

Abstract
ATP depletion and ADP formation are generic detection methods used for the identification of kinase and other ATP-utilizing enzyme inhibitors in high-throughput screening campaigns. However, the most widely used nucleotide detection approaches require high ATP consumption rates or involve the use of coupling enzymes, which can complicate the selection of lead compounds. As an alternative, we have developed the Transcreener® (BellBrook Labs, Madison, WI) platform, which relies on the direct immunodetection of nucleotides. Here we describe the development of antibodies with > 100-fold selectivity for ADP versus ATP, which enable robust detection of initial velocity rates (Z ′ > 0.7 at 10% substrate consumption) at ATP concentrations ranging from 0.1 μ M to 1,000 μ M in a competitive fluorescence polarization (FP) immunoassay. Competitive binding experiments indicate similar affinities for other nucleotide diphosphates, including 2′-deoxy ADP, GDP, and UDP. The antibody-tracer complex and the red-shifted, ratiometric FP signal are stable for at least 24 h at room temperature, providing suitable conditions for high-throughput screening. A method for calculating a kinase ATP Km with this FP immunoassay is also presented. The Transcreener ADP assay provides a simple, generic assay platform for inhibitor screening and selectivity profiling that can be used for any ADP-generating enzyme.
Introduction
Protein kinases are targeted by pharmaceutical companies for a broad range of diseases and disorders, especially cancers.1,2 Clinical success with small molecule kinase inhibitor drugs, most notably imatinib mesylate, a BCR-ABL inhibitor used to treat chronic myelogenous leukemia, has provided validation for the therapeutic targeting of kinases,3 the most intensively screened target class.2,4
Despite its increasing importance, kinase drug discovery has been hampered by a lack of screening assays capable of rapidly accommodating the diverse kinase superfamily. Nonradioactive, homogeneous assays are preferable for HTS, and fluorescence and bioluminescence are the most common readouts.5,6 Binding of tagged phosphopeptides to antibody or immobilized metal ions7,8 and electrophoretic separation of the negatively charged peptides9 are popular HTS assays for kinases, but regardless of the readout, they are not generic, i.e., reagent development is required for different subsets of kinases. The time and money required to develop phosphopeptide-based assay methods for kinases with novel recognition sequences delay the screening of promising new targets. Moreover, there is a growing interest in using native protein acceptor substrates for screening because this approach allows for the identification of more diverse inhibitor types. This approach has resulted in some of the most selective kinase inhibitors discovered to date.10–12 HTS with native protein substrates is either not possible or restricted to varying degrees by phosphopeptide-based detection methods.
As all kinase reactions include the conversion of ATP to ADP, detection of either nucleotide is the only truly generic method to assay kinase activity. Luciferase has been used to monitor ATP consumption,13,14 but this approach requires a high level of substrate turnover to generate adequate signal to background ratios. Coupled enzyme assays that generate a chromogenic or fluorescent product such as NADH or resorufin have been employed to detect ADP produced in kinase reactions,15,16 but these methods are prone to interference from test compounds, especially because one of the coupling enzymes is generally a glycolytic kinase.17,18 To overcome these and other technical hurdles encountered with group transfer enzyme targets, we developed the Transcreener® (BellBrook Labs, Madison, WI) assay platform, which relies on direct detection of the invariant product of a group transfer reaction.19 ADP, which is produced by all members of the kinase family, displaces a fluorescently labeled tracer in a homogeneous, competitive fluorescence polarization (FP) immunoassay (Fig. 1). This platform thus accommodates any kinase and any acceptor substrate. The two key Transcreener ADP assay components are a selective monoclonal anti-ADP antibody and a high-affinity, red-shifted fluorescent ADP tracer. Huss et al.20 first validated the Transcreener technology as an HTS assay format using protein kinase A and kemptide, reporting excellent correlations with values for competitor (inhibitor) concentration that results in a 50% reduction in signal (IC50) determined using the traditional 33P-radioassays.
FIG. 1.

Schematic of the Transcreener ADP assay principle. ADP produced during transfer of phosphate to acceptor substrate is detected using a competitive FP immunoassay. Unlike antibody-bound tracer (high mP), displaced tracer (low mP) spins rapidly in solution, resulting in a lower proportion of emitted polarized light. Because ADP detection relies on detection of the invariant reaction product, the same detection reagents can be used for any kinase and any substrate or any ADP-producing enzyme.
The generic properties of the Transcreener platform enable previously intractable ATP-utilizing drug targets to be screened as well. For example, inhibitor identification for acetyl-coenzyme A carboxylases, which play a key role in fatty acid biosynthesis and oxidation, was difficult because the existing complex radioactive assays, including chromatic purification of malonyl-coenzyme A,21 were not easily adapted to HTS. By monitoring ADP production using the Transcreener ADP Assay, Liu et al.22 successfully identified several inhibitors from a>500,000 compound library.
In this report we present a detailed characterization of the key components of the Transcreener ADP assay, the antibody and tracer, and demonstrate how to obtain the maximal assay signal, especially at high ATP:ADP ratios typically found in kinase reactions. We examine the robustness of the assay at low ATP conversion (mimicking initial velocity [Vinitial] conditions) and investigate signal stability over time. We also demonstrate its use for kinetic analysis by determining the Michaelis-Menten constant (Km) for ATP with a target kinase.
Materials and Methods
Materials
The monoclonal ADP antibodies and Transcreener ADP assay were developed at BellBrook Labs. Alexa Fluor®633 succinimidyl ester was purchased from Invitrogen (Carlsbad, CA) and conjugated to ADP. The GenPlus compound library was from MicroSource (Gaylordsville, CT) and was supplemented with the BIOMOL (Plymouth Meeting, PA) kinase inhibitor library and other kinase inhibitors from EMD Chemicals Inc., an affiliate of Merck KGaA (Darmstadt, Germany). ATP was purchased from Bioline (London, UK) as a component of the NTP Set (catalog number Bio-39052) (Vendor 1) or from Sigma (St. Louis, MO) (catalog number A7699) (Vendor 2). Other nucleotides and basic buffer components were purchased from Sigma or Fisher Scientific (Hampton, NH). The kinase and kinase substrate were supplied by Schering-Plough Research Institute (Oss, The Netherlands).
Standard Plate and Instrumentation Settings
Assays were performed in black Corning® (Corning, NY) 384-well flat-bottom microplates (catalog number 3654) or black Corning 384-well round-bottom low-volume polystyrene non-binding surface microplates (catalog number 3676). Mixing after additions was performed by orbital shaking for 1 min. Unless otherwise noted, 20-μl assays were equilibrated for 1 h at room temperature before the plate was read on the instrument.
FP measurements utilizing the ADP-Alexa Fluor633 tracer were performed on a Tecan (Durham, NC) Safire2™ plate reader using 635 nm excitation (light-emitting diode) and 670 nm emission (20 nm bandwidth) settings. The free tracer reference was set to 20 millipolarization (mP) units, and the buffer (with or without antibody) was used as the buffer blank for both the sample and free tracer reference wells.
High-Performance Liquid Chromatography (HPLC)
Nucleotide purity was assessed by HPLC.23 Aqueous solutions of ADP and ATP (100 μmol) were injected onto a Dionex (Sunnyvale, CA) Carbopac™ PA-1, 4- × 250-mm column equilibrated with 200 mM sodium acetate in 1 mM NaOH. The nucleotides were then eluted by varying the sodium acetate concentration in 1 mM NaOH: 200–550 mM sodium acetate for 10 min, 550 mM sodium acetate for 15 min, 550–800 mM sodium acetate for 10 min, and 800–1,000 mM sodium acetate for 5 min. The flow rate was 1 ml/min. Retention times for ADP of 20.3 min and ATP of 37.2 min were measured.
Assay Development
The antibody-tracer pair used for FP assay development was identified by performing antibody equilibrium binding curves and nucleotide competition binding assays with the ADP-Alexa Fluor633 tracer. Antibodies were titrated twofold in conditions typical of a quenched kinase reaction, i.e., 50 mM HEPES (pH 7.5), 0.01% Brij®-35 (trademark of Akzo Nobel [Amsterdam, The Netherlands]), 1 mM EGTA, 2 mM MgCl2, 0.5% dimethyl sulfoxide, 200 mM NaCl, 10 mM EDTA, and 2 nM ADP-Alexa Fluor633 tracer. Antibodies that bound the tracer with reasonable affinity (dissociation constant [KD] <1 μM) and demonstrated >100 mP shift were then tested for ADP selectivity using competitive binding assays with ADP and ATP in the same final buffer conditions. Twofold serial dilutions of nucleotide (n = 2) were performed in 20 μl of the above equilibrium binding buffer containing antibody at the antibody concentration at 85% of its saturation (EC85). The EC85 is calculated by using the antibody concentration at 50% of its saturation (EC50) and Hill slope values, calculated from fitting the equilibrium binding data to a variable slope sigmoidal dose-response curve (GraphPad Prism, GraphPad Software, San Diego, CA), using the following equation:
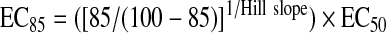 |
Antibodies demonstrating high affinity for ADP (IC50 <1 μM) and great selectivity for ADP versus ATP (>50-fold) were used for further FP assay development. Because nucleotide contaminants can compromise the selectivity of the antibody-tracer pair and subsequently the assay performance, assay components were prepared with quality reagents and purified water. Additional competition binding assays were performed with other nucleotide diphosphates (GDP, UDP, and CDP) and adenosine derivatives (2′-deoxy-ADP, AMP, adenosine 3′,5′-cyclic monophosphate [cAMP], adenosine, and d-ribose) to assess cross-reactivity.
The ADP assay is a one-step, mix-and-read endpoint assay. An equal volume of ADP detection mixture, consisting of the antibody, tracer, and components that stop the enzyme reaction and aid in the stabilization of the FP signal, was added to a 10-μl enzyme reaction and equilibrated for 1 h prior to reading the 384-well plate. To assess optimum assay performance, two ADP detection buffers were evaluated in the preparation of the ADP detection mixture: Buffer 1 consisted of 50 mM HEPES (pH 7.5), 400 mM NaCl, 20 mM EDTA, and 0.02% Brij-35, and Buffer 2 consisted of 25 mM HEPES (pH 7.5), 40 mM EDTA, and 0.02% Brij-35.
To identify the maximum FP assay window (change in mP [ΔmP]) and ideal assay sensitivity, the EC85 antibody concentration was determined in the presence of ATP. Antibody titrations were performed in 10 μl of ADP Detection Buffer 1 containing 4 nM tracer followed by addition of an equal volume of common kinase (CK) buffer (50 mM HEPES [pH 7.5], 4 mM MgCl2, 1 mM EGTA, 0.01% Brij-35, and 1% dimethyl sulfoxide) containing 1 mM dithiothreitol and ATP (ranging from 10 to 500 μM). The antibody concentration in the final 20 μl of mixture was determined by calculating the EC85 from the ATP binding curve (0% ATP conversion).
ATP/ADP Standard Curves
Standard curves were prepared with various ATP/ADP samples and the ADP detection mixture. To simulate an enzyme reaction, standard samples contained decreasing concentrations of ATP and proportionally increasing concentrations of ADP; the adenine concentration remained constant. Stock concentrations of 12 different mixtures of ATP/ADP were generated for the 0.1 μM, 1 μM, 10 μM, 100 μM, and 1,000 μM ATP/ADP standard curves. Each ATP/ADP standard was prepared in the CK buffer and dispensed into wells. An equal volume of ADP Detection Mixture 1 (3–400 μg/ml antibody 1 [Ab1] in Buffer 1) or ADP Detection Mixture 2 (1–866 μg/ml antibody 2 [Ab2] in Buffer 2) was added to the wells. The ADP detection mixture for a given initial ATP concentration was prepared in ADP detection buffer containing 4 nM tracer and twice the EC85 antibody concentrations. ADP concentrations reported in the standard curves reflect the amount produced in the 10-μl enzyme reaction. The data were plotted as ΔmP versus log [ADP] using four-parameter nonlinear regression curve fitting: ΔmP = mPinitial [ATP] − mPsample. Z′ values were calculated using replicates of 24 for each standard point.24 The lower limit of ADP detection (LLD) was determined by calculating the concentration of ADP corresponding to 3 standard deviations of the mean mP value for the 0% ATP conversion control (initial ATP concentration) (n = 24).
Km Determination for ATP
Reaction Vinitial values were measured for a range of ATP concentrations. This was done by performing the kinase reaction in 10 μl (2.5 μl of kinase, 2.5 μl of substrate, and 5 μl of ATP, all in CK buffer) for a number of fixed time points (up to 45 min), with varying ATP concentrations (up to 120 μM). Enzyme reactions were stopped by adding 10 μl of the ADP Detection Mixture 1 (Ab1 in Buffer 1), and the FP signal was read 1 h later. For each ATP concentration, individual detection mixtures were prepared using the appropriate EC85 Ab1 concentration. Using a standard curve for each ATP concentration, ΔmP values were converted to the amount of ADP produced. The Km value was calculated using nonlinear Michaelis-Menten curve-fitting (Graphpad Prism).
Reagent Equilibration and Signal Stability
Binding equilibration rates were determined for the 10 μM ATP/ADP standard curve by adding the ADP Detection Mixture 2 to samples representing different points in an enzyme reaction progress, e.g., 0% conversion, 10% conversion, 20% conversion, etc., and recording the FP signal over time. The FP signal was measured in the kinetic mode every 4 min over a 60-min period at 30°C. FP signal stability for the 10 μM ATP/ADP standard curve was also determined in the endpoint mode over a 24-h period. To assess the stability of the ADP detection mixture before adding to nucleotide mixtures, Ab2 was combined with the tracer in Buffer 2, and the mixture was stored for up to 21 days at −80°C, −20°C, 4°C, 25°C, and 37°C. Aliquots from these samples were then added to freshly prepared ATP/ADP standards, and the FP signal was measured after a 1-h equilibration. For the control sample (Day 0) Ab2 was combined with the tracer in Buffer 2 and added immediately to the ATP/ADP standards.
Control Compound Screen
Compatibility of compounds in the GenPlus compound library (960 compounds), BIOMOL Kinase Inhibitor Library (80 compounds), and other known ATP-utilizing enzyme inhibitors (22 compounds) was determined by incubating 10 μM compound (in 1% dimethyl sulfoxide) with 10 μM ATP or 9 μM ATP/1 μM ADP in the CK buffer. An equal volume of the ADP detection mixture containing 15 μg/ml Ab2 in Buffer 2 was then added to each well. To represent 0% and 100% ATP conversion, 10 μM ATP and 10 μM ADP were added to the CK buffer, respectively (n = 16). Reagents were dispensed into wells using the CyBi®-Well Pipettor from CyBio Ag (Jena, Germany). All compounds were tested in triplicate.
Results
The use of ADP immunodetection as a kinase assay method for measuring Vinitial values requires detection of small amounts of ADP in the presence of much higher concentrations of ATP. For this reason a successful FP immunoassay must demonstrate high binding sensitivity for ADP (<1 μM) and high selectivity for ADP versus ATP (>50-fold) while delivering a large change in polarization between bound and unbound tracer (ΔmP > 100 units).
An anti-ADP antibody satisfying these criteria was demonstrated in tracer equilibrium binding and nucleotide competition experiments. The KD for the tracer complexed with Ab1 was 2 nM (data not shown), and the ΔmP between bound (mP = 245 units) and unbound tracer (mP = 20 units) was 225 mP units (Fig. 2A). Competition binding curves with ADP and ATP (Fig. 2A) indicated ADP displaced the tracer from Ab1 with an IC50 value of 190 nM, whereas ATP was approximately 50-fold (Vendor 1, IC50 = 9 μM) to 180-fold less effective (Vendor 2, IC50 = 33 μM). This variability in selectivity is due to the contamination of the ATP reagent with ADP as shown in Fig. 2B. HPLC analysis of the ATP from Vendor 1 (95% purity) revealed three contaminating peaks (including ADP) in addition to the main ATP peak. The ATP from Vendor 2 contained less ADP, resulting in 99% purity.
FIG. 2.

Impurities in the ATP reagent affected the apparent selectivity of the ADP antibody. (A) Competitive binding assays with ADP (O) and two vendor sources (V1 [▴] and V2 [▪]) of ATP were performed with Ab1 in ADP Detection Mixture 1. The IC50 values were 0.190 mM for ADP, 9 mM for ATPV1, and 33 mM for ATPV2. (B) HPLC chromatogram of ADP (—) and two vendor sources (V1 [–.–.] and V2 [– –]) of ATP. Percentage purity of ATP from Vendor 1 and Vendor 2 was calculated to be 95% and 99%, respectively.
As most kinases and other ATP-utilizing enzymes are screened with the ATP concentration set at the Km, it is important that the assay method accommodates varied initial ATP concentrations. To determine how much antibody was required to maintain the full assay window, we examined the antibody-tracer equilibrium binding curves over a range of initial ATP concentrations (10–500 μM ATP). As shown in Fig. 3A, increasing concentrations of the cross-reacting ATP shifted the binding curves to the right, resulting in an overall range of EC85 values between 1 μg/ml and 140 μg/ml antibody. A linear relationship between the initial ATP concentration and the EC85 antibody concentration was observed (Fig. 3B).
FIG. 3.

Increasing concentrations of ATP required an adjustment of the EC85 antibody concentration. (A) Equilibrium binding curves for ATP concentrations ranging from 10 to 500 μM ATP: 10 μM (▴), 20 μM (♦), 40 μM (▪), 80 μM (+), 100 μM (•), 150 μM (▵), 200 μM (◊), 300 μM (□), 400 μM (×), and 500 μM (○). The 20 μl total volume consisted of 50 μM HEPES (pH 7.5), 0.01% Brij-35, 1 mM EGTA, 2 mM MgCl2, 0.5% dimethyl sulfoxide, 200 mM NaCl, 10 mM EDTA, 0.5 mM dithiothreitol, ATP, and Ab1. (B) Comparisons of initial ATP concentrations in the standard mix with EC85 antibody concentrations from equilibrium binding curves.
The requirement for adjusting antibody as ATP is varied raised the question of whether it would be possible to determine the Km for ATP with the Transcreener ADP assay. A practical method is shown in Fig. 4 in which 10 successive endpoint assays were used to measure enzyme Vinitial at each of 10 different ATP concentrations ranging from 1 μM to 120 μM. Ten-point standard curves representing conversion of ATP to ADP were constructed for each initial ATP concentration and used to quantify ADP formation. Vinitial values were determined by fitting the time curve data in the linear phase of the assay (Fig. 4A). A representative example of a plot of Vinitial versus ATP concentration is shown in Fig. 4B. The Km for this test kinase was determined to be 72 ± 3.3 μM (standard error of the mean). This Transcreener assay was used for screening purposes and compound profiling. Data were compared to those obtained with a more traditional enzyme-linked immunosorbent assay-based phosphoprotein detection assay for the same kinase and the same natural protein substrate. For instance, similar EC50 values were found for the 50 most active compounds in this project (J.J.H., unpublished data). The main difference between the two methods is that the Transcreener is a homogeneous assay format and can therefore easily be used in an automated miniaturized fashion.
FIG. 4.

The ATP Km for a target kinase was determined by measuring Vinitial values for a range of ATP concentrations using multiple standard curves. (A) Kinase reactions were performed in 10 μl (2.5 μl of kinase, 2.5 μl of substrate, and 5 μl of ATP, all in CK buffer) for a number of fixed time points (up to 45 min), with varying ATP concentrations (up to 120 μM). Kinase reactions were stopped by adding 10 μl of the ADP Detection Mixture 1 (containing two times the EC85 Ab1 concentrations), and mP values were measured after a 1-h equilibration. Polarization values were converted to the amount of ADP produced using a standard curve for each ATP concentration: 1 μM ATP (▪), 2 μM ATP (▴), 5 μM ATP (▾), 10 μM ATP (♦), 20 μM ATP (•), 40 μM ATP (□), 60 μM ATP (▵), 80 μM ATP (▿), 100 μM ATP (◊), 120 μ ATP (○). (B) A representative Michaelis-Menten plot of the initial rates versus ATP concentration; in replicate experiments, the Km value was determined to be 72 6 3.3 μM (standard error of the mean).
Limitations in the assay sensitivity and ADP detection range led us to explore new ADP antibodies in the hope of improving ADP affinity and selectivity. Using the same immunogen, we recently discovered an antibody (Ab2) that bound the ADP-Alexa Fluor633 tracer with affinity (KD = 0.9 nM) similar to Ab1. In competition binding experiments Ab2 demonstrated 12-fold higher sensitivity for ADP than Ab1 (IC50 = 15 nM vs. 180 nM) but showed similar selectivity for ADP versus ATP (93-fold vs. 130-fold) (Buffer 1, Table 1). Removal of NaCl from the ADP detection buffer (Buffer 2) further enhanced the ADP sensitivity approximately twofold (IC50 = 10 nM) with no change in selectivity (note that the NaCl was included with Ab1 in ADP Detection Mixture 1 because it stabilized the FP signal in the presence of ATP) (data not shown).
Table 1.
ADP and ATP IC50 Values for Ab1 and Ab2 in Different Buffers
| |
IC50 (nM) |
|
|
|
|||
|---|---|---|---|---|---|---|---|
| |
Ab1 |
Ab2 |
Times Fold Improvement in Sensitivity for Ab2 vs. Ab1 |
||||
| Competitor | Buffer 1 | Buffer 2 | Buffer 1 | Buffer 2 | Buffer 1 | Buffer 2 | Buffer 2: Buffer 1 |
| ATP | 17,300 | 5,280 | 1,970 | 915 | 9 | 6 | 19 |
| ADP | 180 | 40 | 15 | 10 | 12 | 4 | 18 |
Buffer 1 consists of 50 mM HEPES (pH 7.5), 400 mM NaCl, 20 mM EDTA, and 0.02% Brij-35. Buffer 2 consists of 25 mM HEPES (pH 7.5), 40 mM EDTA, and 0.02% Brij-35.
Competitive binding assays with other nucleotide diphosphates revealed GDP (IC50 = 10 nM), 2′-deoxy-ADP (IC50 = 20 nM), and UDP (IC50 = 65 nM) competed with tracer with similar effects as ADP (Table 2). CDP was the least effective nucleotide diphosphate (IC50 = 200 nM). Relative to ATP, higher concentrations of AMP and cAMP were required to displace the tracer (IC50 = 16,000 nM and 11,000 nM, respectively). d-Ribose, adenine, and adenosine did not displace the tracer at the highest concentration tested (5 mM).
Table 2.
Comparison of ADP Antibody IC50 Values for Various Nucleotides and Nucleotide Derivatives
| IC50 (nM) | |||
|---|---|---|---|
| Competitor | Ab1/Buffer 1 | Ab2/Buffer 2 | Fold Difference |
| ATP | 17,300 | 915 | 19 |
| ADP | 180 | 10 | 18 |
| GDP | 150 | 10 | 15 |
| 2′-Deoxy-ADP | 310 | 20 | 16 |
| UDP | 720 | 65 | 11 |
| CDP | 2,500 | 200 | 13 |
| AMP | 608,000 | 16,000 | 38 |
| cAMP | 266,000 | 11,000 | 24 |
| Adenosine | >900,000 | >900,000 | NA |
| Adenine | >900,000 | >900,000 | NA |
| d-Ribose | >900,000 | >900,000 | NA |
NA, not applicable.
ATP/ADP standard curves predict the response of the two antibodies for measurement of enzyme activity over a range of initial ATP concentrations (Fig. 5). The nucleotide concentrations in the standard curves mimicked enzyme reactions: ATP was decreased proportionately as ADP was added, keeping the total nucleotide concentration constant. As with other competitive binding assays, the change in signal is not linear with product formation, instead producing a sigmoidal response with the logarithm of product formation. These data show Ab2 was able to detect 10% conversion of 0.1 μM ATP (ΔmP = 70 units), whereas Ab1 was approximately 10-fold less sensitive. Significant improvements in assay window at 10% ATP conversion were also realized with Ab2 relative to Ab1 at 1 μM ATP (ΔmP = 145 mP units vs. 70 mP units) and 10 μM ATP (ΔmP = 145 mP units vs. 126 mP units). Lower concentrations of ADP were detectable with Ab2 versus Ab1 at both 0.1 μM and 1 μM ATP (Table 3), whereas similar concentrations were detectable with both antibodies at the higher ATP concentrations (>10 μM). Z′ values exceeded 0.7 at ≤10% ATP conversion for the entire range of starting ATP concentrations, except at the lowest concentration with Ab1 (Table 3).
FIG. 5.

ATP/ADP standard curves prepared with Ab1 and Ab2. To mimic ADP generated during an enzyme reaction, standard curves for 0.1 μM, 1 μM, 10 μM, 100 μM, and 1,000 μM ATP were prepared by keeping the adenine concentration constant: Ab1/0.1 μM ATP (•), Ab2/0.1 μM ATP (▪), Ab1/1 μM ATP (□), Ab2/1 μM ATP (□), Ab1/10 μM ATP (♦), Ab2/10 μM ATP (◊), Ab1/100 μM ATP (▴), Ab2/100 μM ATP (▵), Ab1/1,000 μM ATP (▾), and Ab2/1,000 μM ATP (▿). Ab1 and Ab2 were prepared in ADP Detection Buffer 1 and ADP Detection Buffer 2, respectively. The EC85 antibody concentration for both ADP antibodies was used for each initial ATP concentration. Error bars represent standard deviations of the mean (n = 24). ΔmP = mPinitial ATP − mP ATP/ADP*.
Table 3.
LLD, ADP IC50 Values, and Percentage ATP Conversion to Achieve Z′ > 0.7 with Select ADP Antibodies
| |
LLD (μM) |
IC50 (μM) |
% ATP Conversion to Achieve Z′ > 0.7 |
|||
|---|---|---|---|---|---|---|
| ATP (μM)a | Ab1 | Ab2 | Ab1 | Ab2 | Ab1 | Ab2 |
| 0.1 | 0.022 | 0.0009 | 0.09 | 0.02 | >60 | 5 |
| 1 | 0.008 | 0.0032 | 0.15 | 0.05 | 7 | 3 |
| 10 | 0.026 | 0.0261 | 0.45 | 0.50 | 3 | 3 |
| 100 | 0.159 | 0.221 | 2.86 | 5.78 | 2 | 3 |
| 1,000 | 1.90 | 2.37 | 26.22 | 57.76 | 3 | 3 |
Concentration of ATP in the 10 ml of standard mix.
Implementation of an assay in HTS typically requires stable reagents during dispensing and sustained signal stability after addition of quenching and detection reagents. To assess these parameters we first determined the binding equilibrium rate for the assay reagents by adding antibody-tracer complex to ATP/ADP standards and measuring FP immediately and over time. As shown in Fig. 6A all of the reactions reached 90% equilibrium in <10 min and were fully equilibrated after 30 min. The FP signal was sustained for at least 24 h after addition of the Ab2-tracer complex to the ATP/ADP standards (Fig. 6B). Assay performance was not compromised by storing precombined Ab2 and tracer at −80°C or −20°C for 21 days (the longest incubation time evaluated) before use (Fig. 6C). In fact, storage of the Ab2-tracer complex at 4°C, 25°C, or 37°C for the same time period caused only a slight downward shift of the 10 μM ATP/ADP standard curve; the assay window at 10% ATP conversion decreased <15% as compared to the Day 0 control (ΔmP = 129 units vs. 153 units).
FIG. 6.

Stability of the FP signal and the ADP detection mixture over time. (A) Equilibration of FP signals for ATP/ADP standards representing different percentages of ATP conversions (10 μM ATP/ADP standard curve). Plates were incubated at room temperature and read at 4-min intervals for 1 h after addition of ADP Detection Mixture 2. Error bars represent standard deviations of the mean (n = 16). (B) Long-term signal stability of the ATP/ADP standard samples. The 10 μM ATP/ADP standard curve was prepared with ADP Detection Mixture 2, and the FP signal was recorded at 0 h (○), 1 h (•), 4 h (▾), 8 h (♦), and 24 h (▴). Error bars represent standard deviations of the mean (n = 24). (C) 10 μM ATP/ADP standard curves prepared with stock ADP detection mixtures. ADP detection mixtures were prepared and stored for 21 days at the designated temperatures prior to adding to freshly prepared ATP/ADP standards: Day 0 control (♦), −80°C (u), −20°C (▵), 4°C (▿), 25°C (◊), and 37°C (○). The Day 0 control (standard curve) was prepared with fresh ADP Detection Mixture 2. Error bars represent standard deviations of the mean (n = 4).
To quantify potential fluorescent compound interference with the Transcreener ADP assay, a collection of 1,062 compounds from the GenPlus compound library, BIOMOL Kinase Inhibitor Library, and other inhibitors were screened at a concentration of 10 μM (in 1% dimethyl sulfoxide) in the presence of 1 μM ADP plus 9 μM ATP in CK buffer (Fig. 7). A polarization shift of 158 mP units was achieved relative to the 10 μM ATP control wells (0% ATP conversion), resulting in a Z′ of 0.89 (n = 384). Only three compounds (out of 1,062) fell outside the band of 3 standard deviations on either side of the mean. These compounds were identified as lonidamin, palmitoyl-dl-carnitine, and dirithromycin. A fourth compound, hypericin, did not register an FP signal because its fluorescence intensity was sixfold higher than the control wells. The intensity of dirithromycin, a bright-blue compound, was more than twofold higher than the control wells. To determine if these same compounds had an effect on the antibody-tracer complex in the absence of ADP, a similar control screen was performed at 10 μM ATP; lonidamin and dirithromycin interfered with the antibody-tracer, but palmitoyl-dl-carnitine did not. These results indicate that immunodetection of ADP with the red-shifted Alexa Fluor633 tracer is very robust, reducing the likelihood of false-positives.
FIG. 7.

Control compound screen for assessing assay interference. Test compounds at 10 μM (1% dimethyl sulfoxide) were incubated with 10 mM ATP and a mixture of 1 μM ADP and 9 μM ATP (to represent 10% ATP conversion) in the CK reaction buffer: 0% ATP conversion (•), 10% ATP conversion (▴), 0% ATP conversion control (○), 10% ATP conversion control (▵), and 100% ATP conversion control (u). An equal volume of ADP Detection Mixture 2 (20 mM HEPES [pH 7.5], 40 mM EDTA, 0.02% Brij-35, 4 nM ADP-Alexa Fluor633 tracer, and 15 μg/ml Ab2) was then added to each well. The Z′ value for the 10% ATP conversion control was 0.89 (n = 384 from 24 plates). Compounds were tested in triplicate. Controls shown on the graph are from one plate (n = 16).
Discussion
In this report, we characterize the critical parameters for successful enzyme assays based on immunodetection of ADP using FP. The key reagents are an antibody that specifically recognizes ADP in the presence of excess ATP and a red-shifted fluorescent ADP tracer.
Though antibodies to AMP and cAMP have been produced previously,25,26 to our knowledge this was the first example of an antibody to an ADP hapten. We show here that Ab1 has high binding affinity to the ADP–Alexa Fluor633 tracer (KD = 2 nM), demonstrating a maximal change in polarization between bound and unbound tracer (ΔmP = 225 units), and had high selectivity for ADP versus ATP (>100-fold), satisfying the initial requirements for a successful ADP FP immunoassay. The ADP versus ATP selectivity was greater than first reported (52-fold),19 which can be largely contributed to the reduced levels of contaminating ADP in the ATP reagent (Fig. 2). For this reason care must be taken in preparing assay reagents that are free of contaminating nucleotides and nucleotidases.
The Transcreener ADP assay was formatted similar to other FP immunoassays: antibody is present at near saturating amounts, and tracer is present at or below the KD for the complex. The total polarization shift and the detection range of the assay were determined by the antibody EC85 concentration. This antibody concentration provided a good compromise between sensitivity and maximal polarization value; the use of subsaturating antibody allows more sensitive detection but yields a submaximal initial polarization value, whereas the use of a higher concentration of antibody results in higher initial polarization but also decreases sensitivity because more analyte is required to displace the tracer. To maintain maximum assay window at elevated ATP concentrations, additional antibody was required to absorb the additional cross-reacting nucleotide (Fig. 3).
The >100-fold selectivity for ADP versus ATP attained with Ab1 and red-shifted tracer is sufficient to allow Vinitial measurements over a broad range of starting ATP concentrations. To observe accurate inhibitor potencies, Vinitial conditions must be maintained, i.e., substrates must not be consumed to the point that reaction rate slows significantly, especially when using a single time point (incubation) data. These requirements need to be balanced with the need to generate a sufficient signal in the assay. Most screeners set 10–30% substrate conversion as a practical limit.15 Additionally, drug target screening is done over a wide range of ATP concentrations—usually the Km for a particular enzyme—in order to normalize for the competitive effects of inhibitors that bind at the ATP site.27 We demonstrated that by adjusting the Ab1 concentration, <10% ATP conversion could be detected at starting ATP concentrations ranging from 1 μM to 1,000 μM with a Z′ of greater than 0.7 (Fig. 5 and Table 3). Of the protein kinase Km ATP values reported in the literature, the vast majority fall between 2 μM and 300 μM,27 so the assay meets the kinetic constraints for screening over the reported range of kinase ATP affinities. We have also shown here that Transcreener technology is well suited to measure Vinitial values to determine the Km for ATP (Fig. 4). Note that to do so, it is necessary to determine the optimal antibody concentration for each ATP concentration that is used in the Km determination, and thus to perform a standard curve for each ATP concentration (Fig. 5).
The FP immunoassay utilizing Ab1 has been used to monitor protein and lipid kinase activity and to demonstrate correct pharmacology with known inhibitors. Huss et al.20 reported excellent concordance in IC50 values for protein kinase A inhibitors determined with the Transcreener assay and the traditional radioactive filter-binding assay. The assay was also shown to be resistant to low-wavelength fluorescent compound interference and has been subsequently used to validate >14 protein and lipid kinases. Because the nucleotide immunoassay does not detect the phosphorylated product, it can accommodate any substrate, including peptides, proteins, carbohydrates, and lipids. We recently evaluated lipid substrate dispersal methods and the effect of chain lengths on the activity and inhibition of phosphoinositide 3-kinase isoforms.28 HTS campaigns with ATP-dependent drug targets such as acetyl-coenzyme A carboxylase,22 nucleotide-dependent phosphatases,29 and an ATP group transferase30 exhibited less compound interference with the Transcreener platform than fluorescence and absorbance-based assays that detect inorganic phosphate.
The discovery of a new antibody combined with modified buffer conditions has substantially expanded the sensitivity and utility of the Transcreener assay. Enzyme Vinitial values can now be measured with starting ATP concentrations spanning four logarithms (0.1–1,000 μM ATP) because of the 18-fold improvement in ADP sensitivity achieved with Ab2 (Fig. 5 and Table 2). In addition, greater mP shifts can be obtained at lower percentage ATP conversions for 1 μM and 10 μM ATP when using Ab2 versus Ab1 (Fig. 5), resulting in higher quality assays (Z′ > 0.7) (Table 3). This increased sensitivity is largely due to the inherent antibody affinity for the tracer and ADP but is also partly due to the absence of NaCl in the ADP detection buffer (Table 1). Unlike Ab1, Ab2 did not require NaCl for signal stabilization in the presence of ATP. An increase in ionic strength has been shown to decrease the affinity of binding observed with other antibody–antigen complexes, which is attributed to the presence of salt bridges.31,32 These results support further studies to improve ADP affinity and possibly ATP selectivity by optimizing the detection buffer composition.
The nucleotide selectivity profiles of both antibodies, ADP = G DP > UDP > CDP > ATP > AMP (Table 2), indicates that binding appears to be localized to the diphosphate moiety. Addition of a phosphate to ADP disrupts antibody binding less than removal of phosphates. Adenine and d-ribose did not compete with the ADP tracer. Additionally, the 2′ hydroxyl group on ADP is not important in binding as 2′-deoxy-ADP was recognized as well as ADP. The application of this generic assay for dinucleotide detection can be expanded to other XDP-producing drug targets, including GTPases, but may be complicated with complex enzyme systems, i.e., crude extracts.
Formatting an assay for the rigors of HTS requires stable reagents, sustainable assay window over extended periods of time, and low compound interference. We have found that once the binding equilibrium has been established (∼30 min), the FP signal is stable in the plate for up to 24 h after addition of the ADP detection mixture (Fig. 6A and B). The ADP detection mixture can be stored for at least 3 weeks at ≤4°C without compromising assay performance (Fig. 6C), which can further simplify the assay design. Only four compounds out of 1,062 (0.4%) (>100 of which are known ATP-competitive inhibitors) were found to interfere with ADP detection when the control compound screen was performed in the presence of 1 μM ADP/9 μM ATP (Fig. 7). This is consistent with the results of Liu et al.,22 who reported 0.5% compound interference with the Transcreener assay when a 500,000 compound library was screened with acetyl-coenzyme A carboxylase at 100 μM ATP. Other studies have shown that the use of red tracers for FP-based assays eliminates much of the interference seen with green tracers.33,34 The improved Transcreener ADP assay provides Z′ values >0.7 at low ATP conversion (<10%) across a range of ATP concentrations (0.1–1,000 μM). This enables screening of virtually any ADP-producing enzyme at or below the ATP Km, under Vinitial conditions, so that accurate ATP-competitive inhibitor potencies are observed. The increased sensitivity is especially useful for drug targets possessing low catalytic activity or possessing ATP Km values of <1 μM. Performing inhibitor dose-dependency experiments over four logarithms of initial ATP concentrations also allows for the classification of noncompetitive inhibitors. This generic ADP immunodetection assay should greatly accelerate efforts in kinase drug discovery and to identify new inhibitors of other known ADP-producing drug targets, i.e., the protein chaperone heat shock protein 70, as well as GTPases.
Abbreviations
- Ab1
antibody 1
- Ab2
antibody 2
- cAMP
adenosine 39,59-cyclic monophosphate
- CK
common kinase
- DmP
change in millipolarization
- EC50 and EC85
antibody concentration at 50% and 85% of its saturation, respectively
- FP
fluorescence polarization
- HPLC
high-performance liquid chromatography
- IC50
competitor (inhibitor) concentration that results in a 50% reduction in signal
- KD
dissociation constant
- Km
Michaelis-Menten constant
- LLD
lower limit of ADP detection
- mP
millipolarization
- Vinitial
initial velocity.
Acknowledgments
This work was supported by SBIR grants CA110535-01A1, 2R44 CA110535-02, and 5R44 CA110535-03 from the National Institutes of Health. The technology used in this study is covered by U.S. Patents 7,332,278, 7,355,010, and 7,378,505 and by U.S. Patent Application numbers 11/353,500 and 11/958,965 and International Patent Application PCT/US04/002618.
Disclosure Statement
K.M.K.-L., T.A.K., A.L.K., T.A.W., M.D.K., B.R.L., T.J.W., C.A.P., and R.G.L. are employees of BellBrook Labs. S.A.T.V.K., G.J.R.Z., and J.J.H. are employees of the Schering-Plough Research Institute.
References
- 1.Sridhar R. Hanson-Painton O. Cooper DR. Protein kinases as therapeutic targets. Pharm Res. 2000;17:1345–1353. doi: 10.1023/a:1007507224529. [DOI] [PubMed] [Google Scholar]
- 2.Cohen P. Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 3.Dancey J. Sausville EA. Issues and progress with protein kinase inhibitors for cancer treatment. Nat Rev Drug Discov. 2003;2:296–313. doi: 10.1038/nrd1066. [DOI] [PubMed] [Google Scholar]
- 4.High Throughput Screening 2007 New Strategies Success Rates Use of Enabling Technologies. HighTech Business Decisions. San Jose; CA: 2007. [Google Scholar]
- 5.Xu K. Stern AS. Levin W. Chua A. Vassilev LT. A generic time-resolved fluorescence assay for serine/threonine kinase activity: application to Cdc7/Dbf4. J Biochem Mol Biol. 2003;36:421–425. doi: 10.5483/bmbrep.2003.36.4.421. [DOI] [PubMed] [Google Scholar]
- 6.Eglen RM. Singh R. Beta galactosidase enzyme fragment complementation as a novel technology for high throughput screening. Comb Chem High Throughput Screen. 2003;6:381–387. doi: 10.2174/138620703106298473. [DOI] [PubMed] [Google Scholar]
- 7.Zaman GJ. Garritsen A. de Boer T. van Boeckel CA. Fluorescence assays for high-throughput screening of protein kinases. Comb Chem High Throughput Screen. 2003;6:313–320. doi: 10.2174/138620703106298563. [DOI] [PubMed] [Google Scholar]
- 8.Gaudet EA. Huang KS. Zhang Y. Huang W. Mark D. Sportsman JR. A homogeneous fluorescence polarization assay adaptable for a range of protein serine/threonine and tyrosine kinases. J Biomol Screen. 2003;8:164–175. doi: 10.1177/1087057103252309. [DOI] [PubMed] [Google Scholar]
- 9.Xue Q. Wainright A. Gangakhedkar S. Gibbons I. Multiplexed enzyme assays in capillary electrophoretic single-use microfluidic devices. Electrophoresis. 2001;22:4000–4007. doi: 10.1002/1522-2683(200110)22:18<4000::AID-ELPS4000>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 10.Burke JR. Pattoli MA. Gregor KR. Brassil PJ. MacMaster JF. Mclntyre KW, et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003;278:1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- 11.Davidson W. Frego L. Peet GW. Kroe RR. Labadia ME. Lukas SM, et al. Discovery and characterization of a substrate selective p38alpha inhibitor. Biochemistry. 2004;43:11658–11671. doi: 10.1021/bi0495073. [DOI] [PubMed] [Google Scholar]
- 12.Dudley DT. Pang L. Decker SJ. Bridges AJ. Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh P. Harden BJ. Lillywhite BJ. Broad PM. Identification of kinase inhibitors by an ATP depletion method. Assay Drug Dev Technol. 2004;2:161–169. doi: 10.1089/154065804323056503. [DOI] [PubMed] [Google Scholar]
- 14.Koresawa M. Okabe T. High-throughput screening with quantitation of ATP consumption: a universal non-radioisotope, homogeneous assay for protein kinase. Assay Drug Dev Technol. 2004;2:153–160. doi: 10.1089/154065804323056495. [DOI] [PubMed] [Google Scholar]
- 15.Walters WP. Namchuk M. Designing screens: how to make your hits a hit. Nat Rev Drug Discov. 2003;2:259–266. doi: 10.1038/nrd1063. [DOI] [PubMed] [Google Scholar]
- 16.Charter NW. Kauffman L. Singh R. Eglen RM. A generic, homogenous method for measuring kinase and inhibitor activity via adenosine 5′-diphosphate accumulation. J Biomol Screen. 2006;11:390–399. doi: 10.1177/1087057106286829. [DOI] [PubMed] [Google Scholar]
- 17.Gibon Y. Blaesing OE. Hannemann J. Carillo P. Hîhne M. Hendriks JH, et al. A robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness. Plant Cell. 2004;16:3304–3325. doi: 10.1105/tpc.104.025973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schirmer A. Kennedy J. Murli S. Reid R. Santi DV. Targeted covalent inactivation of protein kinases by resorcylic acid lactone polyketides. Proc Natl Acad Sci U S A. 2006;103:4234–4239. doi: 10.1073/pnas.0600445103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lowery RG. Kleman-Leyer K. Transcreener: screening enzymes involved in covalent regulation. Expert Opin Ther Targets. 2006;10:179–190. doi: 10.1517/14728222.10.1.179. [DOI] [PubMed] [Google Scholar]
- 20.Huss KL. Blonigen PE. Campbell RM. Development of a Transcreener kinase assay for protein kinase A and demonstration of concordance of data with a filter-binding assay format. J Biomol Screen. 2007;12:578–584. doi: 10.1177/1087057107300221. [DOI] [PubMed] [Google Scholar]
- 21.Harwood HJ., Jr Petras SF. Shelly LD. Zaccaro LM. Perry DA. Makowski MR, et al. Isozyme-nonselective N-substituted bipiperidylcarboxamide acetyl-CoA carboxylase inhibitors reduce tissue malonyl-CoA concentrations, inhibit fatty acid synthesis, and increase fatty acid oxidation in cultured cells and in experimental animals. J Biol Chem. 2003;278:37099–37111. doi: 10.1074/jbc.M304481200. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y. Zalameda L. Kim KW. Wang M. McCarter JD. Discovery of acetylcoenzyme A carboxylase 2 inhibitors: comparison of a fluorescence intensity-based phosphate assay and a fluorescence polarization-based ADP assay for high-throughput screening. Assay Drug Dev Technol. 2007;5:225–235. doi: 10.1089/adt.2006.045. [DOI] [PubMed] [Google Scholar]
- 23.Tomiya N. Ailor E. Lawrence SM. Betenbaugh MJ. Lee YC. Determination of nucleotides and sugar nucleotides involved in protein glycosylation by high-performance anion-exchange chromatography: sugar nucleotide contents in cultured insect cells and mammalian cells. Anal Biochem. 2001;293:129–137. doi: 10.1006/abio.2001.5091. [DOI] [PubMed] [Google Scholar]
- 24.Zhang JH. Chung TD. Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 25.Bredehorst R. Schluter MM. Hilz H. Determination of 5-AMP in the presence of excess 3′(2′)-AMP with the aid of antibodies raised against N6-carboxymethyl-5′-AMP conjugates. Use for the quantitation of pyridine nucleotides and of protein-bound ADP-ribose. Biochim Biophys Acta. 1981;652:16–28. doi: 10.1016/0005-2787(81)90204-5. [DOI] [PubMed] [Google Scholar]
- 26.Richman RA. Kopf GS. Hamet P. Johnson RA. Preparation of cyclic nucleotide antisera with thyroglobulin-cyclic nucleotide conjugates. J Cyclic Nucleotide Res. 1980;6:461–468. [PubMed] [Google Scholar]
- 27.Knight ZA. Shokat KM. Features of selective kinase inhibitors. Chem Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 28.Klink TA. Kleman-Leyer KM. Kopp A. Westermeyer TA. Lowery RG. Evaluating PI3 kinase isoforms using Transcreener ADP assays. J Biomol Screen. 2008;13:476–485. doi: 10.1177/1087057108319864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antczak C. Ciro A. Sanchez G. Djaballah H. Development of a high-density fluorescence polarization-based assay for nucleotide-dependent phosphatases using the Transcreener™ technology. http://www.mskcc.org/mskcc/shared/pdf/Core_Facilities/SBSposter_April2007_TBFP.pdf. [Dec;2008 ]. http://www.mskcc.org/mskcc/shared/pdf/Core_Facilities/SBSposter_April2007_TBFP.pdf
- 30.Zalameda LP. Liu Y. McCarter JD. Application of high throughput fluorescence polarization-based ADP assay for an ATP-dependent group transferase. Poster presented at IBC's 12th Annual DDT World Congress: Building Your Drug Pipeline; Aug;2007 ; Boston, MA. [Google Scholar]
- 31.Roy P. Roth CM. Margolies MN. Yarmush ML. Effect of pressure on antigen-antibody complexes: modulation by temperature and ionic strength. Mol Immunol. 1999;36:1149–1158. doi: 10.1016/s0161-5890(99)00133-9. [DOI] [PubMed] [Google Scholar]
- 32.Tsumoto K. Ogasahara K. Ueda Y. Watanabe K. Yutani K. Kumagai I. Role of salt bridge formation in antigen-antibody interaction. Entropic contribution to the complex between hen egg white lysozyme and its monoclonal antibody HyHEL10. J Biol Chem. 1996;271:32612–32616. doi: 10.1074/jbc.271.51.32612. [DOI] [PubMed] [Google Scholar]
- 33.Vedvik KL. Eliason HC. Hoffman RL. Gibson JR. Kupcho KR. Somberg RL, et al. Overcoming compound interference in fluorescence polarization-based kinase assays using far-red tracers. Assay Drug DevTechnol. 2004;2:193–203. doi: 10.1089/154065804323056530. [DOI] [PubMed] [Google Scholar]
- 34.Simeonov A. Jadhav A. Thomas CJ. Wang Y. Huang R. Southall NT, et al. Fluorescence spectroscopic profiling of compound libraries. J Med Chem. 2008;51:2363–2371. doi: 10.1021/jm701301m. [DOI] [PubMed] [Google Scholar]