus heterogeneity in autosomal recessive congenital cataracts: linkage to 9q and germline HSF4 mutations. Hum Genet. 2005;117:452–9. doi: 10.1007/s00439-005-1309-9. [DOI] [PubMed] [Google Scholar]
- 15.Illig T, Bongardt F, Schopfer A, Holle R, Müller S, Rathmann W, Koenig W, Meisinger C, Wichmann HE, Kolb H, KORA Study Group The endotoxin receptor TLR4 polymorphism is not associated with diabetes or components of the metabolic syndrome. Diabetes. 2003;52:2861–4. doi: 10.2337/diabetes.52.11.2861. [DOI] [PubMed] [Google Scholar]
- 16.Taudien S, Galgoczy P, Huse K, Reichwald K, Schilhabel M, Szafranski K, Shimizu A, Asakawa S, Frankish A, Loncarevic IF, Nobuyoshi S, Siddiqui R, Platzer M. Polymorphic segmental duplications at 8p23.1 challenge the determination of individual defensin gene repertoires and the assembly of a contiguous human reference sequence. BMC Genomics. 2004;5:92. doi: 10.1186/1471-2164-5-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garnier J, Gibrat JF, Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996;266:540–53. doi: 10.1016/s0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- 19.Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, Li J, Huang C, Xue L, Peng Y, Fu Q, Gao L, Zhang J, Li W. Targeted knockout of the mouse βB2-crystallin gene (Crybb2) induces age-related cataract. Invest Ophthalmol Vis Sci. 2008;49:5476–83. doi: 10.1167/iovs.08-2179. [DOI] [PubMed] [Google Scholar]
- 21.Bateman OA, Sarra R, van Genesen ST, Kappé G, Lubsen NH, Slingsby C. The stability of human acidic β-crystallin oligomers and hetero-oligomers. Exp Eye Res. 2003;77:409–22. doi: 10.1016/s0014-4835(03)00173-8. [DOI] [PubMed] [Google Scholar]
- 22.Takemoto LJ. Disulfide bond formation of Cysteine-37 and Cysteine-66 of betaB2 crystallin during cataractogenesis of the human lens. Exp Eye Res. 1997;64:609–14. doi: 10.1006/exer.1996.0247. [DOI] [PubMed] [Google Scholar]
- 23.Slingsby C, Bateman OA. Quarternary interactions in the eye lens β-crystallins: basic and acidic subunits of β-crystallins favor heterologous association. Biochemistry. 1990;29:6592–9. doi: 10.1021/bi00480a007. [DOI] [PubMed] [Google Scholar]
- 24.Magabo KS, Horwitz J, Piatigorsky J, Kantorow M. Expression of βB2-crystallin mRNA and protein in retina, brain and testis. Invest Ophthalmol Vis Sci. 2000;41:3056–60. [PMC free article] [PubMed] [Google Scholar]
- 25.Ganguly K, Favor J, Neuhäuser-Klaus A, Sandulache R, Puk O, Beckers J, Horsch M, Schädler S, Vogt Weisenhorn D, Wurst W, Graw J. Novel allele of Crybb2 in the mouse and its expression in the brain. Invest Ophthalmol Vis Sci. 2008;49:1533–41. doi: 10.1167/iovs.07-0788. [DOI] [PubMed] [Google Scholar]
- 26.Liedtke T, Schwamborn JC, Schröer U, Thanos S. Elongation of axons during regeneration involves retinal crystallin β b2 (crybb2). Mol Cell Proteomics. 2007;6:895–907. doi: 10.1074/mcp.M600245-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Litt M, Carrero-Valenzuela R, LaMorticella DM, Schultz DW, Mitchell TN, Kramer P, Maumenee IH. Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human β-crystallin gene CRYBB2. Hum Mol Genet. 1997;6:665–8. doi: 10.1093/hmg/6.5.665. [DOI] [PubMed] [Google Scholar]
- 28.Gill D, Klose R, Munier FL, McFadden M, Priston M, Billingsley G, Ducrey N, Schorderet DF, Héon E. Genetic heterogeneity of the Coppock-like cataract: a mutation in CRYBB2 on chromosome 22q11.2. Invest Ophthalmol Vis Sci. 2000;41:159–65. [PubMed] [Google Scholar]
- 29.Vanita, Reis A, Jung M, Singh D, Sperling K, Singh JR, Burger J. A unique form of autosomal dominant cataract explained by gene conversion between β-crystallin B2 and its pseudogene. J Med Genet. 2001;38:392–6. doi: 10.1136/jmg.38.6.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao K, Tang X, Shentu X, Wang K, Rao H, Xia K. Progressive polymorphic congenital cataract caused by a CRYBB2 mutation in a Chinese family. Mol Vis. 2005;11:758–63. [PubMed] [Google Scholar]
- 31.Bateman JB, von-Bischhoffshaunsen FR, Richter L, Flodman P, Burch D, Spence MA. Gene conversion mutation in crystallin, beta-B2 (CRYBB2) in a Chilean family with autosomal dominant cataract. Ophthalmology. 2007;114:425–32. doi: 10.1016/j.ophtha.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 32.Li FF, Zhu SQ, Wang SZ, Gao C, Huang SZ, Zhang M, Ma X. Nonsense mutation in the CRYBB2 gene causing autosomal dominant progressive polymorphic congenital coronary cataracts. Mol Vis. 2008;14:750–5. [PMC free article] [PubMed] [Google Scholar]
- 33.Liu BF, Liang JJN. Interaction and biophysical properties of human lens Q155* βB2-crystallin mutant. Mol Vis. 2005;11:321–7. [PubMed] [Google Scholar]
- 34.Pauli S, Söker T, Klopp N, Illig T, Engel W, Graw J. Mutation analysis in a German family identified a new cataract-causing allele in the CRYBB2 gene. Mol Vis. 2007;13:962–7. [PMC free article] [PubMed] [Google Scholar]
- 35.Lou D, Tong JP, Zhang LY, Chiang SWY, Lam DSC, Pang CP. A novel mutation in CRYBB2 responsible for inherited coronary cataract. Eye. 2009;23:1213–20. doi: 10.1038/eye.2008.222. [DOI] [PubMed] [Google Scholar]
- 36.Chambers C, Russell P. Deletion mutation in an eye lens β-crystallin. J Biol Chem. 1991;266:6742–6. [PubMed] [Google Scholar]
- 37.Graw J, Löster J, Soewarto D, Fuchs H, Reis A, Wolf E, Balling R, Hrabé de Angelis M. Aey2, a new mutation in the βB2-crystallin-encoding gene of the mouse. Invest Ophthalmol Vis Sci. 2001;42:1574–80. [PubMed] [Google Scholar]
- 38.Gong X, Baldo GJ, Kumar NM, Gilula NB, Mathias RT. Gap junctional coupling in lenses lacking α3 connexin. Proc Natl Acad Sci USA. 1998;95:15303–8. doi: 10.1073/pnas.95.26.15303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baldo GJ, Gong X, Martinez-Wittinghan FJ, Kumar NM, Gilula NB, Mathias RT. Gap junctional coupling in lenses from α8 connexin knockout mice. J Gen Physiol. 2001;118:447–56. doi: 10.1085/jgp.118.5.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White TW. Unique and redundant connexin contributions to lens development. Science. 2002;295:319–20. doi: 10.1126/science.1067582. [DOI] [PubMed] [Google Scholar]
- 41.Martinez-Wittinghan FJ, Sellitto C, White TW, Mathias RT, Paul D, Goodenough DA. Lens gap junctional coupling is modulated by connexin identity and the locus of gene expression. Invest Ophthalmol Vis Sci. 2004;45:3629–37. doi: 10.1167/iovs.04-0445. [DOI] [PubMed] [Google Scholar]
- 42.Gong X, Cheng C, Xia C. Connexins in lens development and cataractogenesis. J Membr Biol. 2007;218:9–12. doi: 10.1007/s00232-007-9033-0. [DOI] [PubMed] [Google Scholar]
- 43.Addison PKF, Berry V, Holden KR, Espinal D, Rivera B, Su H, Srivastava AK, Bhattacharya SS. A novel mutation in the connexin 46 gene (GJA3) causes autosomal dominant zonular pulverulent cataract in a Hispanic family. Mol Vis. 2006;12:791–5. [PubMed] [Google Scholar]
- 44.Hansen L, Yao W, Eiberg H, Funding M, Riise R, Kjaer KW, Hejtmancik JF, Rosenberg T. The congenital “ant-egg” cataract phenotype is caused by a missense mutation in connexin46. Mol Vis. 2006;12:1033–9. [PubMed] [Google Scholar]
- 45.Devi RR, Reena C, Vijayalakshmi P. Novel mutations in GJA3 associated with autosomal dominant congenital cataract in the Indian population. Mol Vis. 2005;11:846–52. [PubMed] [Google Scholar]
- 46.Jiang H, Jin Y, Bu L, Zhang W, Liu J, Cui B, Kong X, Hu L. A novel mutation in GJA3 (connexin46) for autosomal dominant congenital nuclear pulverulent cataract. Mol Vis. 2003;9:579–83. [PubMed] [Google Scholar]
- 47.Guleria K, Sperling K, Singh D, Varon R, Singh JR, Vanita V. A novel mutation in the connexin 46 (GJA3) gene associated with autosomal dominant congenital cataract in an Indian family. Mol Vis. 2007;13:1657–65. [PubMed] [Google Scholar]
- 48.Bennett TM, Mackay DS, Knopf HLS, Shiels A. A novel missense mutation in the gene for gap-junction protein α3 (GJA3) associated with autosomal dominant “nuclear punctuate” cataracts linked to chromosome 13q. Mol Vis. 2004;10:376–82. [PubMed] [Google Scholar]
- 49.Mackay D, Ionides A, Kibar Z, Rouleau G, Berry V, Moore A, Shiels A, Bhattacharya S. Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet. 1999;64:1357–64. doi: 10.1086/302383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guleria K, Vanita V, Singh D, Singh JR. A novel “pearl box” cataract associated with a mutation in the connexin 46 (GJA3) gene. Mol Vis. 2007;13:797–803. [PMC free article] [PubMed] [Google Scholar]
- 51.Rees MI, Watts P, Fenton I, Clarke A, Snell RG, Owen MJ, Gray J. Further evidence of autosomal dominant congenital zonular pulverulent cataract linked to 13q11 (CZP3) and a novel mutation in connexin 46 (GJA3). Hum Genet. 2000;106:206–9. doi: 10.1007/s004390051029. [DOI] [PubMed] [Google Scholar]
- 52.Li Y, Wang J, Dong B, Man H. A novel connexin46 (GJA3) mutation in autosomal dominant congenital nuclear pulverulent cataract. Mol Vis. 2004;10:668–71. [PubMed] [Google Scholar]
- 53.Ma ZW, Zheng J, Yang F, Ji J, Li X, Tang X, Yuan X, Zhang X, Sun H. Two novel mutations of connexin genes in Chinese families with autosomal dominant congenital nuclear cataract. Br J Ophthalmol. 2005;89:1535–7. doi: 10.1136/bjo.2005.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshida M, Harada Y, Kaidzu S, Ohira A, Masuda J, Nabika T. New genetic model rat for congenital cataracts due to a connexin 46 (Gja3) mutation. Pathol Int. 2005;55:732–7. doi: 10.1111/j.1440-1827.2005.01896.x. [DOI] [PubMed] [Google Scholar]
- 55.Gong X, Li E, Klier G, Huang Q, Wu Y, Lei H, Kumar NM, Horwitz J, Gilula NB. Disruption of α3 connexin gene leads to proteolysis and cataractogenesis in mice. Cell. 1997;91:833–43. doi: 10.1016/s0092-8674(00)80471-7. [DOI] [PubMed] [Google Scholar]
- 56.Chang B, Wang X, Hawes NL, Ojakian R, Davisson MT, Lo WK, Gong XA. Gja8 (Cx50) point mutation causes an alteration of alpha 3 connexin (Cx46) in semi-dominant cataracts of Lop10 mice. Hum Mol Genet. 2002;11:507–13. doi: 10.1093/hmg/11.5.507. [DOI] [PubMed] [Google Scholar]
- 57.Xia CH, Cheung D, De Rosa AM, Chang B, Lo WK, White TW, Gong X. Knock-in of α3 connexin prevents severe cataracts caused by an α8 point mutation. J Cell Sci. 2006;119:2138–44. doi: 10.1242/jcs.02940. [DOI] [PubMed] [Google Scholar]
- 58.Gong X, Agopian K, Kumar NM, Gilula NB. Genetic factors influence cataract formation in α3 connexin knockout mice. Dev Genet. 1999;24:27–32. doi: 10.1002/(SICI)1520-6408(1999)24:1/2<27::AID-DVG4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 59.Xia CH, Liu H, Cheung D, Cheng C, Wang E, Du X, Beutler B, Lo WK, Gong X. Diverse gap junctions modulate distinct mechanisms for fiber cell formation during lens development and cataractogenesis. Development. 2006;133:2033–40. doi: 10.1242/dev.02361. [DOI] [PubMed] [Google Scholar]