

Abstract
The Inhibitor of apoptosis (IAP) antagonists Reaper (Rpr), Grim and Hid are central regulators of developmental apoptosis in Drosophila. Ectopic expression of each is sufficient to trigger apoptosis, and hid and rpr have been shown to be important for programmed cell death (PCD). To investigate the role for grim in PCD, a grim null mutant was generated. grim was not a key proapoptotic gene for embryonic PCD, confirming that grim cooperates with rpr and hid in embryogenesis. In contrast, PCD of glial cells in the microchaete lineage required grim, identifying a death process dependent upon endogenous grim. Grim associates with mitochondria and has been shown to activate a mitochondrial death pathway distinct from IAP antagonization; therefore, the Drosophila bcl-2 genes buffy and debcl were investigated for genetic interaction with grim. Loss of buffy led to microchaete glial cell survival and suppressed death in the eye induced by ectopic Grim. This is the first example of a developmental PCD process influenced by buffy, and places buffy in a proapoptotic role. PCD of microchaete glial cells represents an exceptional opportunity to study the mitochondrial proapoptotic process induced by Grim.
Keywords: Drosophila melanogaster, Microchaete bristle, Buffy, Apoptosis, Bcl-2
1. Introduction
Programmed cell death (PCD) is required for normal metazoan development, shaping the embryo as well as adult tissues and organs. Deletion of a small genomic region, called H99, was demonstrated to block virtually all PCD in the Drosophila embryo (White et al., 1994). Three genes were identified in this region, reaper (rpr), grim and head involution defective (hid) that encode proteins Rpr, Hid and Grim, collectively termed the RHG proteins (Chen et al., 1996; Grether et al., 1995; White et al., 1994). Ectopic expression of any one of the RHG proteins is sufficient to induce caspase-dependent apoptosis in vivo. Analysis of individual mutants has demonstrated requirements for hid in shaping many developing tissues and in removing larval tissues during metamorphosis and for rpr in removing excess neurons in the developing ventral nerve cord (Grether et al., 1995; Peterson et al., 2002; Yin and Thummel, 2004; Yu et al., 2002). However, it is not known whether grim functions in, or is required for, developmental PCD due to the lack of a specific mutation in grim.
As in vertebrate apoptosis, cell death signals in Drosophila converge on caspase activation. Ectopic expression studies demonstrate that the RHG proteins activate apoptosis by antagonizing Drosophila Inhibitor of Apoptosis Protein 1 (DIAP1), leading to release of active caspases as well as decreased DIAP1 protein levels (Goyal et al., 2000; Hays et al., 2002; Holley et al., 2002; Ryoo et al., 2002; Vucic et al., 1998; Wang et al., 1999; Wilson et al., 2002; Yokokura et al., 2004; Yoo et al., 2002; Zachariou et al., 2003). Rpr, Hid and Grim all contain an amino-terminal IAP-binding motif (IBM) that binds and inhibits DIAP1. Rpr and Grim also have a second functionally-defined region; an internal domain called a GH3 domain (Claveria et al., 2002; Wing et al., 2001). Deletion of either the IBM or the GH3 domain in Rpr or Grim reduces ectopic killing activity in the Drosophila eye (Claveria et al., 2002; Freel et al., 2008; Wing et al., 2001; Wing et al., 1998). The GH3 domain targets Rpr and Grim to the mitochondria and is sufficient to induce cell death (Chen et al., 2004; Claveria et al., 2002; Olson et al., 2003), suggesting that the GH3 domain mediates a mechanism for cell death that is independent of DIAP1 antagonization and mitochondria (Claveria et al., 2002). However, defining the role played by the mitochondria in Drosophila apoptosis has remained challenging. Although fruit fly Bcl-2 family members exist, apoptosis in Drosophila does not appear to require release of Cytochrome c or other proapoptotic molecules from the mitochondria (Abdelwahid et al., 2007; Brachmann et al., 2000; Colussi et al., 2000; Goyal et al., 2007; Igaki et al., 2000; Quinn et al., 2003; Zhang et al., 2000a, b).
To address how Grim promotes cell death, we investigated the endogenous protein as it functions in PCD. To date, all experimental data on grim consist of ectopic expression studies or loss-of-function in the context of deletions that also remove hid (Chen et al., 1996). We generated the first grim null mutant and show that grim is required for PCD of microchaete glial cells. Additionally, we present evidence that the bcl-2 gene buffy promotes grim-dependent cell death, possibly mediated by a physical interaction between the two proteins at the mitochondria.
2. Results
2.1. grim mutants develop normally
Previous attempts to isolate mutants of grim using standard techniques have been unsuccessful (White et al., 1994). We engineered a small genomic deletion (grimA6C) that completely removed grim and two distal uncharacterized genes (Fig 1A). Deletion of grim was confirmed by genomic PCR and grim-specific RT-PCR (Fig 1B and C). grim lies between hid and rpr and the grimA6C deletion endpoints map 102 kb from hid and 85 kb from rpr. Expression of neither rpr nor hid was affected in the grim deletion mutant (Fig 1D and E).
Fig. 1.
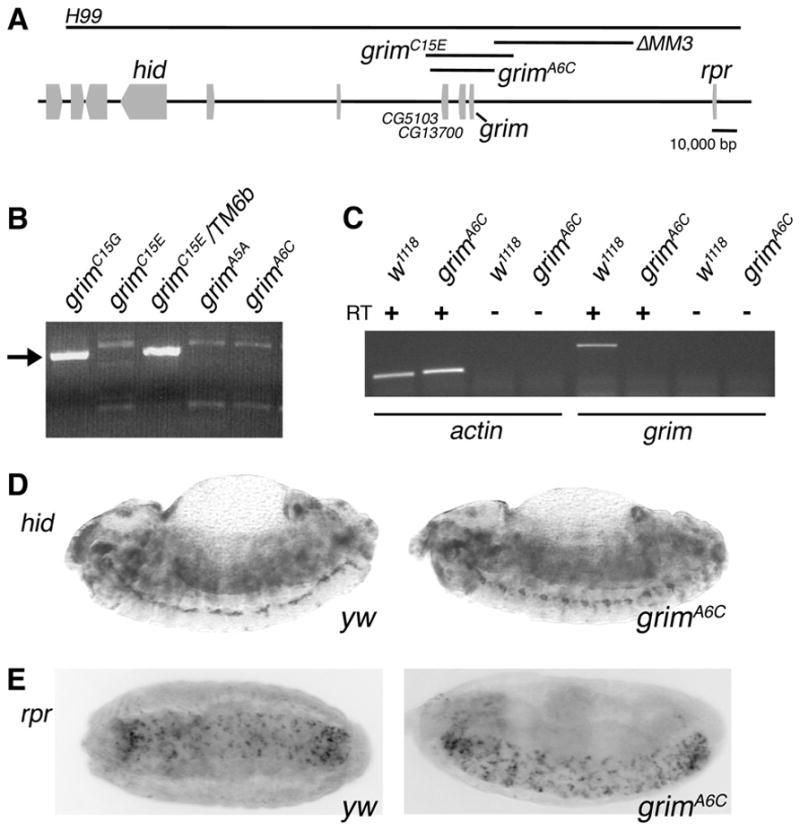
Generation of a grim mutant. (A) Schematic of the H99 region showing the extent of the deletions, with black bars, generated in this study. (B) Genomic PCR to confirm deletion of grim. The grim-specific band is seen in the non-deleted grimC15G isolate and due to the balancer in grimC15E/TM6b. grimA5A, grimA6C, and grimC15E all contain grim deletions. (C) RT-PCR demonstrating that there is no grim transcription in the grimA6C mutant. RNA samples from the indicated genotypes with (+) or without (−) reverse transcriptase (RT) were amplified with actin- or grim-specific primers as noted. actin was used as a control to demonstrate that RNA was supplied and -RT controlled for DNA contamination. (D and E) in situ analysis of hid (upper panels) and rpr (lower panels) indicate that the expression of these genes in embryos is unaffected in the grimA6C mutant. Note that in this and all following figures, grimA6C refers to the homozygous mutant.
grim null mutants are viable and fertile. No developmental delays were observed, nor were the mutants visibly different from wild-type flies. Chen and co-workers previously showed that the distribution of grim mRNA resembled patterns of embryonic PCD (Chen et al., 1996). To extend this information to identify the cell death processes that require grim, a developmental time course of grim expression was determined (Fig 2A). grim transcription is observed throughout embryonic development with its highest level in embryos of mid-stage 13 to early stage 15 (10–12 h). During these stages, PCD is observed in the head region and the developing ventral nerve cord (VNC); both regions containing cells with grim mRNA (Chen et al., 1996). grim transcription is also observed in late 3rd instar larvae and during pupation, two periods in which PCD shapes tissues and organs that will give rise to the adult form. We concentrated on stage 13–15 embryos and midpupation to broadly investigate a role for grim in PCD.
Fig. 2.
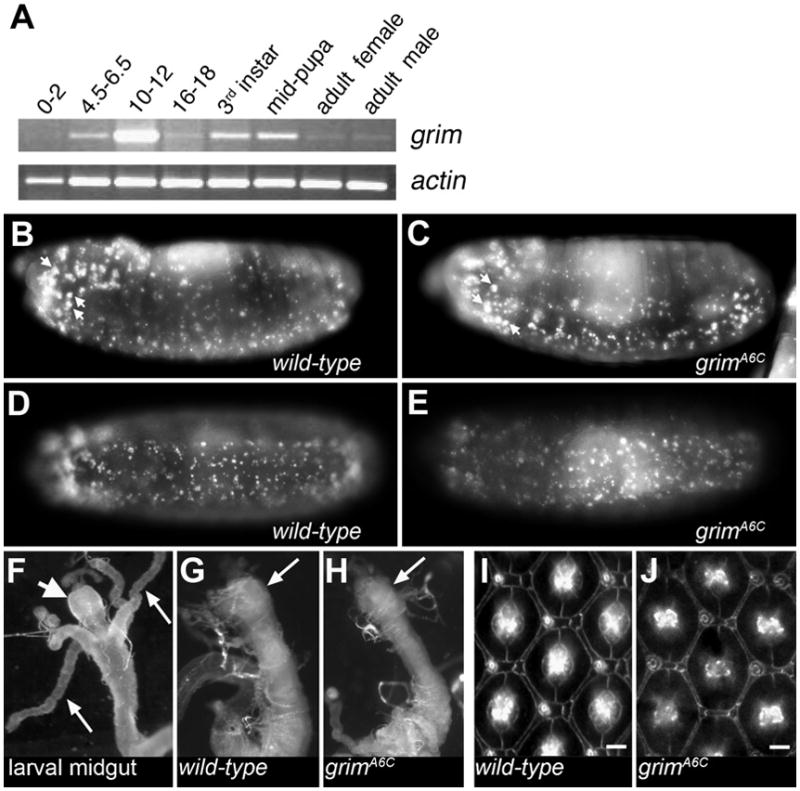
Embryonic PCD in grimA6C mutant embryos is not visibly reduced; nor is midgut histolysis or PCD in the retina affected by loss of grim. (A) Investigation of grim transcription using semi-quantitative RT-PCR at various stages of Drosophila development, beginning with embryos aged as noted (hours). (B–E) Apoptosis in stage 12 grimA6C mutant embryos (C and E) is similar to wild-type (B and D) as demonstrated by Acridine Orange staining. Acridine Orange was previously shown to stain all apoptotic cells in the Drosophila embryo, as well as individual macrophages that have engulfed multiple apoptotic cells (Silva et al., 2007). The larger Acridine Orange spots (such as those identified by arrows) are likely such macrophages. Anterior is to the left. (F–H) Histolysis of the larval gut occurs normally. The proventriculus (short, fat arrow) and gastric caeca (2 of 4 caeca are marked with long, thin arrows) of the larval midgut (F) are destroyed by 5 h APF in wild-type and grimA6C mutant (G and H) as the midgut begins the process of histolysis. Arrows in G and H indicate the region where the gastric caeca were prior to histolysis. The anterior midgut region is shown (with proventriculus being most anterior portion) and this is roughly 1/5 the length of the 3rd instar larva or 0.5–0.6 mm. Images (F–H) were taken at the same magnification. (I and J) Excess interommatidial pigment cells in the retina are properly pruned by PCD in pupae lacking grimA6C (cell membranes are highlighted using anti-armadillo in 42 h APF retina). Each wild-type (I) and grimA6C (J) ommatidium is surrounded by the expected 12 interommatidial cells after death of the excess cells. Bar is ~5 μm. Anterior is to the left.
Embryonic apoptosis in the grimA6C mutant (homozygous and hereafter referred to as the grim mutant) was not visibly reduced; indicating that grim does not play a major role in embryonic PCD, unlike hid (Fig 2B–E). Additionally, the size of the ventral ganglia from grim mutant 3rd instar larvae was not increased in size as has been seen in the rpr mutant due to the inappropriate survival of neurons and neuroblasts (data not shown). During metamorphosis, the proventriculus and gastric caeca of the larval midgut are rapidly destroyed and this process was not altered by deletion of grim (Fig 2F–H). This finding is consistent with a recent study demonstrating that, although caspases are active in the midgut, autophagy rather than apoptosis is required for midgut histolysis (Denton et al., 2009). Another well-defined process of PCD is the pruning of excess pigment cells from the retina (Wolff and Ready, 1991) and this occurred normally in the grim mutant (Fig 2I and J). Thus, in the examples of developmental PCD investigated here, grim is not the sole regulator of developmental death but likely functions redundantly with other cell death genes in the H99 genomic region.
2.2. Death of the microchaete glial cell requires grim
Because grim is expressed during mid-pupal development, we focused on a specific example of PCD during this period: removal of the glial cell from the microchaete lineage of the pupal notum. There are on average 200–250 regularly spaced mechanosensory bristles, called microchaetes, on the dorsal surface of the adult fly thorax (the notum) (Hartenstein and Posakony, 1989). Each microchaete bristle is composed of four distinct cell types: the shaft, socket, sheath and neuron. In the cell lineage that generates these mechanosensory bristles, the sensory organ progenitor (SOP) cell divides at 17–19 h after puparium formation (APF) to form the PIIa and PIIb cells. The PIIb cell then divides unequally to generate a small glial cell and the PIII cell. Following the PIIb division, the PIIa cell divides to form the shaft cell and socket cell. Finally, in the 20–22 h APF interval, the PIII cell divides, giving rise to the sheath and neuron (Gho et al., 1999; Reddy and Rodrigues, 1999). Thus, 5 cells are formed from an initial SOP, but only four will form the final sensory organ; the glial cell derived from PIIb dies after the final PIII division that generates the sheath and neuron (Fichelson and Gho, 2003). Death of the glial cell is apoptotic; nuclear fragmentation, activated caspase and TUNEL staining are observed and death is blocked in H99−/− clones or by overexpression of the caspase inhibitor, p35 (Fichelson and Gho, 2003; Koto et al., 2009). A consequence of enforced survival of the glial cells (i.e. in H99−/−) is precocious axonal outgrowth, and in such instances, no glial cells persisted in the adult notum despite the fact that glial cells were forced to survive. Instead, these glial cells were observed to leave the epithelium through migration (Fichelson and Gho, 2003).
In the study establishing that the glial cell dies by apoptosis, the four cells of the microchaete cluster were identified using an antibody specific for Cut and the glial cell was identified with an antibody specific for Repo (Fichelson and Gho, 2003). Following this procedure, we dissected pupal nota and stained with Cut- and Repo-specific antibodies at various time points to investigate cell death of the glial cell in the microchaete lineage. Prior to the period of glial cell death, Repo-positive glial cells were observed adjacent to four-cell clusters of developing microchaete in both grim null and wild-type pupal nota (Fig 3A). Beginning at roughly 22 h APF individual glial cells start to die, and by 28–29 h APF, virtually all of the glia have died (Fichelson and Gho, 2003 and data not shown). At this point, any glial cells that have not yet died (or that survived due to artificial blockage of cell death) migrate away from the sensory organ cluster. To avoid the migratory period, we investigated microchaete glial cell survival in 26–27 h APF nota. We observed a dramatic increase in glial cell survival in grim mutant nota compared with similarly staged wild-type controls (Fig 3B). Because the cell divisions that give rise to the microchaete sensory organ cells are temporally regulated, and all four Cut-positive cells could be identified in each group, it was unlikely that there was any significant developmental delay in the grim mutant (note that glial cell differentiation occurred normally in H99−/−) (Fichelson and Gho, 2003; Koto et al., 2009). Nevertheless, later time points were investigated and established that the migration of surviving glial cells occurred as expected a few hours later (data not shown). To quantify the effect of loss of grim on the survival of microchaete glial cells, Cut-positive clusters were counted that were adjacent to a Repo-positive glial cell. In wild-type nota at this time point, the majority of glial cells had died; only 26% of the developing microchaete clusters contained a glial cell (6 animals, total of 504 clusters counted; Fig 3C, 1st dataset). In contrast, 87% of the clusters in the grim mutant notum still contained glial cells, indicating a significant reduction in glial cell death (5 animals, total of 231 clusters counted; Fig 3C, 2nd dataset). Even at this time point, a number of surviving glial cells in the grim mutant had migrated away and were no longer associated with a Cut-positive cluster (arrows in Fig 3B), leading to a reduced count of clusters with persisting glial cells.
Fig. 3.

Cell death of the microchaete glial cell requires grim. (A) Overlay of anti-Cut (red) and anti-Repo (green) in 22 h APF nota of the indicated genotypes. At this stage prior to most cell death, the majority of Cut-positive groups are associated with a Repo-positive glial cell. (B) Overlay of anti-Cut (red) and anti-Repo (green) in 27 h APF pupal nota demonstrating that most Cut-positive groups in the grimA6C mutant notum still associate with a Repo-positive glial cell. Compare with the wild-type control notum in which only a few Repo-positive glial cells remain in the field of view. Note that some glial cells have begun to migrate away in both wild-type and grimA6C mutant nota (examples identified by arrows). The images in B are at twice the magnification of A. Bar is ~10 μm and the anterior-posterior axis is horizontal. (C) Graph of the percentage of Cut-positive clusters containing a Repo-positive glial cell. Migrating glial cells (identified by arrows in B) were not counted. Each circle represents the percentage of Cut-positive clusters containing a Repo-positive glial cell in one animal. The thick, long horizontal bar represents the mean and the shorter, thinner horizontal bars indicate SEM. The total number of Cut-positive clusters counted is indicated in a box above each dataset. P < .0001, *P < .05 P values were calculated using an unpaired two-tailed Student’s t test. The P-element line, f04656 is labeled as grimΔ52–57 f04656 and the genomic deletion line is labeled as grimΔ52–57 ΔMM3 to indicate the mutation in grim. Genotypes: da-Gal4/TM6B, UAS-grimRNAi/UAS-grimRNAi, UAS-grimRNAi/+; da-Gal4/+, grimA6C/grimA6C, grimΔ52–57 f04656/grimΔ52–57 f04656 and grimΔ52–57 ΔMM3/grimΔ52–57 ΔMM3. (D) Schematic of Grim protein indicating the 6 amino acids deleted (QRHHHQ) in the glutamine-rich domain of GrimΔ52–57. (E) Apoptosis assay in S2 cells indicating that GrimΔ52–57 kills at roughly 70% of the efficiency of wild-type Grim. Error bars are standard deviations of two experiments. (F) Quantitative PCR analysis of grim transcription in RNA isolated from 22–23 h APF pupae of the indicated homozygous genotypes. Error bars are standard deviations of two experiments. (G) The glutamine-rich domains of the Grim proteins from the indicated Drosophila species. The region is predicted by PSIPRED (Bryson et al., 2005) to form a helix – the confidence level (0–9; 9 being highest) for D. melanogaster is listed above the alignment.
To support the data for the grimA6C mutant, expression of a hairpin RNA targeting grim was used to knockdown grim, thus generating a second allele. UAS-grimRNAi was expressed by means of the ubiquitous da-Gal4 driver. Whereas both the da-Gal4 control (4 animals, total of 314 clusters counted; Fig 3C, 3rd dataset) and the UAS-grimRNAi control (3 animals, total of 141 clusters counted; Fig 3C 4th dataset) displayed normal cell death of microchaete glial cells, knockdown of grim (da-Gal4 > UAS-grimRNAi) reduced death of the glial cell to the same extent as seen in the grim mutant (5 animals, total of 387 clusters counted; Fig 3C, 5th dataset).
The large genomic region upstream of grim contains no identified genes and likely includes promoter elements for grim. We reasoned that removal of this region would generate a third grim allele (ΔMM3; Fig 1A; see below for expression data). Cell death of microchaete glial cells was suppressed in pupal nota from the homozygous ΔMM3 mutant (2 animals, total of 195 clusters counted; Fig 3C, 6th dataset).
We observed an unexpected intermediate glial cell phenotype in the f04656 P-element insertion stock used to generate both grimA6C and ΔMM3 mutations (7 animals, total of 450 clusters counted; Fig 3C, 7th dataset). In this stock, we identified a mutation in grim that results in deletion of 6 amino acids (Δ52–57) within the glutamine-rich region of Grim (Fig 3D). Four of the 6 deleted amino acids are not glutamine but positively-charged arginine (codon CGA) and histidine (CAT); these differ in only one nucleotide from the glutamines in this region (CAA or CAG). Expression of GrimΔ52–57 in cultured Drosophila cells confirmed that the deleted amino acids are required for full proapoptotic activity of Grim (Fig 3E). Significantly, Grim is the only RGH protein to encode a glutamine-rich domain and this domain, of varying length, is found in all Grim homologs from sequenced Drosophila species (Fig 3G and Supplementary Fig 1).
Since both the P-element insertion in f04656 and the genomic ΔMM3 deletion affect the region upstream of grim, we investigated whether either impacted upon grim transcription using quantitative real-time PCR (QPCR). Based on the glial cell death results (Fig 3C), it was expected that ΔMM3 would have a greater reduction in grim transcription relative to the P-element insertion; however, a similar reduction in grim transcription was observed in both genetic backgrounds (Fig 3F). The best explanation for why the phenotype of ΔMM3 is more severe than that of f04656 is that the deletion in ΔMM3 removes glial cell-specific promoter elements leading to a greater reduction in grim expression specifically in glial cells. This would not be detected by QPCR since this data reflect grim transcription at the level of whole 22–23 h APF pupae. Thus, with respect to microchaete glial cell death, it is possible that reduced transcription and the deletion in the glutamine-rich region of Grim both contribute to the phenotype of the f04656 P-element insertion stock. This is further exacerbated by reduced grim transcription in the ΔMM3 deletion, leading to microchaete glial cell survival.
2.3. The Bcl-2 protein Buffy promotes cell death of microchaete glial cells
Mitochondrial localization is a shared feature of Drosophila cell death proteins. Like Rpr, Hid and Grim, the Drosophila Bcl-2 proteins Buffy and Debcl are associated with the mitochondria (Doumanis et al., 2007; Quinn et al., 2003 and DC Purves and CBB, unpublished results). We reasoned that Grim might interact with the Drosophila Bcl-2 proteins at mitochondria and investigated whether the proteins physically interact. A co-immunoprecipitation assay demonstrated that Grim can physically associate with Debcl or Buffy (Fig 4A).
Fig. 4.

Buffy promotes Grim-dependent death. (A) Co-immunoprecipitation assay demonstrating that Myc-Buffy or Myc-Debcl and Grim physically interact in Drosophila S3 cells (IP; immunoprecipitating antibody, IB; immunoblotting antibody). Grim may bind a conserved region of the Bcl-2 proteins (Debcl and Buffy share 36% identity with some regions having >60% identity). (B) Percentage of Cut-positive clusters that are Repo-positive is graphed as in Fig 3C. Genotypes are: debclE26/debclE26, buffyH37/buffyH37, da-Gal4 UAS-buffy/+. **P = .0001. The difference between wild-type and debcl is not significant. P values were calculated using an unpaired two-tailed Student’s t test. (C) Adult eyes representative of ectopic expression of Grim in the indicated genetic backgrounds. Two examples are shown for both buffy and debcl interaction with GMR-grim. All images are of adult males and taken at the same magnification. Genotypes are: GMR-grim/TM6b, buffyH37/buffyH37; GMR-grim/TM6b, debclE26/debclE26; GMR-grim/TM6b. (D) Acridine Orange staining of 3rd instar larval eye disc demonstrating an increased number of apoptotic cells in the region due to GMR-grim expression (arrow). Removal of buffy reduces the number of dying cells due to GMR-grim. Genotypes as in C. Anterior is to the left and the morphogenetic furrow is at the left border of each image.
We next explored whether either buffy or debcl influenced grim-dependent PCD. Dissection of pupal nota from debcl null mutants demonstrated that this Drosophila bcl-2 gene has little effect on microchaete glial cell death (5 animals, total of 238 clusters counted; Fig 4B, 2nd dataset). In contrast, removal of buffy resulted in increased survival of microchaete glial cells (5 animals, total of 231 clusters counted; Fig 4B, 3rd dataset). Importantly, this is the first example of a developmental PCD process that is modified by loss of buffy. The decrease in glial cell death in the absence of buffy demonstrates that in this cellular population, Buffy is proapoptotic. This is in contrast to published studies in which Buffy blocked cell death, including death in the fly eye due to grim overexpression (Quinn et al., 2003; Sevrioukov et al., 2007). Therefore, we tested whether ectopic buffy (da-Gal4 > UAS-buffy) could block cell death of the glial cell, and found that it does not (2 animals, total of 112 clusters counted; Fig 4B, 4th dataset). Thus, even an excess of Buffy does not inhibit Grim proapoptotic function in this endogenous cellular context. Because Buffy performed an unexpected proapoptotic function in PCD of microchaete glial cells, we used another method to investigate the role of endogenous buffy in cell death induced by grim.
2.4. Buffy promotes Grim-dependent cell death
The Drosophila eye offers a unique in vivo experimental system for investigating cell death genes because the animal can survive without an eye. To further test the interaction between grim and the bcl-2 genes, grim was expressed in the developing retina of a buffy null or debcl null mutant. Ectopic grim caused cell death of a large percentage of the developing cells of the fly eye, leading to a small eye. debcl does not significantly contribute to death due to grim (Galindo et al., 2009; Fig 4C). Abrogation of grim-induced death was observed in the absence of buffy, indicating that endogenous buffy is required for efficient grim-induced cell death (Fig 4C). Ectopic buffy can inhibit cell cycle progression (Quinn et al., 2003); however, no compensatory increase in cell division was observed in the buffy mutant (Supplementary Fig 2). To directly assess death due to Grim expression in the eye, we assayed retention of Acridine Orange, a commonly used marker of dying cells in Drosophila (White et al., 2001). The increase in cell death seen by expression of grim is reduced in the absence of buffy (Fig 4D, compare panels 2 and 3). Thus, in the endogenous setting of glial cells in the pupal notum as well as the death paradigm of the eye, Buffy promotes cell death that is induced by Grim.
3. Discussion
3.1. grim is not a central regulator of PCD in the embryo, despite being expressed during embryogenesis
Shaping of the Drosophila embryo involves a series of morphogenetic movements, all of which are accompanied by cell death. Embryos homozygous for the H99 deletion lack developmental apoptosis (White et al., 1994). The three known proapoptotic genes in the H99 region, rpr, hid and grim are expressed during embryogenesis (Chen et al., 1996; Grether et al., 1995; White et al., 1994). Data using overlapping deficiencies, mutants and ectopic expression studies suggest that the combined activities of rpr, hid and grim are required for initiation of PCD during embryogenesis. Our study of the first grim null mutant confirms that grim works cooperatively with the other RHG genes for embryonic PCD.
3.2. Grim is required for microchaete glial cell death and the glutamine-rich domain is important for Grim function
In development of the mechanosensory bristles of the Drosophila notum, an SOP gives rise to the four cells of the sensory organ and a glial cell that is doomed to die. Evaluation of the grim null mutant, knockdown of grim, and a large deletion that removes genomic sequences upstream of grim all demonstrated that PCD of this glial cell is dependent upon grim. Additionally, we identified a small deletion within the glutamine-rich domain of Grim that reduced the proapoptotic activity of Grim. Future investigations that focus on the role of the glutamine-rich domain in Grim function will first require separation of the grimΔ52-57 mutation from the f04656 P-element insertion.
3.3. Buffy is required for efficient death of microchaete glial cells and grim-induced death in the eye
We observed an increase in glial cell survival in the absence of buffy. Death of microchaete glial cells is the first developmental PCD identified that is modified by loss of buffy. buffy played a proapoptotic role in this process as well as in grim-dependent cell death in the eye. Expression studies in the eye have led to the conclusion that over-expressed Debcl is a proapoptotic Bcl-2 protein, whereas over-expressed Buffy moderately blocks the killing activity of Debcl and other pro-death proteins such as Rpr, Grim and Hid (Brachmann et al., 2000; Colussi et al., 2000; Igaki et al., 2000; Zhang et al., 2000b). In stress-induced apoptosis we also found that Buffy inhibits apoptosis (Quinn et al., 2003). Why then, does Buffy appear to be proapoptotic in the current study? To begin, although over-expressed Buffy suppresses cell death due to ectopic Grim in the eye, we found that a high level of Buffy was unable to block PCD of the microchaete glial cell. This suggests that there is an important difference in the killing function of over-expressed Grim relative to a physiological amount of Grim. It is not simply that over-expressed Grim induces Debcl-dependent apoptosis that would be enhanced by loss of Buffy and suppressed by ectopic Buffy. If this were the case, then Grim-induced death should have been suppressed by loss of debcl, which it was not. How ectopic Buffy inhibits over-expressed Grim-dependent cell death (as well as other proapoptotic proteins when over-expressed (Quinn et al., 2003)) may instead relate to its ability to suppress mitochondrial dysfunction, discussed below. But in cells that respond to endogenous levels of Grim, Buffy is not antiapoptotic. This establishes that physiological levels of Grim do not utilize a killing mechanism that can be suppressed by Buffy. Furthermore, ectopic Buffy is not generally inhibitory to PCD in the animal (Sevrioukov et al., 2007 and unpublished results). Secondly, the Drosophila Bcl-2 proteins can function as both pro- and anti-death proteins: Debcl protects cells from CED-3 and serum-deprivation induced cell death and protects neurons from expanded polyglutamine-mediated neurodegeneration whereas Buffy can promote cell death (Brachmann et al., 2000; Senoo-Matsuda et al., 2005). The finding that Bcl-2 proteins have dual functions is not novel (reviewed in Cheng et al., 2006; Danial, 2008). Certain mammalian antiapoptotic Bcl-2 proteins can be converted into proapoptotic proteins that induce Cytochrome c release from mitochondria. CED-9, the C. elegans Bcl-2 protein, also has both pro- and antiapoptotic activity. Conversely, Bax, Bak and Bad are mammalian proapoptotic Bcl-2 proteins that can promote cell survival. In Drosophila, as in mammals, the ability of Bcl-2 proteins to promote or inhibit cell death likely depends on the specific cellular context.
3.4. Buffy, Grim and PCD of microchaete glial cells
Grim can activate two pathways leading to cell death: the first is dependent upon the IBM to block DIAP1 and release active caspases, and the second requires the GH3 domain for mitochondrial targeting. In the case of Rpr, these two pathways are inter-dependent in that DIAP1 degradation promoted by Rpr is significantly more efficient at the mitochondria (Freel et al., 2008). However, this may not be the situation for Grim as there are numerous observations that Grim maintains some killing activity even when unable to bind and antagonize DIAP1 and that the GH3 domain alone targets mitochondria and induces death (Abdelwahid et al., 2007; Chen et al., 1996; Claveria et al., 1998; Haining et al., 1999; McCarthy and Dixit, 1998; Vucic et al., 1998; Wing et al., 2001; Wing et al., 1998). These data are suggestive of the existence of a DIAP1-independent killing mechanism for Grim that is mitochondrial. Expression of mutants lacking the ability to engage one or the other of the two mechanisms demonstrated that the two pathways could cooperate (Claveria et al., 2002). It is not known how these two activities contribute to Grim function in an endogenous PCD setting. Our study defines PCD of microchaete glial cells as the first example of grim-dependent PCD, and a very recent report demonstrates that life and death of these cells does not rely upon DIAP1 antagonization (Koto et al., 2009). In this report, DIAP1 protein turnover was monitored in live animals. DIAP1 protein was not detectable in cells of the microchaete SOP lineage from the 2-cell stage (PIIa and PIIb) through the 5-cell stage (shaft, socket, neuron, sheath and glial cell). Thus, even from birth, there was no DIAP1 protein in the glial cell. The lack of DIAP1 protein was not because RHG proteins induced DIAP1 protein degradation since there was also no detectable DIAP1 in H99−/− clones. Lastly, Rpr was expressed in cells of the SOP lineage and this did not lead to precocious glial cell death (Koto et al., 2009). If IAP protein was present and responsible for maintaining glial cell viability, then Rpr expression should have caused premature cell death. These data demonstrate that Grim does not induce PCD of microchaete glial cells solely through DIAP1 antagonization, and suggest that another mechanism utilized by Grim contributes to glial cell death. The previously described mitochondrial pathway for Grim-dependent death is the most likely candidate and is supported by the genetic and physical interaction of Grim with Buffy. Verification of the dependence of microchaete glial cell death on the GH3 domain of Grim awaits mutational analysis of grim at its endogenous locus.
A mitochondrial pathway activated by Grim could involve mitochondrial permeabilization or alternatively mitochondrial fragmentation and dysfunction, but must ultimately lead to cell death by activating caspases (microchaete glial cells have active caspase activity prior to death and p35 expression forces their survival (Fichelson and Gho, 2003; Koto et al., 2009)). When expressed in vertebrate cells, Grim induced release of Cytochrome c, through a process that required the GH3 domain but was independent of IAP antagonization or Bcl-2 proteins (Claveria et al., 2004). Although Cytochrome c is not released, mitochondrial permeabilization may play a role in Drosophila cell death (mitochondria are clearly affected as Cytochrome c changes confirmation early in death). Mitochondrial fragmentation has been observed in Drosophila cells undergoing PCD and occurs prior to caspase activation, suggesting that fragmentation may be causative (Abdelwahid et al., 2007; Goyal et al., 2007). A large body of work has demonstrated that mitochondrial fission in mammalian cells accompanies apoptosis (reviewed in Autret and Martin, 2009; Perkins et al., 2009).
In our study, although it is possible that Grim and Buffy promote cell death through separate mechanisms, we propose that our results are most consistent with Buffy enhancing or amplifying a mitochondrial death process activated by Grim. The physical interaction between the two proteins supports this. Such a model would explain why loss of buffy has only a partial effect on ectopic Grim-dependent death (likely mostly DIAP1-dependent) and why microchaete cell death was affected less by loss of buffy than by loss of grim (because Grim is augmented by Buffy). Endogenous Buffy must be sufficient for grim-dependent cell death because excess Buffy did not further increase microchaete cell death. Since buffy does not modify ectopic Rpr or Hid in the fly eye (DC Purves and CBB, unpublished results), and since there are situations in which Grim induces cell death in which neither Rpr nor Hid can (Palaga and Osborne, 2002; Vucic et al., 1998; Wing et al., 1998; Yoo et al., 2002; Zachariou et al., 2003; Zhou et al., 1997), the interaction between Grim and Buffy may be unique to Grim.
There is significant evidence that mammalian Bcl-2 family members can influence the dynamics of mitochondrial fission and fusion in both healthy and apoptotic cells, possibly through direct interaction with core components of the mitochondrial fission/fusion machinery (reviewed in Autret and Martin, 2009). Although a role for Buffy in mitochondrial morphogenesis has not been carefully investigated, Buffy expression suppresses mitochondrial phenotypes of PINK1 and Prel mutant animals (Park et al., 2006; Tsubouchi et al., 2009). In the simplest scenario, either Grim induces mitochondrial dysfunction more efficiently through interaction with Buffy (perhaps leading to more mitochondrial fission) or mitochondrial dysfunction is actively amplified by Buffy (perhaps through release of a proapoptotic factor).
PCD of microchaete glial cells is the first example of an in vivo death that utilizes buffy and requires grim and provides an unparalleled opportunity to investigate a cell death mechanism that is likely to elucidate a role for mitochondria in Drosophila PCD.
4. Materials and methods
4.1. Drosophila stocks
All flies were raised on standard cornmeal food at 25 °C. GMR-grim experiments were performed at 29 °C. Stocks used: da-Gal4 (Bloomington Stock Center), debclE26, buffyH37 and UAS-buffy (Sevrioukov et al., 2007), GMR-grim (Chen et al., 1996). yw and w1118 were used as wild-type stocks. UAS-grimRNAi was generated by inserting the entire grim cDNA in both orientations separated by the chloramphenical-resistance gene (cmR) in the forward direction into pUAS-T.
4.2. Generation and analysis of grim null mutants
The grim and ΔMM3 mutants were generated by homologous recombination using FLP recombinase as described (Parks et al., 2004). Three independent crosses were set up to generate the deletion using stocks from Exelixis; cross 1) e01197 and f04656 (no successful mutants), cross 2) f02601 and f00560, (deletes 33,691 bp; grimC15E), and cross 3) f07860 and f04656 (deletes 24,835 bp; grimA6C and grimA5A). Genomic deletions also remove two uncharacterized genes: CG5103, predicted to be one of two transketolase genes in the genome and CG13700, a gene with no conserved domains and no homology to known genes in other organisms. grim mutants were confirmed by genomic PCR. grimC15E, grimA6C and grimA5A were indistinguishable as far as growth, viability and early phenotypic tests. grimA6C was chosen for further experiments as a representative of the smallest deletion. ΔMM3 deletes the region between f04656 and f00984 (53,623 bp). In situ analyses were performed as described (White et al., 1994).
4.3. Immunohistochemistry
Pupae were aged at 25 °C and nota were dissected at 19–26 h APF as described for each experiment. Dissected nota were fixed for 25 min with 4% paraformaldehyde/PBS and permeabilized in PBS/0.2% Triton X-100. Monoclonal antibodies (from Developmental Studies Hybridoma Bank) used: anti-Repo (1:200), anti-22C10 (1:200), anti-Armadillo (concentrated ascites, 1:200). Other antibodies: rabbit anti-Repo (1:500; gift from A. Travers), anti-phosphohistone H3 (Cell Signaling Technology), goat anti-Mouse IgG Alexa 488 and 568 (Molecular Probes). Acridine Orange staining was performed as described (White et al., 1994).
4.4. RT-PCR
Total RNA was collected using TRIZOL reagent (Invitrogen), DNase treated (10 units; Boehringer Mannheim) and TRIZOL re-purified. cDNA was synthesized using Superscript II (Invitrogen). PCR primers: forward: 5′-GATTTTCTGGGAAAGGCA GG-3′; reverse: 5′-GGTTCTGTATTGTAGTTCTG-3′ (Tm 53 °C).
4.5. Quantitative real-time PCR
Four or five pupae of each strain were collected 22–23 h APF. Total RNA was extracted using RNAqueous-4PCR kit (Ambion) and DNase treated. cDNA was synthesized using Advantage RT-for-PCR Kit (Clontech). Quantitative real-time PCR followed manufacturer’s protocols (Applied Biosystems) and was performed in triplicate. The expression levels of the replicates were averaged and normalized against the dRP49 internal control using the ΔΔCT method of analysis. No signal was detected in wells using RNA as template, indicating no genomic DNA contamination. PCR primers: grim: 5′-TCGGAGTTTGGATGCTGGGATCTT-3′ and 5′-AGTCACGTCGTCCTCATCGTTGTT-3′; dRP49: 5′-CTCATGCAGAACCGCGTTTA-3′ and 5′-ACAAATGTGTATTCCGACCA-3′. Reported results are the average of two completely independent experiments.
4.6. Secondary structure prediction
Structure analysis was performed using the PSIPRED server (http://www.bioinf4.cs.ucl.ac.uk:3000/psipred) (Bryson et al., 2005). Most of the residues within the glutamine-rich domain of Grim score 6 for probability of helical formation on a scale of 0–9 with 9 being high.
4.7. S2 death assay
S2 cells were co-transfected with pMT-Grim (Claveria et al., 2002) or pMT-GrimΔ52–57 and pIE-RFP (3:1) and induced with 0.7 mM Cu2SO4 after 48 h. Apoptotic transfected cells (RFP-positive, blebbing and fragmenting) were counted in three fields at 4 or 6 h after induction. The experiment was repeated with similar results.
4.8. Immunoprecipitation
Drosophila S3 cells stably transfected with pMT-Myc-buffy (Myc epitope tag added to the amino terminus of Buffy) and pMT-grim, pMT-Myc-debcl (Myc epitope tag added to the amino terminus of Debcl) and pMT-grim or pMT-grim alone were induced with 0.7 mM CuSO4 overnight with 50 μM zVAD.fmk. Cell lysis and antibody binding, using 10 μg of anti-c-Myc antibody (9E10, DSHB), were carried out in 0.5% Triton X-100 with protease inhibitors. For Western analysis, 10% SDS–PAGE gels were probed with anti-c-Myc and 15% SDS–PAGE gels were probed with rabbit anti-Grim (Claveria et al., 1998). The experiment was performed twice with similar results.
Supplementary Material
Acknowledgments
This research was partially supported by UC LEADS, ID-SURE, UROP Programs (JNW) and a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund (CBB). K.W. was supported by NIH grant GM55568.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.mod.2010.06.001.
References
- Abdelwahid E, Yokokura T, Krieser RJ, Balasundaram S, Fowle WH, White K. Mitochondrial disruption in Drosophila apoptosis. Dev Cell. 2007;12:793–806. doi: 10.1016/j.devcel.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Autret A, Martin SJ. Emerging role for members of the Bcl-2 family in mitochondrial morphogenesis. Mol Cell. 2009;36:355–363. doi: 10.1016/j.molcel.2009.10.011. [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Jassim OW, Wachsmuth BD, Cagan RL. The Drosophila bcl-2 family member dBorg-1 functions in the apoptotic response to UV-irradiation. Curr Biol. 2000;10:547–550. doi: 10.1016/s0960-9822(00)00474-7. [DOI] [PubMed] [Google Scholar]
- Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. Protein structure prediction servers at University College London. Nucleic Acids Res. 2005;33:W36–38. doi: 10.1093/nar/gki410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Ho SI, Shi Z, Abrams JM. Bifunctional killing activity encoded by conserved reaper proteins. Cell Death Differ. 2004 doi: 10.1038/sj.cdd.4401406. [DOI] [PubMed] [Google Scholar]
- Chen P, Nordstrom W, Gish B, Abrams JM. Grim, a novel cell death gene in Drosophila. Genes Dev. 1996;10:1773–1782. doi: 10.1101/gad.10.14.1773. [DOI] [PubMed] [Google Scholar]
- Cheng WC, Berman SB, Ivanovska I, Jonas EA, Lee SJ, Chen Y, Kaczmarek LK, Pineda F, Hardwick JM. Mitochondrial factors with dual roles in death and survival. Oncogene. 2006;25:4697–4705. doi: 10.1038/sj.onc.1209596. [DOI] [PubMed] [Google Scholar]
- Claveria C, Albar JP, Serrano A, Buesa JM, Barbero JL, Martinez AC, Torres M. Drosophila grim induces apoptosis in mammalian cells. Embo J. 1998;17:7199–7208. doi: 10.1093/emboj/17.24.7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claveria C, Caminero E, Martinez AC, Campuzano S, Torres M. GH3, a novel proapoptotic domain in Drosophila Grim, promotes a mitochondrial death pathway. Embo J. 2002;21:3327–3336. doi: 10.1093/emboj/cdf354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claveria C, Martinez AC, Torres M. A Bax/Bak-independent mitochondrial death pathway triggered by Drosophila Grim GH3 domain in mammalian cells. J Biol Chem. 2004;279:1368–1375. doi: 10.1074/jbc.M309819200. [DOI] [PubMed] [Google Scholar]
- Colussi PA, Quinn LM, Huang DC, Coombe M, Read SH, Richardson H, Kumar S. Debcl, a proapoptotic Bcl-2 homologue, is a component of the Drosophila melanogaster cell death machinery [see comments] J Cell Biol. 2000;148:703–714. doi: 10.1083/jcb.148.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN. BAD: undertaker by night, candyman by day. Oncogene. 2008;27 (Suppl 1):S53–S70. doi: 10.1038/onc.2009.44. [DOI] [PubMed] [Google Scholar]
- Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–1746. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doumanis J, Dorstyn L, Kumar S. Molecular determinants of the subcellular localization of the Drosophila Bcl-2 homologues DEBCL and BUFFY. Cell Death Differ. 2007;14:907–915. doi: 10.1038/sj.cdd.4402082. [DOI] [PubMed] [Google Scholar]
- Fichelson P, Gho M. The glial cell undergoes apoptosis in the microchaete lineage of Drosophila. Development. 2003;130:123–133. doi: 10.1242/dev.00198. [DOI] [PubMed] [Google Scholar]
- Freel CD, Richardson DA, Thomenius MJ, Gan EC, Horn SR, Olson MR, Kornbluth S. Mitochondrial localization of Reaper to promote inhibitors of apoptosis protein degradation conferred by GH3 domain-lipid interactions. J Biol Chem. 2008;283:367–379. doi: 10.1074/jbc.M708931200. [DOI] [PubMed] [Google Scholar]
- Galindo KA, Lu WJ, Park JH, Abrams JM. The Bax/Bak ortholog in Drosophila, Debcl, exerts limited control over programmed cell death. Development. 2009;136:275–283. doi: 10.1242/dev.019042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gho M, Bellaiche Y, Schweisguth F. Revisiting the Drosophila microchaete lineage: a novel intrinsically asymmetric cell division generates a glial cell. Development. 1999;126:3573–3584. doi: 10.1242/dev.126.16.3573. [DOI] [PubMed] [Google Scholar]
- Goyal G, Fell B, Sarin A, Youle RJ, Sriram V. Role of mitochondrial remodeling in programmed cell death in Drosophila melanogaster. Dev Cell. 2007;12:807–816. doi: 10.1016/j.devcel.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal L, McCall K, Agapite J, Hartwieg E, Steller H. Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. Embo J. 2000;19:589–597. doi: 10.1093/emboj/19.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grether ME, Abrams JM, Agapite J, White K, Steller H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 1995;9:1694–1708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- Haining WN, Carboy-Newcomb C, Wei CL, Steller H. The proapoptotic function of Drosophila Hid is conserved in mammalian cells. Proc Natl Acad Sci USA. 1999;96:4936–4941. doi: 10.1073/pnas.96.9.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenstein V, Posakony JW. Development of adult sensilla on the wing and notum of Drosophila melanogaster. Development. 1989;107:389–405. doi: 10.1242/dev.107.2.389. [DOI] [PubMed] [Google Scholar]
- Hays R, Wickline L, Cagan R. Morgue mediates apoptosis in the Drosophila melanogaster retina by promoting degradation of DIAP1. Nat Cell Biol. 2002;4:425–431. doi: 10.1038/ncb794. [DOI] [PubMed] [Google Scholar]
- Holley CL, Olson MR, Colon-Ramos DA, Kornbluth S. Reaper eliminates IAP proteins through stimulated IAP degradation and generalized translational inhibition. Nat Cell Biol. 2002;4:439–444. doi: 10.1038/ncb798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T, Kanuka H, Inohara N, Sawamoto K, GNE, Okano H, Miura M. Drob-1, a Drosophila member of the Bcl-2/CED-9 family that promotes cell death. Proc Natl Acad Sci USA. 2000;97:662–667. doi: 10.1073/pnas.97.2.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koto A, Kuranaga E, Miura M. Temporal regulation of Drosophila IAP1 determines caspase functions in sensory organ development. J Cell Biol. 2009;187:219–231. doi: 10.1083/jcb.200905110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JV, Dixit VM. Apoptosis induced by Drosophila reaper and grim in a human system Attenuation by inhibitor of apoptosis proteins (cIAPs) J Biol Chem. 1998;273:24009–24015. doi: 10.1074/jbc.273.37.24009. [DOI] [PubMed] [Google Scholar]
- Olson MR, Holley CL, Gan EC, Colon-Ramos DA, Kaplan B, Kornbluth S. A GH3-like domain in reaper is required for mitochondrial localization and induction of IAP degradation. J Biol Chem. 2003;278:44758–44768. doi: 10.1074/jbc.M308055200. [DOI] [PubMed] [Google Scholar]
- Palaga T, Osborne B. The 3D’s of apoptosis: death, degradation and DIAPs. Nat Cell Biol. 2002;4:E149–151. doi: 10.1038/ncb0602-e149. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Parks AL, Cook KR, Belvin M, Dompe NA, Fawcett R, Huppert K, Tan LR, Winter CG, Bogart KP, Deal JE, et al. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat Genet. 2004;36:288–292. doi: 10.1038/ng1312. [DOI] [PubMed] [Google Scholar]
- Perkins G, Bossy-Wetzel E, Ellisman MH. New insights into mitochondrial structure during cell death. Exp Neurol. 2009;218:183–192. doi: 10.1016/j.expneurol.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson C, Carney GE, Taylor BJ, White K. Reaper is required for neuroblast apoptosis during Drosophila development. Development. 2002;129:1467–1476. doi: 10.1242/dev.129.6.1467. [DOI] [PubMed] [Google Scholar]
- Quinn L, Coombe M, Mills K, Daish T, Colussi P, Kumar S, Richardson H. Buffy, a Drosophila Bcl-2 protein, has anti-apoptotic and cell cycle inhibitory functions. Embo J. 2003;22:3568–3579. doi: 10.1093/emboj/cdg355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy GV, Rodrigues V. A glial cell arises from an additional division within the mechanosensory lineage during development of the microchaete on the Drosophila notum. Development. 1999;126:4617–4622. doi: 10.1242/dev.126.20.4617. [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Bergmann A, Gonen H, Ciechanover A, Steller H. Regulation of Drosophila IAP1 degradation and apoptosis by reaper and ubcD1. Nat Cell Biol. 2002;4:432–438. doi: 10.1038/ncb795. [DOI] [PubMed] [Google Scholar]
- Senoo-Matsuda N, Igaki T, Miura M. Bax-like protein Drob-1 protects neurons from expanded polyglutamine-induced toxicity in Drosophila. Embo J. 2005;24:2700–2713. doi: 10.1038/sj.emboj.7600721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrioukov EA, Burr J, Huang EW, Assi HH, Monserrate JP, Purves DC, Wu JN, Song EJ, Brachmann CB. Drosophila Bcl-2 proteins participate in stress-induced apoptosis, but are not required for normal development. Genesis. 2007;45:184–193. doi: 10.1002/dvg.20279. [DOI] [PubMed] [Google Scholar]
- Silva E, Au-Yeung HW, Van Goethem E, Burden J, Franc NC. Requirement for a Drosophila E3-ubiquitin ligase in phagocytosis of apoptotic cells. Immunity. 2007;27:585–596. doi: 10.1016/j.immuni.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Tsubouchi A, Tsuyama T, Fujioka M, Kohda H, Okamoto-Furuta K, Aigaki T, Uemura T. Mitochondrial protein Preli-like is required for development of dendritic arbors and prevents their regression in the Drosophila sensory nervous system. Development. 2009;136:3757–3766. doi: 10.1242/dev.042135. [DOI] [PubMed] [Google Scholar]
- Vucic D, Kaiser WJ, Miller LK. Inhibitor of apoptosis proteins physically interact with and block apoptosis induced by Drosophila proteins HID and GRIM. Mol Cell Biol. 1998;18:3300–3309. doi: 10.1128/mcb.18.6.3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SL, Hawkins CJ, Yoo SJ, Muller HA, Hay BA. The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell. 1999;98:453–463. doi: 10.1016/s0092-8674(00)81974-1. [DOI] [PubMed] [Google Scholar]
- White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H. Genetic control of programmed cell death in Drosophila. Science. 1994;264:677–683. doi: 10.1126/science.8171319. [DOI] [PubMed] [Google Scholar]
- White K, Lisi S, Kurada P, Franc N, Bangs P. Methods for studying apoptosis and phagocytosis of apoptotic cells in Drosophila tissues and cell lines. Methods Cell Biol. 2001;66:321–338. doi: 10.1016/s0091-679x(01)66015-1. [DOI] [PubMed] [Google Scholar]
- Wilson R, Goyal L, Ditzel M, Zachariou A, Baker DA, Agapite J, Steller H, Meier P. The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nat Cell Biol. 2002;4:445–450. doi: 10.1038/ncb799. [DOI] [PubMed] [Google Scholar]
- Wing JP, Schwartz LM, Nambu JR. The RHG motifs of Drosophila Reaper and Grim are important for their distinct cell death-inducing abilities. Mech Dev. 2001;102:193–203. doi: 10.1016/s0925-4773(01)00316-1. [DOI] [PubMed] [Google Scholar]
- Wing JP, Zhou L, Schwartz LM, Nambu JR. Distinct cell killing properties of the Drosophila reaper, head involution defective, and grim genes. Cell Death Differ. 1998;5:930–939. doi: 10.1038/sj.cdd.4400423. [DOI] [PubMed] [Google Scholar]
- Wolff T, Ready DF. Cell death in normal and rough eye mutants of Drosophila. Development. 1991;113:825–839. doi: 10.1242/dev.113.3.825. [DOI] [PubMed] [Google Scholar]
- Yin VP, Thummel CS. A balance between the diap1 death inhibitor and reaper and hid death inducers controls steroid-triggered cell death in Drosophila. Proc Natl Acad Sci USA. 2004;101:8022–8027. doi: 10.1073/pnas.0402647101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokokura T, Dresnek D, Huseinovic N, Lisi S, Abdelwahid E, Bangs P, White K. Dissection of DIAP1 functional domains via a mutant replacement strategy. J Biol Chem. 2004;279:52603–52612. doi: 10.1074/jbc.M409691200. [DOI] [PubMed] [Google Scholar]
- Yoo SJ, Huh JR, Muro I, Yu H, Wang L, Wang SL, Feldman RM, Clem RJ, Muller HA, Hay BA. Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat Cell Biol. 2002;4:416–424. doi: 10.1038/ncb793. [DOI] [PubMed] [Google Scholar]
- Yu SY, Yoo SJ, Yang L, Zapata C, Srinivasan A, Hay BA, Baker NE. A pathway of signals regulating effector and initiator caspases in the developing Drosophila eye. Development. 2002;129:3269–3278. doi: 10.1242/dev.129.13.3269. [DOI] [PubMed] [Google Scholar]
- Zachariou A, Tenev T, Goyal L, Agapite J, Steller H, Meier P. IAP-antagonists exhibit non-redundant modes of action through differential DIAP1 binding. Embo J. 2003;22:6642–6652. doi: 10.1093/emboj/cdg617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Holzgreve W, De Geyter C. Evolutionarily conserved Bok proteins in the Bcl-2 family. FEBS Lett. 2000a;480:311–313. doi: 10.1016/s0014-5793(00)01921-9. [DOI] [PubMed] [Google Scholar]
- Zhang H, Huang Q, Ke N, Matsuyama S, Hammock B, Godzik A, Reed JC. Drosophila pro-apoptotic bcl-2/Bax homologue reveals evolutionary conservation of cell death mechanisms. J Biol Chem. 2000b;275:27303–27306. doi: 10.1074/jbc.M002846200. [DOI] [PubMed] [Google Scholar]
- Zhou L, Schnitzler A, Agapite J, Schwartz LM, Steller H, Nambu JR. Cooperative functions of the reaper and head involution defective genes in the programmed cell death of Drosophila central nervous system midline cells. Proc Natl Acad Sci USA. 1997;94:5131–5136. doi: 10.1073/pnas.94.10.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.