

Abstract
Excitotoxicity and/or microglial reactivity might underlie neurologic dysfunction in HIV patients. The HIV regulatory protein Tat is both neurotoxic and pro-inflammatory, suggesting that Tat might participate in the pathogenesis of HIV-associated neurocognitive disorders (HAND). The present study was undertaken to evaluate if Tat can increase extracellular glutamate, and was specifically designed to determine the degree to which, and the mechanisms by which Tat could drive microglial glutamate release. Data show that application of Tat to cultured primary microglia caused dose-dependent increases in extracellular glutamate that were exacerbated by morphine, which is known to worsen Tat cytotoxicity. Tat-induced glutamate release was decreased by inhibitors of p38 and p42/44 MAPK, and by inhibitors of NADPH oxidase and the xc− cystine-glutamate antiporter. Furthermore, Tat increased expression of the catalytic subunit of xc− (xCT), but Tat-induced increases in xCT mRNA were not affected by inhibition of NADPH oxidase or xc− activity. Together, these data describe a specific and biologically significant signaling component of the microglial response to Tat, and suggest that excitotoxic neuropathology associated with HIV infection might originate in part with Tat-induced activation of microglial glutamate release.
Keywords: excitotoxicity, glia, inflammation, morphine, HIV-associated neurocognitive disorders, oxidative stress
INTRODUCTION
The Centers for Disease Control and Prevention estimate that over 1 million individuals in the United States are infected with the human immunodeficiency virus (HIV), which affects nearly 33 million people world-wide. While widespread availability of antiretroviral therapies in developed countries has led to remarkable improvements in overall health outcomes, HIV-associated neurocognitive disorders (HAND) remain a significant public health concern (reviewed in [1]). Although HAND is not universal among HIV infected persons, HIV remains the most common cause of neurologic impairment in patients under 50 [1]. The viral and/or host factor(s) responsible for HAND have not been clearly identified, but observations that mRNA levels of HIV viral regulatory protein Tat are elevated in patients with dementia [32], and that Tat is actively secreted by infected cells [10] suggest a role for Tat in the progression of HAND. Furthermore, Tat has pro-inflammatory and neurotoxic properties in microglia and macrophages [30], [20], [31], and Tat levels correlate positively with the development of HIV-and chimeric simian-human immunodeficiency virus (SHIV)-induced encephalitis [14].
Brain-resident microglia and macrophages likely participate in HAND via a variety of mechanisms, including serving as viral reservoirs and by releasing neurotoxic cytokines and pro-oxidants [16], [23]. Microglial activation is extensive in brains from patients with neurologic impairment [11] and data from our labs and others have shown that Tat can specifically and significantly increase macrophage/microglial reactivity and microglial-mediated neurotoxicity [20], [31]. Further data show that these processes depend in part on Tat-induced activation of NADPH oxidase [31], [33]. NADPH oxidase is a key component of the oxidative burst activity, and is a superoxide-producing enzyme system consisting of membrane (gp91phox and p22phox) and cytosolic (p47phox, p67phox, and p40phox) components [7]. Accumulating experimental evidence suggests that activation of NADPH oxidase in microglia can lead to increased extracellular glutamate via activation of the xc− cystine-glutamate antiporter [4]. Upregulation of xc− antiporter activity in immunocompetent cells is thought to be a self-protective mechanism to increase intracellular cysteine and glutathione so that the cells are not damaged by their own oxidative burst [4], [25]. This study was thus designed to determine if Tat causes the release of physiologically significant levels of glutamate from microglia via activation of xc−.
MATERIALS AND METHODS
Materials
Recombinant Tat 1-72 was produced at the University of Kentucky Tat Core facility as described previously [30], [31]. Apocynin, DL-2-aminoadipic acid, LPS from E. coli serotype O111:B4, and morphine sulfate were purchased from Sigma-Aldrich (St. Louis, MO), while 4-carboxypheylglycine, SB 239063, LY 294002, P 600124, UO126, and UO124 were purchased from Tocris Bioscience (Ellisville, MO). Primer pairs for 18S were from Invitrogen (Carlsbad, California) and xCT primers were from IDT (Coralville, IA).
Cell culture and treatment
Primary microglia were isolated from mixed glial cultures derived from neonatal Sprague-Dawley rat pups as described previously [31]. Cells were pelleted and subcultured at 5 × 105 cells/ml, and used within 24 hours after subculturing. All treatments were performed under serum-free conditions.
Glutamate measures
The concentration of glutamate in the culture medium was determined by a colorimetric enzymatic assay kit (BioVision, Mountain View, CA) according to the manufacturer’s instructions.
xc− expression
Conventional two-step, real-time PCR analysis of cystine-glutamate antiporter expression in microglia was conducted by measuring expression of the catalytic subunit (xCT) of the xc− transporter. mRNA levels were quantified using the 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) against18S. The primers used were as follows: xCT forward, 5′-CCCAGATATGCATCGTCCTT-3′ and reverse, 5′-ACAACCATGAAGAGGCAGGT-3′; and 18S forward, 5′-AGTCCCTGCCCTTTGTACACA-3′ and reverse, 5′-GATCCGAGGGCCTCACTAAAC-3′.
Statistical analyses
All data were analyzed using one-way ANOVA, followed by Tukey’s post-hoc analysis to determine statistical significance; p values < 0.05 were designated as statistically significant, and are indicated in the text as *, **, or *** corresponding to p values < 0.05, < 0.01, or < 0.001, respectively.
RESULTS
1. Tat increases microglial glutamate release
Previous reports have shown that Tat can significantly increase microglia reactivity and microglial-mediated neurotoxicity [20], [31] in a manner specific to the viral protein. Furthermore, reports have demonstrated a role for excitotoxicity in the effects of Tat in whole brain [27], but the direct relationship of Tat-induced microglial activation and elevations in extracellular glutamate has not been evaluated. Initial experiments were thus designed to evaluate if Tat could specifically trigger the release of glutamate from primary cultured microglia. Microglia were treated with 10–100 nM Tat 1-72 for 24 hours, and glutamate concentration in the medium was measured. Data show that Tat caused significant, dose-dependent increases in extracellular glutamate (Fig 1). LPS (100–1000 ng/ml) was used as a positive control based on previous reports [4], and data verified the effects of LPS on glutamate release (Fig 1). Cell viability was examined by documenting the ability of vehicle-, Tat-, and LPS-treated cells to convert 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) to formazan as described previously [30]. Neither LPS nor Tat induced cytotoxicity (data not shown), indicating that glutamate release was not caused by cell lysis or injury. Furthermore, glutamate levels in stock solutions of 100 nM Tat or 1000 ng/ml of LPS were beneath the threshold of detection in the assay used, showing that amino acid contaminants in the stock solutions did not artificially affect the data.
Figure 1. Effects of Tat and LPS on glutamate release from cultured primary microglia.
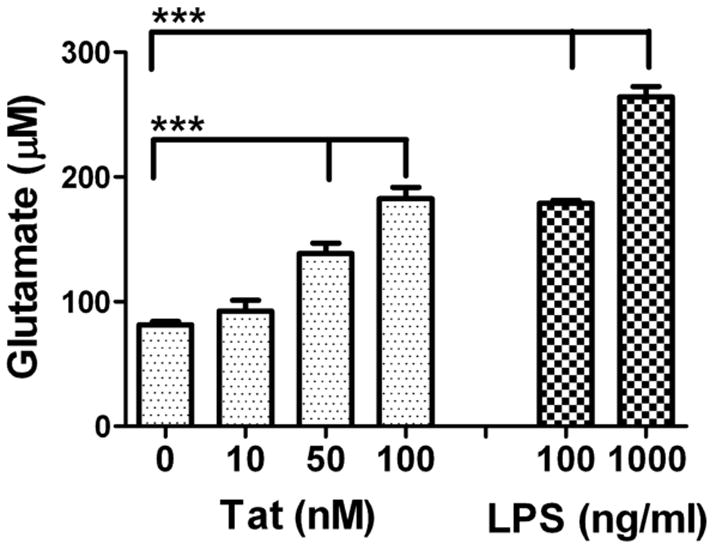
Microglia were exposed to Tat 1-72 or LPS, and glutamate accumulation in the medium was measured after 24 hours. Data were compiled from 4 separate experiments, and are means and SEM of 8–18 samples per group. Data were analyzed by 1-way ANOVA, and *** indicates statistically significant (p<0.001) increases in extracellular glutamate measured in Tat- and LPS-treated dishes.
To test whether alternate inflammatory stimuli triggered glutamate, microglia were treated with Tat in the presence of increasing doses of morphine, which has been shown to specifically increase Tat-induced free radical production and markers of inflammatory activation in microglia [30], [5]. Cells were also treated with the cytokine IFNγ and with ATP. IFNγ expression is increased in brains of HIV-1-infected patients and SIV-infected macaques [28], while ATP enhances the migratory properties of microglia, particularly in the context of CNS injury [17]. Data show that Tat-induced glutamate release was exacerbated by high concentrations (1000 nM) of morphine (Table 1), in keeping with the known immunomodulatory properties of this potent opiate agonist (reviewed in [13]), but neither IFNγ nor ATP caused glutamate release (Table 1).
Table 1. Effects of different immune stimuli on glutamate release from cultured primary microglia.
Primary rodent microglia were isolated and grown as described in Methods. Cells were exposed to Tat 1-72 in the presence of absence of increasing concentrations morphine, or to LPS, ATP, or the cytokine IFNγ. Glutamate accumulation in the medium was measured after 24 hours.
| Treatment | Glu (μM) |
|---|---|
| Vehicle | 60.1 ± 4.1 |
| Tat (100 nM) | 223.4 ± 8.4*** |
| Tat/morphine (100 nM) | 244.8 ± 5.1*** |
| Tat/morphine (500 nM) | 250.3 ± 8.5*** |
| Tat/morphine (1000 nM) | 262.1 ± 16.8*** # |
| Morphine (1000 nM) | 72.9 ± 4.4 |
| LPS (100 μg/ml) | 303.1 ± 7.6*** |
| ATP (100 μM) | 88.7 ± 3.6 |
| IFNγ (50 U/ml) | 65.6 ± 2.1 |
Data are means and SEM of 4–12 samples per group, and were analyzed by 1-way ANOVA.
indicates statistically significant (p<0.001) increases in extracellular glutamate measured in Tat- and LPS-treated dishes, while
indicates the statistically significant increase (p<0.05) in glutamate release from cells treated with Tat in the presence of 1000 nM morphine compared to cells treated with Tat alone.
To determine the intracellular mechanisms whereby Tat triggers glutamate release, inhibitors of p38MAPK (SB 239063, 5 μM), phosphatidylinositol 3-kinase (LY 294002, 10 μM), c-Jun N-terminal kinase (SP 600124, 1μM), and p42/p44 MAPK (UO126, 5 μM) were applied 30 minutes before 100 nM Tat. UO124 (5 μM), a negative control for UO126, was also used, and glutamate levels were evaluated after 24 hours. Data show that SB 239063 and UO126 pretreatment significantly reduced Tat-induced glutamate release (Table 2), suggesting that activation of both p38 and p42/p44 MAPK participate in the effects of Tat. None of the inhibitors significantly affected basal glutamate levels or MTT conversion (data not shown), suggesting that the inhibitory effects of SB 239063 and UO126 were not related to cell death or injury, although higher doses (25 μM) of SB 239063 and UO126 were cytotoxic over 24 hours (data not shown).
Table 2. Effects of signal transduction pathway inhibitors on Tat-induced glutamate release from cultured primary microglia.
Primary rodent microglia were isolated and grown as described in Methods. Cells were exposed to Tat 1-72 in the presence or absence of inhibitors of p38MAPK (SB 239063, 5 μM), phosphatidylinositol 3-kinase (LY 294002), c-Jun N-terminal kinase (SP 600124), p42/44 MAPK (UO126), and UO124 (negative control for UO126). Glutamate accumulation in the medium was measured after 24 hours.
| Treatment | Glu (μM) |
|---|---|
| Vehicle | 56.2 ± 2.1 |
| Tat (100 nM) | 223.8 ± 10.2*** |
| Tat/SB 239063 (5 μM) | 175.6 ± 12.8## |
| Tat/LY 294002 (10 μM) | 253.8 ± 4.5 |
| Tat/SP 600124 (1 μM) | 204.1 ± 6.7 |
| Tat/UO126 (5 μM) | 130.8 ± 4.9### |
| Tat/UO124 (5 μM) | 211.4 ± 7.7 |
Data are means and SEM of 6–12 samples per group collected over 2 experiments, and were analyzed by 1-way ANOVA.
indicates statistically significant (p<0.001) increases in extracellular glutamate measured in Tat-treated dishes, while
indicate statistically significant decreases (p<0.01, and p<0.001, respectively) in glutamate release from cells treated with Tat in the presence of SB 239063 or UO126 compared to cells treated with Tat alone.
2. Tat-induced glutamate release is blocked by inhibition of xc− and NADPH oxidase
Although microglia express glutamate transporters [21], data suggests microglial-mediated glutamate release is via the xc− cystine-glutamate antiporter [4], [25]. Indeed, upregulation of the xc− antiporter in immunocompetent cells is associated with NADPH oxidase-driven oxidative burst activity [4], which is also activated by Tat [31]. Accordingly, experiments were designed to determine the role of the xc− cystine-glutamate antiporter in Tat-induced glutamate release. Microglia were treated with 100 nM Tat in the presence or absence of the xc− antiporter inhibitors DL-amino adipic acid (DL-AAA) or 4-carboxypheylglycine (4-CPG) for 24 hours. Data show that Tat-induced glutamate release was significantly reduced by 4-CPG or DL-AAA (Fig. 2). To determine if NADPH oxidase participated in glutamate release, additional cells were treated with Tat in the presence or absence of the NADPH oxidase inhibitor apocynin. Data indicate that apocynin was likewise able to prevent the actions of Tat on glutamate release (Fig. 2).
Figure 2. Effects of NADPH oxidase and glutamate/cystine antiporter inhibitors on Tat-induced glutamate release from cultured primary microglia.
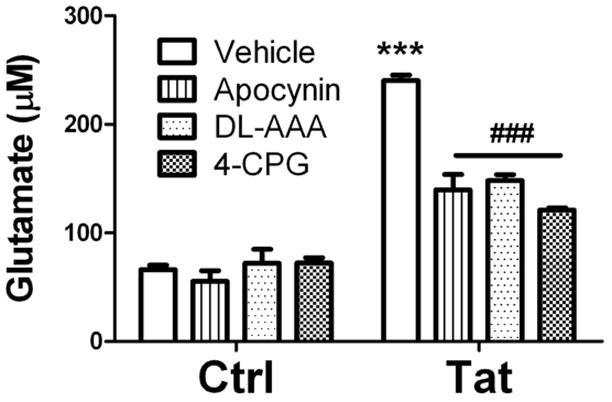
Microglia were treated with the NADPH oxidase inhibitor apocynin (200 μM), the xc− antiporter inhibitor DL-aminoadipic acid (DL-AAA, 2.5 mM), or the xc− antiporter inhibitor (S)-4-carboxyphenylglycine (4-CPG, 50 μM) in the presence or absence of 100 nM Tat-1-72. Glutamate accumulation in the medium was measured after 24 hours. Data were compiled from 2 separate experiments, and are means and SEM of 8–12 samples per group. Data were analyzed by 1-way ANOVA, and *** indicates statistically significant (p<0.001) increases in extracellular glutamate released from cells treated with Tat alone, while ### indicates the statistically significant decrease (p<0.001) in glutamate release from cells treated with Tat in the presence of apocynin, DL-AAA, or 4-CPG.
3. Tat-induced xc− expression is not blocked by NADPH oxidase inhibition
To further determine if Tat-induced increases in microglial glutamate release were mediated via increased synthesis of the glutamate antiporter, we sought to determine if relative expression of xCT mRNA was affected by Tat treatment. The effects of NADPH oxidase inhibitors and xc− antiporter blockers on xCT mRNA were also determined. Microglia were pretreated with apocynin or 4-CGP, and then challenged with vehicle or Tat. After 6 hours, the cells were harvested for RNA isolation, and xCT mRNA levels were evaluated by quantitative real time PCR. Data show that Tat treatment caused a significant increase in xCT mRNA relative to 18S (Fig. 3), but that treatment with apocynin or 4-CPG alone did not affect basal levels of xCT mRNA (data not shown). Furthermore, neither apocynin nor 4-CPG was able to significantly decrease Tat-induced xCT expression (Fig. 3).
Figure 3. Effects of NADPH oxidase and glutamate/cystine antiporter inhibitors on Tat-induced regulation of Xc antiporter expression in cultured primary microglia.
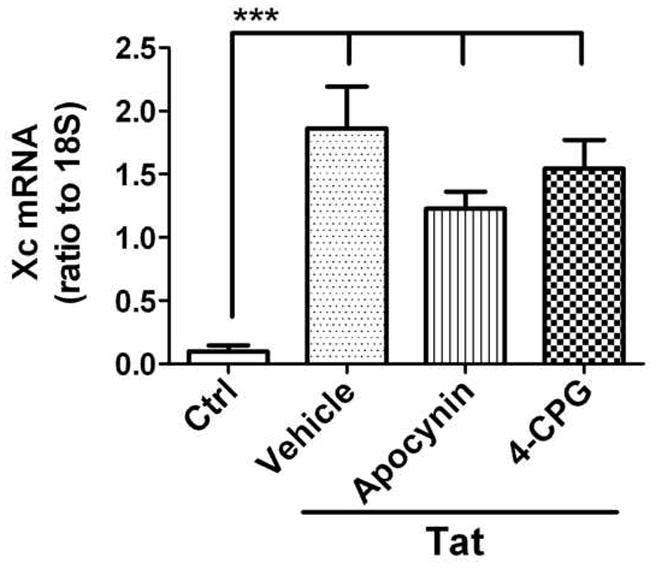
Microglia were treated with the NADPH oxidase inhibitor apocynin (200 μM) or the xc− antiporter inhibitor (S)-4-carboxyphenylglycine (4-CPG, 50 μM) in the presence or absence of 100 nM Tat-1-72. xCT mRNA levels were measured by real time PCR after 6 hours. Data indicate the level of xCT mRNA relative to 18S rRNA in each sample, and are means and SEM of 8–12 samples per group compiled from 4 separate experiments. Data were analyzed by 1-way ANOVA, and *** indicates statistically significant (p<0.001) increases in xCT mRNA expression in cells treated with Tat (alone or in the presence of apocynin or 4-CPG) as compared to control cells.
DISCUSSION
Data demonstrate the specific and biologically significant effects of the HIV regulatory protein Tat on microglial glutamate release. Specifically, Tat triggers dose-dependent glutamate release, which is enhanced by high levels of morphine. Tat-induced glutamate release is associated with increased expression of the xc− glutamate-cystine antiporter and is blunted by inhibitors of p38 and p42/44 MAPK, as well as xc− and NADPH oxidase inhibitors. Finally, glutamate release does not appear to be a generalized microglial response to pro-inflammatory stimuli, as neither IFNγ nor ATP were able to elicit significant glutamate release. Collectively, these data support a specific and clinically important role for Tat in directing excitotoxic inflammatory signaling in microglia, and suggest that Tat may play a more extensive role in excitotoxicity in the HIV-infected CNS then previously appreciated.
AIDS is a multisystem disease and the CNS is highly vulnerable to HIV (reviewed in [1]). Indeed, HIV-associated neurocognitive disorders (HAND) continue to be common, and as many as one fifth of patients with AIDS could develop indications of HAND [12]. Thus, there is considerable impetus to resolve the viral and/or host mechanisms that drive the progression of HAND. The viral regulatory protein Tat has repeatedly been implicated in the progression of HIV neuropathogenesis (reviewed in [18]). For example, Tat expression or delivery to brain alters histological indices of microglial inflammation and synaptic density, and impairs behavioral performance [15], [9]. While the mechanisms of Tat-mediated toxicity are not fully resolved, evidence indicates that the neurotoxic effects of Tat could be glutamate-mediated, which is in keeping with the data reported in this manuscript. Indeed, some of the evidence that excitotoxicity mediates neuronal dysfunction in HIV patients comes from studies of the neurotoxic actions of viral proteins such as Tat and gp120 [22]. Findings that the NMDA antagonist memantine, an uncompetitive NMDA receptor antagonist, prevents calcium changes in neurons and astrocytes, protects neurons from HIV viral protein-induced cell death [22], and improves synaptic function in murine HIV encephalitis [2], also support an excitotoxic component of HAND. Early results of clinical trials using memantine as a potential adjunctive therapy in HIV are encouraging [34], but final results have not yet been reported on the clinical efficacy of memantine in preventing or mitigating HAND.
Microglia are viral reservoirs and sites of HIV-1 replication, but could further drive HAND by serving as sources of neurotoxic cytokines and pro-oxidants (reviewed in [16]). Data suggest that microglia could be key players in the development of excitotoxicity in HIV brains via activation of the xc− antiporter. When exposed to proinflammatory stimuli, the microglial cytotoxins that are responsible for collateral or “bystander” damage may very likely act on microglia themselves to produce autonomous damage, and studies suggest that microglial activation can be associated with compromised viability [19]. A significant aspect of this vulnerability likely comes from the oxidative stress of the respiratory burst, as activation of NADPH oxidase is a common response of microglia to viral or bacterial stimuli. A major mammalian cellular defense against oxidative stress is covalent attachment of glutathione to lipid peroxidation products by glutathione S-transferases (GSTs) such as GSTA4-4 and GST5.8 [3], leading to decreases in cellular glutathione reserves. The xc− cystine-glutamate transporter mediates Na+-independent, electroneutral exchange of cystine and glutamate, and acts to restore glutathione reserves [24]. The present findings indicate that Tat increases expression and activity of xc− in brain resident microglia, raising the possibility that Tat-mediated activation of xc− could participate in HIV-related pathology. In further support of this scenario are published reports that HIV infection is associated with decreased levels of circulating and tissue glutathione [8], [29], and that Tat depletes glutathione in animal models [6]. Because glutathione not only participates as an antioxidant but also is important for conjugation of various drugs and xenobiotics necessary for their subsequent elimination, glutathione deficiency in HIV-1 infection could contribute not just to oxidative damage, but also to the heightened drug toxicity often associated with HIV infection [26]. Collectively, these data strongly support further investigation into the role of Tat and microglial xc− activation in the development of HAND, as well as in other end-stage, AIDS-related neurologic syndromes.
Research Highlights.
HIV-Tat triggers the release of significant amounts of the excitatory amino acid glutamate from cultured microglia
Tat-induced glutamate release involves activation of p38 and p42/44 MAPK signal transduction pathway
Tat-induced glutamate release depends on activation of NADPH oxidase, and is mediated through the xc− cystine-glutamate antiporter
Acknowledgments
This work was supported by grants from the NIH (NS46267, DA19398, and AG05119).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ances BM, Ellis RJ. Dementia and neurocognitive disorders due to HIV-1 infection. Semin Neurol. 2007;27:86–92. doi: 10.1055/s-2006-956759. [DOI] [PubMed] [Google Scholar]
- 2.Anderson ER, Gendelman HE, Xiong H. Memantine protects hippocampal neuronal function in murine human immunodeficiency virus type 1 encephalitis. J Neurosci. 2004;24:7194–7198. doi: 10.1523/JNEUROSCI.1933-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Awasthi YC, Yang Y, Tiwari NK, Patrick B, Sharma A, Li J, Awasthi S. Regulation of 4-hydroxynonenal-mediated signaling by glutathione S-transferases. Free Radic Biol Med. 2004;37:607–619. doi: 10.1016/j.freeradbiomed.2004.05.033. [DOI] [PubMed] [Google Scholar]
- 4.Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bokhari SM, Yao H, Bethel-Brown C, Fuwang P, Williams R, Dhillon NK, Hegde R, Kumar A, Buch SJ. Morphine enhances Tat-induced activation in murine microglia. J Neurovirol. 2009;22:1–10. doi: 10.1080/13550280902913628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi J, Liu RM, Kundu RK, Sangiorgi F, Wu W, Maxson R, Forman HJ. Molecular mechanism of decreased glutathione content in human immunodeficiency virus type 1 Tat-transgenic mice. J Biol Chem. 2000;275:3693–3698. doi: 10.1074/jbc.275.5.3693. [DOI] [PubMed] [Google Scholar]
- 7.DeLeo FR, Quinn MT. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J Leukoc Biol. 1996;60:677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- 8.Droge W. Cysteine and glutathione deficiency in AIDS patients: a rationale for the treatment with N-acetyl-cysteine. Pharmacology. 1993;46:61–65. doi: 10.1159/000139029. [DOI] [PubMed] [Google Scholar]
- 9.Duncan MJ, Bruce-Keller AJ, Conner C, Knapp PE, Xu R, Nath A, Hauser KF. Effects of chronic expression of the HIV-induced protein, transactivator of transcription, on circadian activity rhythms in mice, with or without morphine. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1680–1687. doi: 10.1152/ajpregu.90496.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan R, Wingfield P, Gallo RC. Release, uptake, and affects of extracellular human immunodeficiency virus type-1 Tat protein on cell growth and viral replication. J Virol. 1993;67:277–287. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glass J, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantification of human immunodeficiency virus in the brain; correlation with dementia. Ann Neurol. 1995;38:755–762. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- 12.Grant I, Sacktor H, McArthur J. HIV neurocognitive disorders. In: Gendelman HE, Grant I, Everall I, Lipton SA, Swindells S, editors. The Neurology of AIDS. Oxford University Press; London: 2005. pp. 357–373. [Google Scholar]
- 13.Hauser KF, El-Hage N, Buch S, Nath A, Tyor WR, Bruce-Keller AJ, Knapp PE. Impact of opiate-HIV-1 interactions on neurotoxic signaling. J Neuroimmune Pharmacol. 2006;1:98–105. doi: 10.1007/s11481-005-9000-4. [DOI] [PubMed] [Google Scholar]
- 14.Hudson L, LJ, Nath A, Jones M, Raghavan R, Narayan O, Male D, Everall I. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol. 2000;6:145–155. doi: 10.3109/13550280009013158. [DOI] [PubMed] [Google Scholar]
- 15.Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol. 2003;162:1693–1707. doi: 10.1016/S0002-9440(10)64304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell J, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Lambert C, Ase AR, Séguéla P, Antel JP. Distinct migratory and cytokine responses of human microglia and macrophages to ATP. Brain Behav Immun. 2010 doi: 10.1016/j.bbi.2010.02.010. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 18.Li W, Li G, Steiner J, Nath A. Role of Tat protein in HIV neuropathogenesis. Neurotox Res. 2009;16:205–220. doi: 10.1007/s12640-009-9047-8. [DOI] [PubMed] [Google Scholar]
- 19.Liu B, Wang K, Gao HM, Mandavilli B, Wang JY, Hong JS. Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J Neurochem. 2001;77:182–189. doi: 10.1046/j.1471-4159.2001.t01-1-00216.x. [DOI] [PubMed] [Google Scholar]
- 20.Minghetti L, Visentin S, Patrizio M, Franchini L, Ajmone-Cat MA, Levi G. Multiple actions of the human immunodeficiency virus type-1 Tat protein on microglial cell functions. Neurochem Res. 2004;29:965–978. doi: 10.1023/b:nere.0000021241.90133.89. [DOI] [PubMed] [Google Scholar]
- 21.Nakajima K, Tohyama Y, Kohsaka S, Kurihara T. Ability of rat microglia to uptake extracellular glutamate. Neurosci Lett. 2001;307:171–174. doi: 10.1016/s0304-3940(01)01943-7. [DOI] [PubMed] [Google Scholar]
- 22.Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47:186–194. [PubMed] [Google Scholar]
- 23.Nelson P, Soma L, Lavi E. Microglia in diseases of the central nervous system. Ann Med. 2002;34:491–500. doi: 10.1080/078538902321117698. [DOI] [PubMed] [Google Scholar]
- 24.Palacin M, Estevez R, Bertran J, Zorzano A. Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev. 1998;78:969–1054. doi: 10.1152/physrev.1998.78.4.969. [DOI] [PubMed] [Google Scholar]
- 25.Piani D, Fontana A. Involvement of the cystine transport system Xc in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J Immunol. 1994;153:3578–3585. [PubMed] [Google Scholar]
- 26.Rieder MJ, Krause R, Bird IA, Dekaban GA. Toxicity of sulfonamide-reactive metabolites in HIV-infected, HTLV-infected, and noninfected cells. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;8:134–140. [PubMed] [Google Scholar]
- 27.Self RL, Smith KJ, Butler TR, Pauly JR, Prendergast MA. Intra-cornu ammonis 1 administration of the human immunodeficiency virus-1 protein trans-activator of transcription exacerbates the ethanol withdrawal syndrome in rodents and activates N-methyl-D-aspartate glutamate receptors to produce persisting spatial learning deficits. Neuroscience. 2009;163:868–876. doi: 10.1016/j.neuroscience.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shapshak P, Duncan R, Minagar A, Veda PRdl, Stewart RV, Goodkin K. Elevated expression of IFN-gamma in the HIV-1 infected brain. Front Biosci. 2004;9:1073–1081. doi: 10.2741/1271. [DOI] [PubMed] [Google Scholar]
- 29.Staal F. Glutathione and HIV infection: reduced, or increased oxidized? Eur J Clin Invest. 1998;28:194–196. doi: 10.1046/j.1365-2362.1998.00268.x. [DOI] [PubMed] [Google Scholar]
- 30.Turchan-Cholewo J, Dimayuga FO, Gupta S, Keller JN, Knapp PE, Hauser KF, Bruce-Keller AJ. Morphine and HIV-Tat increase microglial-free radical production and oxidative stress: possible role in cytokine regulation. J Neurochem. 2009;108:202–215. doi: 10.1111/j.1471-4159.2008.05756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turchan-Cholewo J, Dimayuga VM, Gupta S, Gorospe RM, Keller JN, Bruce-Keller AJ. NADPH oxidase drives cytokine and neurotoxin release from microglia and macrophages in response to HIV-Tat. Antioxid Redox Signal. 2009;11:193–204. doi: 10.1089/ars.2008.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiley CA, Baldwin M, Achim CL. Expression of HIV regulatory and structural mRNA in the central nervous system. AIDS. 1996;10:843–847. doi: 10.1097/00002030-199607000-00007. [DOI] [PubMed] [Google Scholar]
- 33.Williams R, Yao H, Peng F, Yang Y, Bethel-Brown C, Buch S. Cooperative induction of CXCL10 involves NADPH oxidase: Implications for HIV dementia. Glia. 2010;58:611–621. doi: 10.1002/glia.20949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Y, Navia BA, Marra CM, Singer EJ, Chang L, Berger J, Ellis RJ, Kolson DL, Simpson D, Miller EN, Lipton SA, Evans SR, Schifitto G A.A.C.T.G.A. Team. Memantine for AIDS dementia complex: open-label report of ACTG 301. HIV Clin Trials. 2010;11:59–67. doi: 10.1310/hct1101-59. [DOI] [PMC free article] [PubMed] [Google Scholar]