

Introduction
With our growing understanding of the pathways involved in cell proliferation and signalling, targeted therapies for the treatment of cancer are entering the clinical arena. New and emerging targets are proteins that participate in DNA repair processes. Inhibition of proteins in these pathways can sensitize cells to DNA damaging agents, such as chemotherapeutics and ionizing radiation, the main tools currently available to improve the outcome of patients with advanced cancers. The efficacy of these treatment methods is directly related to the ability of the agent to specifically induce cytotoxic damage in tumor cells rather than in normal cells. This specific targeting is accomplished through molecular and cellular features of the cancer cell, such as its higher proliferative rate, which may be directly attacked using inhibitors of the cell cycle. Besides compounds that block cell division at the level of mitotic spindle formation (e.g. vinca alkaloids and taxanes), and growth signal inhibitors, which operate through hormonal manipulation by means of therapeutic antibodies or drugs, a growing number of cell cycle inhibitors encompass DNA damaging agents. Genomic damage may cause cell cycle arrest either directly or indirectly as a consequence of abortive DNA replication during the S-phase of the cell cycle. Notably, the ability of cancer cells to efficiently recognize and remove cytotoxic DNA damage plays a key role in therapeutic resistance, thus profoundly affecting therapeutic efficacy [1–3]. Moreover, since some DNA repair pathways are impaired or inactivated in some types of cancers, a detailed comprehension and strategic manipulation of DNA repair mechanisms could improve the efficacy of DNA damage-based anticancer therapies.
DNA repair pathways [such as base excision repair (BER), nucleotide excision repair (NER), translesion synthesis bypass (TLS), homologous recombination (HR), and nonhomologous end joining (NHEJ)] (Fig. 1) can enable tumor cells to survive DNA damage that is induced by common cancer therapy; therefore, inhibitors of specific DNA repair pathways might prove efficacious when used in combination with DNA-damaging chemotherapeutic drugs (Table 1). In addition, alterations in DNA repair pathways that arise during tumor development can make some cancer cells reliant on a reduced set of DNA repair pathways for survival. Indeed, DNA repair pathways are highly interconnected and collaborative, and while a particular DNA lesion can be processed by multiple repair pathways, a single repair process is capable of repairing multiple DNA lesions. Moreover, DNA enzymology, epigenetic regulation, and transcriptional regulatory mechanisms need to be highly interconnected and strictly coordinated to guarantee proper relief from the consequences of DNA damage. Therefore, knowledge of the fine-tuning mechanisms able to specifically control the activity of DNA repair enzymes is of the utmost importance both for cancer diagnosis and cancer treatment.
Fig. 1.
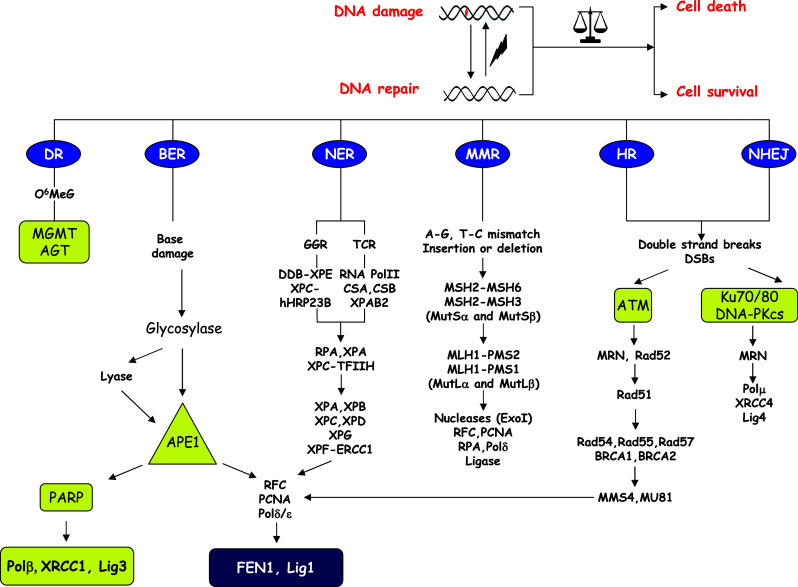
Schematic overview of DNA repair systems and emerging anticancer targets. Repairing toxic DNA lesions induced during most common cancer treatments relies on proper DNA repair response, which has the ability to dictate the cellular outcome, e.g., cell death or cell survival. Triggering of a specific repair pathway (DR, BER, NER, MMR, HR, and NHEJ pathways) depends on the nature of the DNA damage. The main components and the signaling pathways of each repair response are indicated. Yellow boxes highlight emerging anticancer targets that are discussed in this issue. The availability of other components of these pathways to serve as anticancer targets requires further testing. DR Direct reversal, BER base excision repair, NER nucleotide excision repair, GGR global genome repair, TCR transcription coupled repair, MMR mismatch repair, HR homologous recombination, MRN Mre11-Rad50-Nbs1 complex, NHEJ non-homologous end joining, O 6 MeG O6-methylguanine
Table 1.
List of the known repair pathways involved in repairing toxic DNA lesions induced by most common cancer treatments
| Cancer treatment | Toxic lesion | Major repair pathways involved |
|---|---|---|
| Radiomimetics and radiotherapy | Single-strand breaks, double-strand breaks, base damage | NHEJ, BER, HR |
| Ionizing radiation | ||
| Bleomycin | ||
| Monofunctional alkylators | Base damage, bulky adducts, replication lesions | DR, BER, HR, NER |
| Temozolomide | ||
| Nitrosourea compounds | ||
| Alkylsulphonates | ||
| Bifunctional alkylators | Double-strand breaks, DNA crosslinks, replication lesions, bulky adducts | HR, NER |
| Cisplatin | ||
| Mitomycin C | ||
| Nitrogen mustards | ||
| Psoralen | ||
| Antimetabolites | Base damage, replication lesions | BER |
| 5-Fluorouracil | ||
| Thiopurines | ||
| Folate analogues | ||
| Topoisomerase inhibitors | Single-strand breaks, double-strand breaks, replication lesions | HR, NHEJ |
| Camptothecins | ||
| Etoposide | ||
| Replication inhibitors | Double-strand breaks, replication lesions | HR, NHEJ |
| Aphidicolin | ||
| Hydroxyrea |
The present multi-author review consists of nine contributions that cover some of the most important aspects of biochemical and functional regulation of key DNA repair enzymes, as well as the current state-of-art strategies to target these proteins in future cancer treatment paradigms.
The first paper, by Hegde et al., provides a thorough functional and structural overview of the base excision repair (BER) pathway. BER is endowed with the responsibility of repairing small not-distorting DNA lesions, such as oxidized or aberrant bases and single strand breaks (SSBs), which are produced during cell metabolism or as a consequence of treatment with chemotherapeutic agents (e.g., bleomycin, temozolomide) [4, 5]. Since these lesions are often mutagenic and have etiological linkage to sporadic cancers, the BER pathway is important in both the pathogenesis and treatment of cancer. The contribution by Hegde et al. in this issue focuses also on a novel structural aspect of the main enzymes in BER, i.e., the DNA glycosylases and apurinic/apyrimidinic (AP) endonuclease, where the disordered segments of these proteins operate as sites of molecular regulation of their activities.
A key enzyme in BER is the AP endonuclease (APE1), which acts as the predominant AP site incision enzyme in mammalian cells. APE1 is a multifunctional ubiquitous and essential protein playing a role both in the pathogenesis of cancer and in resistance to DNA-interactive drugs, such as monofunctional alkylators and antimetabolites. For these reasons, APE1 is acquiring more and more interest as a novel and promising candidate target for anticancer treatment. APE1, which is altered in more aggressive tumors, is a pleiotropic protein playing a critical role not only in DNA repair but also in regulating apoptosis, cell proliferation and an adaptive cell response to oxidative stress [6]. These observations, by the way, would be compatible with the caveat that in principle APE1 cannot be fully and stably knocked down, being essential for survival of cancer cells as well as normal cells, and therefore implying that specific inhibition of its activities only in cancer cells would be required for designing new efficient candidate drugs [7–10]. Thus, understanding the fine tuning mechanisms responsible for its functional regulation and its alteration in cancer cells is of the utmost importance in developing effective inhibitors. The review by Tell et al. in this issue describes the molecular basis for the multifunctional roles of this protein and is followed by an updated description of the post-translational modifications known to have a regulatory impact in redirecting APE1 toward a specific function (C. Busso et al., this issue). The fourth contribution, by Wilson and Simeonov, describes the current state of efforts to design potent and selective inhibitors against APE1 DNA repair functions.
BER, by virtue of its handling of extremely toxic pathway intermediates, is coupled to cell death and recombination mechanisms, both of which are activated should BER stall or fail. Our understanding of these back-up mechanisms in BER and various linkages to other processes, such as replication, is in its earliest stage, and our understanding of the components of the BER sub-pathways themselves is still limited [11, 12]. Because of this lack of information, we are unable to make accurate predictions on clinical therapeutic approaches to either enhance or interfere with BER. Similarly, we are not able to make accurate predictions on the biological effects of environmental stressors that trigger BER. A deeper understanding of BER will eventually allow us to conduct more meaningful clinical interventions and also use BER capability in risk analyses and prevention of toxicant-induced disease. In the fifth review, Wilson et al. will cover historical and recent information on BER and DNA polymerase β and discuss approaches currently underway toward use of small molecule inhibitors to manipulate BER and potentiate cytotoxicity.
The following article by F. Mégnin-Chanet et al. is an overview of the PARP family of enzymes as essential players in repairing SSBs. Further discussion covers the emerging evidence that PARPs are ideal candidate targets for single agent therapies acting through the principle of synthetic lethality with homologous recombination-deficient (e.g., BRCA) tumors [3, 13, 14].
Alkylating agent-induced DNA damage, besides being repaired via BER, is also acted upon by O6-methylguanine-DNA methyltransferase (MGMT), a suicide protein that specifically removes methyl (or alkyl) groups from the O6 position of guanine or O4 thymine. Since these adducts are formed as a consequence of a number of common alkylator drugs, MGMT levels represent a key mechanism of anticancer drug resistance, particularly in malignant gliomas and melanomas [15, 16]. The contribution by Kaina and Christmann focuses on MGMT inhibitors as promising tools for cancer therapy.
One of the most important classes of drugs used in cancer chemotherapy forms DNA interstrand crosslinks (ICLs), which are extremely cytotoxic lesions in that they block replication and transcription in eukaryotic cells [17]. However, to date, relatively little information is available on the molecular mechanisms responsible for their removal within cells, in part due to the limited availability of well-defined substrates. A. Guainazzi and Schärer describe, in the eighth contribution, a survey of the current understanding of the biological responses triggered by ICLs, including translesion synthesis (TLS) bypass, homologous recombination (HR), and nucleotide excision repair (NER) pathways [18, 19]. The review focuses particularly on the use of synthetic adducts that have facilitated recent studies and on how these findings may be translated into the design of new therapeutic agents for cancer chemotherapy.
Double strand breaks (DSBs) are considered one of the most harmful and lethal forms of DNA damage and are also a main cause of genomic instability associated with the onset of cancer [20]. DSBs, which may result as the consequence of many chemotherapeutic agents that induce damage that blocks DNA replication, are repaired through homology-directed repair (HR) or nonhomologous end joining (NHEJ) [21]. The last review by F.V. Rassool and A.E. Tomkinson focuses on the molecular aspects of DSB repair, their alterations in cancer, and on the future directions for cancer therapies that target these pathways.
Acknowledgments
This article was supported financially by grants from MIUR (FIRB RBRN07BMCT_008 and PRIN 2008CCPKRP_003) to G.T and the Intramural Research Program of the National Institutes of Health, National Institute on Aging (D.M.W. III).
References
- 1.Helleday T. Amplifying tumour-specific replication lesions by DNA repair inhibitors—a new era in targeted cancer therapy. Eur J Cancer. 2008;44:921–927. doi: 10.1016/j.ejca.2008.02.044. [DOI] [PubMed] [Google Scholar]
- 2.Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis. 2010;31:955–960. doi: 10.1093/carcin/bgq064. [DOI] [PubMed] [Google Scholar]
- 3.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 4.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitra S, Hazra TK, Roy R, Ikeda S, Biswas T, Lock J, Boldogh I, Izumi T. Complexities of DNA base excision repair in mammalian cells. Mol Cells. 1997;7:305–312. [PubMed] [Google Scholar]
- 6.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal. 2009;11:601–620. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nyland RL, Luo M, Kelley MR, Borch RF. Design and synthesis of novel quinine inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/ref-1) J Med Chem. 2010;53:1200–1210. doi: 10.1021/jm9014857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo M, He H, Kelley MR, Georgiadis MM. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid Redox Signal. 2010;12:1247–1269. doi: 10.1089/ars.2009.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, 2nd, Borch RF, Qiao X, Georgiadis MM, Kelley MR. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxid Redox Signal. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simeonov A, Kulkarni A, Dorjsuren D, Jadhav A, Shen M, McNeill DR, Austin CP, Wilson DM., 3rd Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease APE1. PLoS ONE. 2009;4:e5740. doi: 10.1371/journal.pone.0005740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Memisoglu A, Samson L. Base excision repair in yeast and mammals. Mutat Res. 2000;451:39–51. doi: 10.1016/s0027-5107(00)00039-7. [DOI] [PubMed] [Google Scholar]
- 12.Russo MT, De Luca G, Degan P, Bignami M. Different DNA repair strategies to combat the threat from 8-oxoguanine. Mutat Res. 2007;614:69–76. doi: 10.1016/j.mrfmmm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Hakmé A, Wong HK, Dantzer F, Schreiber V. The expanding field of poly(ADP-ribosyl)ation reactions. ‘Protein Modifications: Beyond the Usual Suspects’ Review Series. EMBO Rep. 2008;9:1094–1100. doi: 10.1038/embor.2008.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 15.Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W, Hegi ME. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol. 2010;6:39–51. doi: 10.1038/nrneurol.2009.197. [DOI] [PubMed] [Google Scholar]
- 16.Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst) 2007;6:1079–1099. doi: 10.1016/j.dnarep.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 17.Schärer OD. DNA interstrand crosslinks: natural and drug-induced DNA adducts that induce unique cellular responses. Chembiochem. 2005;6:27–32. doi: 10.1002/cbic.200400287. [DOI] [PubMed] [Google Scholar]
- 18.Hinz JM (2010) Role of homologous recombination in DNA interstrand crosslink repair. Environ Mol Mutagen. doi:10.1002/em.20577 [DOI] [PubMed]
- 19.Sarkar S, Davies AA, Ulrich HD, McHugh PJ. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J. 2006;25:1285–1294. doi: 10.1038/sj.emboj.7600993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 21.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]