

Abstract
Background & Aims
Classical ferroportin disease is characterized by hyperferritinemia, normal transferrin saturation, and iron overload in macrophages. A non-classical form is characterized by additional hepatocellular iron deposits and a high transferrin saturation. Both forms demonstrate autosomal dominant transmission and are associated with ferroportin gene (SLC40A1) mutations. SLC40A1 encodes a cellular iron exporter expressed in macrophages, enterocytes, and hepatocytes. The aim of the analysis is to determine the penetrance of SLC40A1 mutations and to evaluate in silico tools to predict the functional impairment of ferroportin mutations as an alternative to in vitro studies.
Methods
We conducted a systematic review of the literature and meta-analysis of the biochemical presentation, genetics, and pathology of ferroportin disease.
Results
Of the 176 individuals reported with SLC40A1 mutations, 80 were classified as classical phenotype with hyperferritinemia and normal transferrin saturation. The non-classical phenotype with hyperferritinemia and elevated transferrin saturation was present in 53 patients. The remaining patients had normal serum ferritin or the data were reported incompletely. Despite an increased hepatic iron concentration in all biopsied patients, significant fibrosis or cirrhosis was present in only 11%. Hyperferritinemia was present in 86% of individuals with ferroportin mutations. Bio-informatic analysis of ferroportin mutations showed that the PolyPhen score has a sensitivity of 99% and a specificity of 67% for the discrimination between ferroportin mutations and polymorphisms.
Conclusions
In contrast to HFE hemochromatosis, ferroportin disease has a high penetrance, is genetically heterogeneous and is rarely associated with fibrosis. Non-classical ferroportin disease is associated with a higher risk of fibrosis and a more severe overload of hepatic iron.
Abbreviations: AUROC, area under the receiver operator curve; ER, endoplasmic reticulum; HIC, hepatic iron concentration; JAK2, Janus Kinase 2; MRI, magnetic resonance imaging; PolyPhen, polymorphism phenotyping; ROC, receiver operator curve; SIFT, sorting intolerant from tolerant
Keywords: SLC40A1, Hemochromatosis type IV, Iron, Bio-informatics, SLC11A3, IREG1, FPN1, PolyPhen
Introduction
Ferroportin disease is a clinically and genetically heterogeneous iron overload syndrome [1]. Its clinical presentation has been documented in individual case reports and small case series. The natural course has been reported in one small series [2]. Hyperferritinemia, a normal to low transferrin saturation and Kupffer cell iron storage, presenting as hepatic and spleen iron overload, are considered characteristic features of classical ferroportin disease [3]. Increased transferrin saturation and hepatocellular iron overload, in addition to hyperferritinemia and macrophage iron loading, is considered characteristic for the non-classical phenotype [3,4].
A genotype–phenotype correlation was suggested to explain the clinical heterogeneity of the disease where most mutations (e.g. A77D, D157G, V162del, N174I, Q182H, Q248H, and G323V) are associated with classical ferroportin disease [5–8]. Distinct mutations (e.g. N144H, Y64N, C326Y/S, S338R, Y501C) have been found in patients who presented with the non-classical phenotype [5,9–12].
Ferroportin is the only known mammalian iron exporter and is expressed in macrophages and the basolateral membrane of enterocytes and hepatocytes [13–15]. In classical ferroportin disease, macrophage iron overload results from cellular iron export deficiency, i.e. loss of ferroportin function [6]. The D157G mutation leads to hepcidin-independent, constitutive binding of Janus Kinase 2 (JAK2) and thus triggers ferroportin down-regulation [16]. The concept that other loss of function mutations of ferroportin result in intracellular retention of the iron export pump has been challenged recently [17,18], but alternative mechanisms leading to iron transport deficiency have not been fully elucidated.
Ferroportin inactivation is mediated by the peptide hormone hepcidin [19,20]. Distinct mutations render ferroportin resistant to inactivation by hepcidin and cause a gain of iron export function, which results in hyper-absorption of dietary iron.
Hepcidin is secreted by hepatocytes into the circulation in response to pro-inflammatory cytokines, hepatocellular iron loading, and endoplasmic reticulum stress [21,22]. Circulating hepcidin directly interacts with ferroportin and induces its internalization and degradation [23]. The free thiol group of cysteine 326 of ferroportin is essential for hepcidin binding and ferroportin gene mutations affecting residue 326 result in hepcidin resistance [7,24]. Patients with ferroportin disease due to hepcidin resistance mutations, exhibit a non-classical phenotype [8,25,26].
Functional studies in cells overexpressing ferroportin variants have been used to differentiate between gain-of-function and loss-of-function mutations and benign sequence variants. The functional relevance of ferroportin mutations can also be deduced from mutation segregation studies within families with ferroportin disease. Association studies of SLC40A1 polymorphisms with serum iron indices in population studies indirectly provide evidence for their functional relevance [27–29].
Bio-informatic tools were used to complement the genetic and functional studies. SIFT [30] and PolyPhen [31] provide an in silico prediction of the functional consequences of missense mutations. The use of these tools in differentiating between diseases-associated ferroportin mutations from benign sequence variants was assessed. The clinical presentation and penetrance of ferroportin gene mutations were assessed from a systematic review of the literature.
Methods
Search strategy
A systematic literature search was undertaken in Medline from 1996 to June 2009 according to the following search strategy: #1 (‘SLC40A1’ or ‘SLC11A3’ or ‘ferroportin’ or ‘IREG’ or ‘IREG1’ or ‘IREG-1’ or ‘ferroportin’ or ‘ferroportin1’ or ‘ferroportin-1’ or ‘FPN’ or ‘FPN1’ or ‘FPN-1’).mp. [mp = title, original title, abstract, name of substance word, subject heading word]; #2 (‘mutation’ or ‘variant’).mp. [mp = title, original title, abstract, name of substance word, subject heading word]; # 3: #1 and #2. This search retrieved 143 references and was supplemented by manual searching for references of the relevant articles identified. After exclusion of review articles and non-human studies, data from 49 studies were included. This search was complemented by an Embase search using the search strategy: #1 (?SLC40A1? or ?SLC11A3? or ?ferroportin? or ?IREG? or ?IREG1? or ?IREG-1? or ?ferroportin? or ?ferroportin1? or ?ferroportin-1? or ?FPN? or ?FPN1? or ?FPN-1?).mp. [mp = title, original title, abstract, name of substance word, subject heading word]; #2 (?mutation? or ?variant?).mp. [mp = title, original title, abstract, name of substance word, subject heading word]; # 3: #1 and #2.
Prediction of the functional effect of amino acid substitutions by in silico analysis
Two algorithms were used to evaluate the impact of amino acid substitutions on the function of ferroportin: polymorphism phenotyping (PolyPhen) [31–33] and sorting intolerant from tolerant (SIFT) [30]. Genetic variants of SLC40A1, identified in patients with suspected iron overload, were collected from the systematic review of the literature. In addition, single nucleotide polymorphisms of SLC40A1 were collected from the literature and from ENSEMBL (http://www.ensembl.org/) and the NCBI databases (http://www.ncbi.nlm.nih.gov/). A SLC40A1 variant was considered a mutation in case its frequency in the control population was undetermined, or lower than 1:50, and the variant was not reported in a SNP database [27–29,34,35].
PolyPhen
PolyPhen (= Polymorphism Phenotyping) is an automatic tool for prediction of possible impact of an amino acid substitution on the structure and function of a human protein. The predictive value of the PolyPhen score was demonstrated in diabetes mellitus [36] and cancer genetics [37]. This prediction is based on empirical rules which are applied to the sequence, phylogenetic, and structural information characterizing the substitution [31]. PolyPhen was employed, as available at http://coot.embl.de/PolyPhen/ (version August 12, 2008), using the NCBI protein accession number Q9NP59 (SLC40A1) for ferroportin. In all cases, the prediction basis was only the default position-specific independent counts (PSIC) score derived from multiple sequence alignment of observations. PolyPhen scores of >2.0 indicated protein function as ‘probably damaging’ and scores of <1.5 as ‘benign’. A PolyPhen score of 1.5–2.0 is classified as ‘possibly’ damaging.
A phylogenetic sequence alignment of selected ferroportin protein sequences is shown in Fig. 2.
Fig. 2.

Phylogenetic alignment of ferroportin protein sequences of various species: Disease-associated ferroportin gene mutations are highlighted in light blue. Single nucleotide polymorphisms are shown in dark blue.
SIFT
SIFT (= Sorting Intolerant from Tolerant) is a sequence-homology-based tool that presumes that important amino acids will be conserved in the protein family. Changes at well-conserved positions tend to be predicted as deleterious. SIFT is a multi-step procedure that uses multiple alignment information to predict tolerated and deleterious substitutions for every position of the query sequence [30]. The SIFT Blink algorithm was applied to compare amino acid sequences of different organisms chosen from precomputed NCBI BLAST searches. For query in SIFT Blink, GI: 49065554 (SLC40A1) was used to analyze the protein sequence by using the parameter ‘best BLAST hit to each organism’ and omitting sequences >90% identical to query. Edited alignments were reanalyzed by the appropriate module of SIFT (http://blocks.fhcrc.org/sift/SIFT_aligned_seqs_submit.html). Results were reported as ‘affects protein function’ or ‘tolerated’ according to this analysis.
To evaluate the bio-informatic tools to distinguish between benign and disease sequence variants, the SLC40A1 polymorphisms were used as true negatives and genetic variants of SLC40A1 identified in patients with ferroportin disease as true positives. The PolyPhen and SIFT scores were then analyzed in a ROC curve for their sensitivity and specificity.
Statistical analysis
For statistical analysis the software package for social sciences (SPSS v15.0) was used. To test for correlation between parameters, Pearson correlation was carried out for normally distributed parameters and Spearman rank correlation for non-normally distributed parameters. Normality of distribution was tested by Kolmogorov–Smirnov curve-fitting. Differences between groups were analyzed by Kruskal Wallis test for non-Gaussian distributed variables. Correlations and differences were considered significant if p <0.05. Sensitivity and specificity of SIFT and PolyPhen scores were calculated form the ROC curve analysis using mutations identified in patients with ferroportin disease as true positives and polymorphisms reported in the SNP database (http://www.ncbi.nlm.nih.gov/sites/entrez) as true negatives.
Results
High biochemical penetrance and variable clinical presentation of SLC40A1 mutations
One hundred and seventy-six individuals with ferroportin gene mutations were reported in the literature, and their demographic and biochemical data are listed in Table 1. The diagnosis of ferroportin disease was most frequently made in the 3rd to 4th decade of life and 43% of patients were female.
Table 1.
Summary of demographics and clinical characteristics of 176 patients with ferroportin disease. (Note not all parameters were reported for each patient.)
 |
∗Phlebotomy was reported for 82 patients. Grams iron removed was reported for 20 patients.
To describe the disease presentation and the penetrance of underlying mutations, serum iron parameters, liver biopsy findings, MRI, comorbidities, and phlebotomy status were assessed. These were reported incompletely for several patients (Table 1).
Forty-five comorbidities were reported in 29 patients (viral hepatitis 3, steatosis 12, obesity 5, diabetes 8, impaired glucose tolerance 2, hyperlipidemia 2, increased alcohol consumption 2, and sarcoidosis 2). Mean age and mean serum ferritin were significantly higher in patients with comorbidities, than in patients without comorbidities, (52 ± 15 years vs. 39 ± 20 years p = 0.002 and 3133 ± 3270 μg/L vs. 1965 ± 2067 μg/L, p = 0.017). Fibrosis ⩾F1 (Metavir) was significantly more common in patients with comorbidities than in patients without comorbidities (12 of 44 vs. 10 of 18; Fisher’s exact test p <0.05).
The biochemical penetrance of ferroportin disease was 86% (142 of 166 patients), defined as hyperferritinemia >300 μg/L in males and >200 μg/L in females. We found a significant correlation between serum ferritin and age (r2 = 0.344, n = 158, p <0.001). The phenotype of hyperferritinemic patients was classified as classical if transferrin saturation was normal and as non-classical if transferrin saturation was increased (males >50%, females >45%). Classical disease was present in 80 patients as opposed to 53 patients with non-classical disease, whose mean age at presentation was significantly higher (44 vs. 37 years, p <0.05) (Table 2). Serum iron parameters for the remaining 43 patients were either incompletely reported or normal (16 patients). Differences between classical and non-classical phenotype are evident in Table 2 where mean hemoglobin, ferritin, and hepatic iron concentration (HIC) were significantly lower in the classical disease cohort.
Table 2.
Characteristics of classical vs. non-classical phenotype of ferroportin disease as defined by the presence of hyperferritinemia with normal transferrin saturation (classical) versus patients with hyperferritinemia and elevated transferrin saturation (non-classical). Data are shown as means ± standard deviation. Differences were tested for significance using student’s t-test.
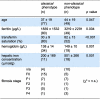 |
Liver iron was increased in 30 of 31 reported patients, with a mean HIC of 310 μmol/g (normal <25 μmol/g) [38]. Liver fibrosis ⩾F2 or liver cirrhosis was present in 7 of 53 biopsied patients (13%), and correlated with age (mean 53.3 vs. 36.2 years, p <0.001). Cirrhosis was reported in 4 of 176 patients, of whom 2 had the N144H mutation, one patient the C326S and one the I180T mutation [25,39,40]. One patient with ferroportin disease was diagnosed with hepatocellular carcinoma which was possibly linked with occult hepatitis B virus infection [2].
In search of factors that discriminate ferroportin disease severity, the correlation between patient demographics, serum iron parameters, and liver biopsy findings was determined. The stage of liver fibrosis correlated significantly with age (r = 0.54, n = 53, p <0.001), but neither with HIC nor with serum ferritin. HIC and transferrin saturation (r = 0.528, p = 0.003, n = 29), and HIC and serum ferritin (r = 0.423, p = 0.018, n = 31) showed a moderate correlation.
Genotype–phenotype correlation partly explains variability in clinical presentation
Of 31 SLC40A1 mutations, 6 and 5 were unequivocally associated with the classical or the non-classical phenotype, respectively (Table 2). Variable phenotypes in individual patients with the same mutation were reported for 9 mutations (Fig. 1 and Table 3).
Fig. 1.

Genotype to phenotype correlation of ferroportin disease: patients with ferroportin disease identified from the systematic meta-analysis were grouped according to the SLC40A1 mutation. Box-Whisker blots of (A) serum ferritin, and (B) transferrin saturation in mutations reported in more than 5 patients are shown. Boxes represent 25th and 75th percentile, Whiskers range and horizontal lines represent the median. Outliers are shown as circles. Grey, dark blue, and light blue boxes indicate that all patients reported with the respective mutation were classified as classical, non-classical, or variable biochemical phenotype, respectively, i.e. all patients had low or normal transferrin saturation (grey), increased transferrin saturation (dark blue), or different patients with the same mutation had variable transferrin saturation (light blue). Any outliers are marked with a circle and extreme cases with an asteriks.
Table 3.
Molecular genetics of SLC40A1 mutations. The frequency of mutations highlighted with ∗ in the control population can be inferred from studies, in which control populations have been screened for the presence of another mutation, affecting the same residue.
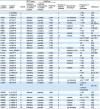 |
∗∗Predicted G468S.
MRI findings of liver and spleen iron were reported for 8 ferroportin mutations (c.-188A>G, G490D, I152F, L233P, R178G, V162del, Y64 N, Y501C) [12,41–45,5,46] where the small number of patients prohibits assessment of the diagnostic performance of MRI for the classification of ferroportin disease.
How to differentiate between SLC40A1 mutations and polymorphisms
Ferroportin disease is genetically heterogeneous with 36 different SLC40A1 mutations reported [8–10,12,26–29,38–41,45–68] as summarized in Table 3. In addition, 9 ferroportin gene polymorphisms were reported, 3 of which were associated with increased serum ferritin in various populations (Table 4) [29,34]. The allele frequencies of 29 disease-associated ferroportin mutations had been determined in matched populations and was found to be <1:100. It remains unclear whether L233V and D270P, each of which have been identified in single patients with iron overload, are disease-causing mutations or represent benign sequence variants [43,65].
Table 4.
SLC40A1 non-synonymous single nucleotide polymorphisms. SNPs, which have been associated with high serum iron parameters are highlighted in bold and italics.
aAs reported in the NCBI SNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?chooseRs=coding&go=Go&locusId=30061).
bhttp://www.ensembl.org/Homo_sapiens/Variation/Summary?v=ENSSNP12180430;vdb=variation.
To determine whether bio-informatic tools PolyPhen and SIFT [30,32,69] can discriminate between disease-causing SLC40A1 mutations and benign sequence variants, their ability to classify known ferroportin sequence variants accordingly was tested. The predicted functional impairment of individual sequence variants from the PolyPhen and SIFT tools were used as outcome variables for a receiver-operator curve analysis. The PolyPhen score had a high diagnostic accuracy with a calculated area under the ROC curve (AUROC) of 0.798 (p = 0.014) (Fig. 3). Using the descriptive output of PolyPhen (i.e. probably damaging, possibly damaging or benign) as predictors of the functional consequence of particular gene variants, the sensitivity of the PolyPhen score was 99% and the specificity 67% (Table 2 and Fig. 3). SIFT analysis predicted that 23 of 32 disease associated missense mutations ‘affects’ protein function and 9 are ‘tolerated’. Of 9 reported polymorphisms 7 were classified as ‘tolerated’ (AUROC 0.605 p = 0.37). The bio-informatics PolyPhen tool has a higher sensitivity and specificity than the SIFT score for the discrimination between missense mutations and single nucleotide polymorphisms of SLC40A1.
Fig. 3.

Diagnostic performance analysis (ROC curve) of PolyPhen and SIFT scores for the prediction of the functional consequence of ferroportin gene variants. Genetic variants of SLC40A1 reported as polymorphisms were used as true negatives and genetic variants of SLC40A1 identified in patients with ferroportin disease were used as true positives. ROC curves are shown with the sensitivity plotted along the abscissa, and 1 – specificity plotted along the ordinate.
Serum ferritin concentration (r = 0.226, n = 136, p = 0.008), and transferrin saturation (r = 0.198, n = 128, p = 0.025) were correlated with the PolyPhen score, suggesting that ferroportin disease is more severe when phylogenetically more conserved residues are affected by the mutation.
Discussion
Ferroportin disease is the commonest genetic iron overload syndrome after HFE associated hemochromatosis and has a variable clinical presentation. This review of the literature and meta-analysis shows that hyperferritinemia and increased hepatic iron concentration are characteristic signs of the disease. In contrast, elevated transferrin saturation and splenic iron overload are variably present. Hyperferritinemia was found in 86% of individual with ferroportin mutations, but incomplete reporting is a limitation for the accurate assessment of disease penetrance. The observed correlation between age and serum ferritin supports the concept that ferroportin disease is a progressive iron overload disorder. It is therefore possible that patients detected through cross-sectional study designs have not yet had time to develop the ‘full-blown’ disease. Hence, it is difficult to assess the true penetrance without further follow-up data. By comparison, in genetic hemochromatosis hyperferritinemia was present in 55–86% of C282Y homozygotes [70,71] and cirrhosis was present in 36% of more severely affected C282Y homozygous individuals with ferritin >1000 μg/L [72].
To assess the pathology of ferroportin disease, the findings of liver biopsies were reviewed. However, liver biopsies were performed in only a few patients and the results were inconsistently reported. This prohibits a systematic classification of ferroportin disease based on liver biopsy results. In patients who underwent liver biopsies for ferroportin disease, hepatic iron concentration was invariably increased and the penetrance of liver fibrosis ⩾F2 was 13%. Age, hepatic iron concentration, and comorbidities were associated with fibrosis stage and are thus potential risk factors for fibrosis in ferroportin disease.
A simple biochemical classification strategy based on transferrin saturation was used as an alternative to liver biopsy. Transferrin saturation is low to normal in classical ferroportin disease and increased in patients with the non-classical phenotype. An arbitrary threshold for transferrin saturation of 45% for females and 50% for males was used to distinguish classical from the non-classical phenotype. The value of this simple classification is supported by our meta-analysis, which shows that patients with the classical phenotype have lower hemoglobin concentrations. This may explain why patients with classical ferroportin disease appear to tolerate therapeutic venesection less well than hemochromatosis patients. MRI was proposed as an alternative to liver biopsy to discriminate between classical and non-classical ferroportin disease. However, no correlation between parameters of disease severity and MRI findings has been reported [73].
The genotype to phenotype correlation suggests that ferroportin disease has a multifactorial cause because not all mutations were unambiguously correlated with the classical or non-classical phenotype in all reported patients with a particular mutation. Our meta-analysis suggests that Y64N, V72F, and Y501C confer hepcidin resistance because the phenotypic presentation as non-classical disease is similar to the C326Y mutation [7,74,75]. Similarly, classical mutations D157N, D181V, G80V, Q182H, R489K, and V162del may impair the iron export function of ferroportin through incorrect folding, subcellular mislocalization, or direct inhibition of the iron transport function [6,18,75].
We have assessed PolyPhen and SIFT [76] as alternatives to predict the effect of ferroportin mutations on protein function, because functional studies of ferroportin mutants [6,17,18,68,75] have shown conflicting results [17,18]. Our analysis shows that PolyPhen is a sensitive tool to identify disease-causing gene variants by classifying mutations identified in patients with iron overload as ‘possibly’ or ‘probably’ damaging. The sensitivity of the PolyPhen score is 99% and the specificity 67%. These findings suggest that the functional effect of ferroportin mutations can be predicted in silico and that these tools may help to interpret the relevance of newly-identified mutations in the future.
The systematic review of the molecular genetics and the clinical presentation of ferroportin disease highlights the heterogeneity of the disease and the difficulties in securing the diagnosis. Similar to HFE hemochromatatosis, age, and comorbidities appear to be important risk factors for the development of fibrosis in patients with ferroportin disease [77]. PolyPhen helps to score newly-identified, presumed SLC40A1 mutations; identification of a genetic variant in SLC40A1 is not sufficient to confirm ferroportin disease in patients with hyperferritinemia.
Conflict of interest
The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Acknowledgements
The authors thank Dr. Marion Breitschopf from the Library of the Medical University of Innsbruck for her help with the EMBASE literature search.
Footnotes
This work was supported by the Austrian Science Funds FWF Project P19579 to H.Z.
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jhep.2010.05.016.
Supplementary data
Results from histology, clinical and biochemical data of individual patients from whom a liver biopsy was reported.
References
- 1.Ponka P. Rare causes of hereditary iron overload. Semin Hematol. 2002;39:249–262. doi: 10.1053/shem.2002.35638. [DOI] [PubMed] [Google Scholar]
- 2.Corradini E., Ferrara F., Pollicino T., Vegetti A., Abbati G.L., Losi L. Disease progression and liver cancer in the ferroportin disease. Gut. 2007;56:1030–1032. doi: 10.1136/gut.2007.122549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis. 2004;32:131–138. doi: 10.1016/j.bcmd.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Zoller H., Cox T.M. Hemochromatosis: genetic testing and clinical practice. Clin Gastroenterol Hepatol. 2005;3:945–958. doi: 10.1016/s1542-3565(05)00607-5. [DOI] [PubMed] [Google Scholar]
- 5.Drakesmith H., Schimanski L.M., Ormerod E., Merryweather-Clarke A.T., Viprakasit V., Edwards J.P. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood. 2005;106:1092–1097. doi: 10.1182/blood-2005-02-0561. [DOI] [PubMed] [Google Scholar]
- 6.Schimanski L.M., Drakesmith H., Merryweather-Clarke A.T., Viprakasit V., Edwards J.P., Sweetland E. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood. 2005;105:4096–4102. doi: 10.1182/blood-2004-11-4502. [DOI] [PubMed] [Google Scholar]
- 7.De Domenico I., Nemeth E., Nelson J.M., Phillips J.D., Ajioka R.S., Kay M.S. The hepcidin-binding site on ferroportin is evolutionarily conserved. Cell Metabol. 2008;8:146–156. doi: 10.1016/j.cmet.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Lok C.Y., Merryweather-Clarke A.T., Viprakasit V., Chinthammitr Y., Srichairatanakool S., Limwongse C. Iron overload in the Asian community. Blood. 2009;114:20–25. doi: 10.1182/blood-2009-01-199109. [DOI] [PubMed] [Google Scholar]
- 9.Wallace D.F., Dixon J.L., Ramm G.A., Anderson G.J., Powell L.W., Subramaniam V.N. A novel mutation in ferroportin implicated in iron overload. J Hepatol. 2007;46:921–926. doi: 10.1016/j.jhep.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 10.Viprakasit V., Merryweather-Clarke A.T., Chinthammitr Y., Schimanski L., Drakesmith H., Srichairatanakool S. Molecular diagnosis of the first ferroportin mutation (C326Y) in the Far East causing a dominant form of inherited iron overload. ASH Annu Meeting Abstr. 2004;104:3204. [Google Scholar]
- 11.Sham R.L., Phatak P.D., Nemeth E., Ganz T. Hereditary hemochromatosis due to resistance to hepcidin: high hepcidin concentrations in a family with C326S ferroportin mutation. Blood. 2009;114:493–494. doi: 10.1182/blood-2009-04-216226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Letocart E., Le Gac G., Majore S., Ka C., Radio F.C., Gourlaouen I. A novel missense mutation in SLC40A1 results in resistance to hepcidin and confirms the existence of two ferroportin-associated iron overload diseases. Br J Haematol. 2009;147:379–385. doi: 10.1111/j.1365-2141.2009.07834.x. [DOI] [PubMed] [Google Scholar]
- 13.Donovan A., Brownlie A., Zhou Y., Shepard J., Pratt S.J., Moynihan J. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 14.McKie A.T., Marciani P., Rolfs A., Brennan K., Wehr K., Barrow D. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 15.Abboud S., Haile D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275:19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 16.De Domenico I., Lo E., Ward D.M., Kaplan J. Human mutation D157G in ferroportin leads to hepcidin-independent binding of Jak2 and ferroportin downregulation. Blood. 2010;115:2956–2959. doi: 10.1182/blood-2009-10-251306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rice A.E., Mendez M.J., Hokanson C.A., Rees D.C., Bjorkman P.J. Investigation of the biophysical and cell biological properties of ferroportin, a multipass integral membrane protein iron exporter. J Mol Biol. 2009;386:717–732. doi: 10.1016/j.jmb.2008.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wallace D.F., Harris J.M., Subramaniam V.N. Functional analysis and theoretical modeling of ferroportin reveals clustering of mutations according to phenotype. Am J Physiol Cell Physiol. 2010;298:C75–C84. doi: 10.1152/ajpcell.00621.2008. [DOI] [PubMed] [Google Scholar]
- 19.Pigeon C., Ilyin G., Courselaud B., Leroyer P., Turlin B., Brissot P. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 20.Park C.H., Valore E.V., Waring A.J., Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 21.Lee P., Peng H., Gelbart T., Beutler E. The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta 2-microglobulin-deficient hepatocytes. Proc Natl Acad Sci USA. 2004;101:9263–9265. doi: 10.1073/pnas.0403108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vecchi C., Montosi G., Zhang K., Lamberti I., Duncan S.A., Kaufman R.J. ER stress controls iron metabolism through induction of hepcidin. Science. 2009;325:877–880. doi: 10.1126/science.1176639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nemeth E., Tuttle M.S., Powelson J., Vaughn M.B., Donovan A., Ward D.M. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 24.Fernandes A., Preza G.C., Phung Y., De Domenico I., Kaplan J., Ganz T. The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood. 2009;114:437–443. doi: 10.1182/blood-2008-03-146134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sham R.L., Phatak P.D., West C., Lee P., Andrews C., Beutler E. Autosomal dominant hereditary hemochromatosis associated with a novel ferroportin mutation and unique clinical features. Blood Cells Mol Dis. 2005;34:157–161. doi: 10.1016/j.bcmd.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 26.Rivard S.R., Lanzara C., Grimard D., Carella M., Simard H., Ficarella R. Autosomal dominant reticuloendothelial iron overload (HFE type 4) due to a new missense mutation in the FERROPORTIN 1 gene (SLC11A3) in a large French-Canadian family. Haematologica. 2003;88:824–826. [PubMed] [Google Scholar]
- 27.Gordeuk V.R., Caleffi A., Corradini E., Ferrara F., Jones R.A., Castro O. Iron overload in Africans and African-Americans and a common mutation in the SCL40A1 (ferroportin 1) gene. Blood Cells Mol Dis. 2003;31:299–304. doi: 10.1016/s1079-9796(03)00164-5. [DOI] [PubMed] [Google Scholar]
- 28.McNamara L., Gordeuk V.R., MacPhail A.P. Ferroportin (Q248H) mutations in African families with dietary iron overload. J Gastroenterol Hepatol. 2005;20:1855–1858. doi: 10.1111/j.1440-1746.2005.03930.x. [DOI] [PubMed] [Google Scholar]
- 29.Beutler E., Barton J.C., Felitti V.J., Gelbart T., West C., Lee P.L. Ferroportin 1 (SCL40A1) variant associated with iron overload in African-Americans. Blood Cells Mol Dis. 2003;31:305–309. doi: 10.1016/s1079-9796(03)00165-7. [DOI] [PubMed] [Google Scholar]
- 30.Ng P.C., Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sunyaev S., Lathe W., 3rd, Bork P. Integration of genome data and protein structures: prediction of protein folds, protein interactions and “molecular phenotypes” of single nucleotide polymorphisms. Curr Opin Struct Biol. 2001;11:125–130. doi: 10.1016/s0959-440x(00)00175-5. [DOI] [PubMed] [Google Scholar]
- 32.Sunyaev S., Ramensky V., Koch I., Lathe W., 3rd, Kondrashov A.S., Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 33.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duca L., Delbini P., Nava I., Vaja V., Fiorelli G., Cappellini M.D. Mutation analysis of hepcidin and ferroportin genes in Italian prospective blood donors with iron overload. Am J Hematol. 2009;84:592–593. doi: 10.1002/ajh.21465. [DOI] [PubMed] [Google Scholar]
- 35.Sussman N.L., Lee P.L., Dries A.M., Schwartz M.R., Barton J.C. Multi-organ iron overload in an African-American man with ALAS2 R452S and SLC40A1 R561G. Acta Haematol. 2008;120:168–173. doi: 10.1159/000181183. [DOI] [PubMed] [Google Scholar]
- 36.Song Y., Hsu Y.H., Niu T., Manson J.E., Buring J.E., Liu S. Common genetic variants of the ion channel transient receptor potential membrane melastatin 6 and 7 (TRPM6 and TRPM7), magnesium intake, and risk of type 2 diabetes in women. BMC Med Genet. 2009;10:4. doi: 10.1186/1471-2350-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Easton D.F., Deffenbaugh A.M., Pruss D., Frye C., Wenstrup R.J., Allen-Brady K. A systematic genetic assessment of 1433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–883. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pelucchi S., Mariani R., Salvioni A., Bonfadini S., Riva A., Bertola F. Novel mutations of the ferroportin gene (SLC40A1): analysis of 56 consecutive patients with unexplained iron overload. Clin Genet. 2008;73:171–178. doi: 10.1111/j.1399-0004.2007.00950.x. [DOI] [PubMed] [Google Scholar]
- 39.Njajou O.T., de Jong G., Berghuis B., Vaessen N., Snijders P.J., Goossens J.P. Dominant hemochromatosis due to N144H mutation of SLC11A3: clinical and biological characteristics. Blood Cells Mol Dis. 2002;29:439–443. doi: 10.1006/bcmd.2002.0581. [DOI] [PubMed] [Google Scholar]
- 40.Bach V., Remacha A., Altes A., Barcelo M.J., Molina M.A., Baiget M. Autosomal dominant hereditary hemochromatosis associated with two novel Ferroportin 1 mutations in Spain. Blood Cells Mol Dis. 2006;36:41–45. doi: 10.1016/j.bcmd.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 41.Jouanolle A.M., Douabin-Gicquel V., Halimi C., Loreal O., Fergelot P., Delacour T. Novel mutation in ferroportin 1 gene is associated with autosomal dominant iron overload. J Hepatol. 2003;39:286–289. doi: 10.1016/s0168-8278(03)00148-x. [DOI] [PubMed] [Google Scholar]
- 42.Cunat S., Giansily Blaizot M., Bismuth M., Blanc F., Dereure O., Larrey D. Global sequencing approach for characterizing the molecular background of hereditary iron disorders. Clin Chem. 2007;53:2060–2069. doi: 10.1373/clinchem.2007.090605. [DOI] [PubMed] [Google Scholar]
- 43.Girelli D., De Domenico I., Bozzini C., Campostrini N., Busti F., Castagna A. Clinical, pathological, and molecular correlates in ferroportin disease: a study of two novel mutations. J Hepatol. 2008;49:664–671. doi: 10.1016/j.jhep.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim F.L., Dooley J.S., Roques A.W., Grellier L., Dhillon A.P., Walker A.P. Hepatic iron concentration, fibrosis and response to venesection associated with the A77D and V162del “loss of function” mutations in ferroportin disease. Blood Cells Mol Dis. 2008;40:328–333. doi: 10.1016/j.bcmd.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 45.Zoller H., McFarlane I., Theurl I., Stadlmann S., Nemeth E., Oxley D. Primary iron overload with inappropriate hepcidin expression in V162del ferroportin disease. Hepatology. 2005;42:466–472. doi: 10.1002/hep.20775. [DOI] [PubMed] [Google Scholar]
- 46.Speletas M., Kioumi A., Loules G., Hytiroglou P., Tsitouridis J., Christakis J. Analysis of SLC40A1 gene at the mRNA level reveals rapidly the causative mutations in patients with hereditary hemochromatosis type IV. Blood Cells Mol Dis. 2008;40:353–359. doi: 10.1016/j.bcmd.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 47.Liu W., Shimomura S., Imanishi H., Iwamoto Y., Ikeda N., Saito M. Hemochromatosis with mutation of the ferroportin 1 (IREG1) gene. Intern Med (Tokyo, Japan) 2005;44:285–289. doi: 10.2169/internalmedicine.44.285. [DOI] [PubMed] [Google Scholar]
- 48.Corradini E., Montosi G., Ferrara F., Caleffi A., Pignatti E., Barelli S. Lack of enterocyte iron accumulation in the ferroportin disease. Blood Cells Mol Dis. 2005;35:315–318. doi: 10.1016/j.bcmd.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 49.Montosi G., Donovan A., Totaro A., Garuti C., Pignatti E., Cassanelli S. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest. 2001;108:619–623. doi: 10.1172/JCI13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pietrangelo A., Montosi G., Totaro A., Garuti C., Conte D., Cassanelli S. Hereditary hemochromatosis in adults without pathogenic mutations in the hemochromatosis gene. N Engl J Med. 1999;341:725–732. doi: 10.1056/NEJM199909023411003. [DOI] [PubMed] [Google Scholar]
- 51.Subramaniam V.N., Wallace D.F., Dixon J.L., Fletcher L.M., Crawford D.H. Ferroportin disease due to the A77D mutation in Australia. Gut. 2005;54:1048–1049. doi: 10.1136/gut.2005.069021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Domenico I., McVey Ward D., Nemeth E., Ganz T., Corradini E., Ferrara F. Molecular and clinical correlates in iron overload associated with mutations in ferroportin. Haematologica. 2006;91:1092–1095. [PMC free article] [PubMed] [Google Scholar]
- 53.Njajou O.T., Vaessen N., Oostra B., Heutink P., Van Duijn C.M. The hemochromatosis N144H mutation of SLC11A3 gene in patients with type 2 diabetes. Mol Genet Metab. 2002;75:290–291. doi: 10.1006/mgme.2002.3299. [DOI] [PubMed] [Google Scholar]
- 54.Njajou O.T., Vaessen N., Joosse M., Berghuis B., van Dongen J.W., Breuning M.H. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat Genet. 2001;28:213–214. doi: 10.1038/90038. [DOI] [PubMed] [Google Scholar]
- 55.Arden K.E., Wallace D.F., Dixon J.L., Summerville L., Searle J.W., Anderson G.J. A novel mutation in ferroportin1 is associated with haemochromatosis in a Solomon Islands patient. Gut. 2003;52:1215–1217. doi: 10.1136/gut.52.8.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hetet G., Devaux I., Soufir N., Grandchamp B., Beaumont C. Molecular analyses of patients with hyperferritinemia and normal serum iron values reveal both L ferritin IRE and 3 new ferroportin (slc11A3) mutations. Blood. 2003;102:1904–1910. doi: 10.1182/blood-2003-02-0439. [DOI] [PubMed] [Google Scholar]
- 57.Cazzola M., Cremonesi L., Papaioannou M., Soriani N., Kioumi A., Charalambidou A. Genetic hyperferritinaemia and reticuloendothelial iron overload associated with a three base pair deletion in the coding region of the ferroportin gene (SLC11A3) Br J Haematol. 2002;119:539–546. doi: 10.1046/j.1365-2141.2002.03946.x. [DOI] [PubMed] [Google Scholar]
- 58.Politou M., Kalotychou V., Pissia M., Rombos Y., Sakellaropoulos N., Papanikolaou G. The impact of the mutations of the HFE gene and of the SLC11A3 gene on iron overload in Greek thalassemia intermedia and beta(s)/beta(thal) anemia patients. Haematologica. 2004;89:490–492. [PubMed] [Google Scholar]
- 59.Roetto A., Merryweather-Clarke A.T., Daraio F., Livesey K., Pointon J.J., Barbabietola G. A valine deletion of ferroportin 1: a common mutation in hemochromastosis type 4. Blood. 2002;100:733–734. doi: 10.1182/blood-2002-03-0693. [DOI] [PubMed] [Google Scholar]
- 60.Wallace D.F., Pedersen P., Dixon J.L., Stephenson P., Searle J.W., Powell L.W. Novel mutation in ferroportin1 is associated with autosomal dominant hemochromatosis. Blood. 2002;100:692–694. doi: 10.1182/blood.v100.2.692. [DOI] [PubMed] [Google Scholar]
- 61.Wallace D.F., Browett P., Wong P., Kua H., Ameratunga R., Subramaniam V.N. Identification of ferroportin disease in the Indian subcontinent. Gut. 2005;54:567–568. doi: 10.1136/gut.2004.060988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cremonesi L., Forni G.L., Soriani N., Lamagna M., Fermo I., Daraio F. Genetic and clinical heterogeneity of ferroportin disease. Br J Haematol. 2005;131:663–670. doi: 10.1111/j.1365-2141.2005.05815.x. [DOI] [PubMed] [Google Scholar]
- 63.Morris T.J., Litvinova M.M., Ralston D., Mattman A., Holmes D., Lockitch G. A novel ferroportin mutation in a Canadian family with autosomal dominant hemochromatosis. Blood Cells Mol Dis. 2005;35:309–314. doi: 10.1016/j.bcmd.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 64.Bozzini C., Campostrini N., Trombini P., Nemeth E., Castagna A., Tenuti I. Measurement of urinary hepcidin levels by SELDI-TOF-MS in HFE-hemochromatosis. Blood Cells Mol Dis. 2008;40(3):347–352. doi: 10.1016/j.bcmd.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 65.Zaahl M.G., Merryweather-Clarke A.T., Kotze M.J., van der Merwe S., Warnich L., Robson K.J. Analysis of genes implicated in iron regulation in individuals presenting with primary iron overload. Hum Genet. 2004;115:409–417. doi: 10.1007/s00439-004-1166-y. [DOI] [PubMed] [Google Scholar]
- 66.Lee P.L., Gelbart T., West C., Barton J.C. SLC40A1 c.1402G→a results in aberrant splicing, ferroportin truncation after glycine 330, and an autosomal dominant hemochromatosis phenotype. Acta Haematol. 2007;118:237–241. doi: 10.1159/000112830. [DOI] [PubMed] [Google Scholar]
- 67.Koyama C., Wakusawa S., Hayashi H., Ueno T., Suzuki R., Yano M. A Japanese family with ferroportin disease caused by a novel mutation of SLC40A1 gene: hyperferritinemia associated with a relatively low transferrin saturation of iron. Intern Med (Tokyo, Japan) 2005;44:990–993. doi: 10.2169/internalmedicine.44.990. [DOI] [PubMed] [Google Scholar]
- 68.Griffiths W.J., Mayr R., McFarlane I., Hermann M., Halsall D.J., Zoller H. Clinical presentation and molecular pathophysiology of autosomal dominant hemochromatosis caused by a novel ferroportin mutation. Hepatology. 2009;51:788–795. doi: 10.1002/hep.23377. [DOI] [PubMed] [Google Scholar]
- 69.Xi T., Jones I.M., Mohrenweiser H.W. Many amino acid substitution variants identified in DNA repair genes during human population screenings are predicted to impact protein function. Genomics. 2004;83:970–979. doi: 10.1016/j.ygeno.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 70.Andersen R.V., Tybjaerg-Hansen A., Appleyard M., Birgens H., Nordestgaard B.G. Hemochromatosis mutations in the general population: iron overload progression rate. Blood. 2004;103:2914–2919. doi: 10.1182/blood-2003-10-3564. [DOI] [PubMed] [Google Scholar]
- 71.Allen K.J., Gurrin L.C., Constantine C.C., Osborne N.J., Delatycki M.B., Nicoll A.J. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221–230. doi: 10.1056/NEJMoa073286. [DOI] [PubMed] [Google Scholar]
- 72.Beaton M., Guyader D., Deugnier Y., Moirand R., Chakrabarti S., Adams P. Noninvasive prediction of cirrhosis in C282Y-linked hemochromatosis. Hepatology. 2002;36:673–678. doi: 10.1053/jhep.2002.35343. [DOI] [PubMed] [Google Scholar]
- 73.Pietrangelo A., Corradini E., Ferrara F., Vegetti A., De Jong G., Luca Abbati G. Magnetic resonance imaging to identify classic and nonclassic forms of ferroportin disease. Blood Cells Mol Dis. 2006;37:192–196. doi: 10.1016/j.bcmd.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 74.Fernandes A., Preza G.C., Phung Y., De Domenico I., Kaplan J., Ganz T. The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood. 2009;114:437–443. doi: 10.1182/blood-2008-03-146134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Drakesmith H., Schimanski L.M., Ormerod E., Merryweather-Clarke A.T., Viprakasit V., Edwards J.P. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood. 2005;106:1092–1097. doi: 10.1182/blood-2005-02-0561. [DOI] [PubMed] [Google Scholar]
- 76.Miltenberger-Miltenyi G., Schwarzbraun T., Loscher W.N., Wanschitz J., Windpassinger C., Duba H.C. Identification and in silico analysis of 14 novel GJB1, MPZ and PMP22 gene mutations. Eur J Hum Genet. 2009;17:1154–1159. doi: 10.1038/ejhg.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Olynyk J.K., St Pierre T.G., Britton R.S., Brunt E.M., Bacon B.R. Duration of hepatic iron exposure increases the risk of significant fibrosis in hereditary hemochromatosis: a new role for magnetic resonance imaging. Am J Gastroenterol. 2005;100(4):837–841. doi: 10.1111/j.1572-0241.2005.41287.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Results from histology, clinical and biochemical data of individual patients from whom a liver biopsy was reported.