

Abstract
Engulfment and cell motility 1 (ELMO1), a bipartite guanine nucleotide exchange factor (GEF) for the small GTPase Rac 1, was identified as a susceptibility gene for glomerular disease. Here, we reported that ELMO1 interacted with COX-2 in human mesangial cells. Furthermore, we identified ELMO1 as a posttranslational regulator of COX-2 activity. We demonstrated that COX-2 cyclooxygenase activity increased fibronectin promoter activity. The protein-protein interaction between ELMO1 and COX-2 increased the cyclooxygenase activity of COX-2 and, correspondingly, fibronectin expression. We also found that ET625, the dominant negative form of ELMO1 lacking Rac1 activity, interacted with COX-2, increased cyclooxygenase activity of COX-2 and enhanced COX-2-mediated fibronectin upregulation. To further rule out Rac1 as an ELMO1-mediated regulator of COX-2 activity, we employed the constitutive active Rac1, Rac1Q63E, and demonstrated that Rac1 signaling has no effect on COX-2-mediated fibronectin promoter activity. These results suggest that ELMO1 contributes to the development of glomerular injury through serving as a regulator of COX-2 activity. The interaction of ELMO1 with COX-2 could play an important role in the development and progression of renal glomerular injury.
1. Introduction
Renal glomerulosclerosis is the final major lesion of many primary and secondary glomerular diseases such as glomerulonephritis and diabetic nephropathy. It is characterized by glomerular accumulated deposits of extracellular matrix (ECM) proteins. Recent studies have indicated that renal glomerular mesangial cells play a pivotal role in the development and progression of glomerular injury[1]. Exposure to pathological stimuli can initiate the activation of quiescent mesangial cells and stimulate mesangial cells to enhance the production of ECM proteins such as collagens, fibronectin and proteinase inhibitors[2, 3], which results in the abnormal accumulation ECM in glomerular mesangium and irreversible glomerular injury in the end.
Cyclooxygenase (COX), also known as PGH synthase, is a membrane-bound, bifunctional enzyme that catalyzes the conversion of arachidonic acid to PGG2 by cyclooxygenase activity, and of PGG2 to PGH2 by peroxidase activity[4]. It is the rate-limiting step in biosynthesis of the biologically active and physiologically important prostaglandins. Two isoforms of COX have been identified: COX-1 and COX-2. COX-1 is constitutively expressed in most tissues. In contrast, COX-2 operates as an inducible enzyme, and its expression is stimulated by various cytokines, growth factors, and other inflammatory stimuli[5-9]. The pathogenic importance of COX-2 in glomerular disease has been addressed in a variety of clinical and experimental studies. Increased glomerular COX-2 expression has been reported in many glomerular diseases such as diabetic nephropathy, lupus nephritis, and IgA nephropathy[10-12]. Consequently, selective inhibition of COX-2 was reported to retard the progression of glomerular injury[11, 13-17]. However, the precise mechanisms that cause glomerular injury have not yet been elucidated.
Engulfment and cell motility 1 (ELMO1) is an evolutionarily conserved protein with no obvious catalytic domains. ELMO1 and Dock180 form a complex, which functions as an unconventional guanine nucleotide exchange factor (GEF) for the small GTPase Rac 1, and thereby regulating the actin cytoskeleton during phagocytosis and cell migration through Rac1 activation[18, 19]. Recent studies have reported that ELMO1 is closely associated with susceptibility to glomerular disease[20-22]. However, the contribution of ELMO1 to the pathogenesis of glomerular injury remains unclear.
In the present study, we reported that ELMO1 interacted with COX-2 in human mesangial cells. Furthermore, we reported that the interaction of ELMO1 with COX-2 promoted COX-2 cyclooxygenase activity as demonstrated by increased COX-2-mediated fibronectin promoter activity. These results suggest that ELMO1 contributes to the development of glomerular injury through serving as a regulator of COX-2 activity. The interaction of ELMO1 with COX-2 could play an important role in the development and progression of renal glomerular disease.
2. Materials and methods
2.1. Cell culture
HMC were isolated and maintained in RPMI 1640 containing 16.7% FBS and 100ug/ml penicillin and streptomycin as previously described[3]. HMC at passages 5 through 8 were used. THMC were maintained in RPMI 1640 containing 10% FBS and antibiotics[23].
2.2. Plasmid constructs and mutagenesis
Fn-Luc construct containing human fibronectin promoter sequences from −998 to +28 was obtained from HMC genomic DNA by PCR amplification. The forward (5′-GATAGGTACCTTTGTGTGTAACTGGCACGGTG-3′) and reverse primers (5′-GCGTGCTAGCCCCTGTGCAGCACAGCCGGCG-3′) contain KpnI and NheI restriction site, respectively. The amplification product was subcloned into the pGL3 basic luciferase vector (Promega). The dominant negative ELMO1, ET625 (aa. 1-625), was generated from the ELMO1 cDNA template with a PCR-based strategy. All constructs were sequenced to confirm fidelity and the presence of the appropriate mutations.
2.3. Transient transfection and adenovirus infection
Transfection was performed with the Lipofectamine 2000 transfection reagent (Invitrogen), and adenovirus infection was performed as previously described[23]. Briefly, for COX-2 and ELMO interaction, THMC was firstly transfected with plasmid expressing wild-type ELMO1 or mutant ELMO1. After 6hr, the cells were then infected by AdCOX-2WT at a multiplicity of infection (M.O.I.) of 200 and were cultured for 48h. In all of the experiments, empty vectors were added to keep equal plasmid concentration and adenovirus titer among different samples.
2.4. Fibronectin promoter activity assay
THMC were transfected with Fn-luc promoter by Lipofectamine 2000. 6hr after transfection, the cells were infected with wild-type or mutant AdCOX-2 or stimulated with PGE2. After 24hr of incubation, cells were lysed with reporter lysis buffer, and luminescence was measured in a luminometer. In all transfections, the expression vector pSV-β-galactosidase served as an internal control to correct for transfection efficiency.
2.5. Immunoprecipitation and immunoblotting
Cells were lysed in immunoprecipitation buffer (20mM Tris-Cl, 150mM NaCl, 1% Triton X-100 and 10% glycerol; pH 7.5) and precipitated with the indicated antibodies conjugated with agarose for 2 h at 4°C. The beads were then washed, eluted or boiled, and subjected to SDS-PAGE. For immunoblotting, the proteins were electrophoretically transferred onto PVDF membranes and blotted with primary antibodies. The primary antibodies were then incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies and detected by chemiluminescence.
2.6. Immunofluorescence
THMC plated on coverslips were fixed with 4% paraformaldehyde and permeabilized with 1% Triton X-100. Tissues or cells were then blocked and subsequently stained with appropriate primary antibodies. Immunostaining was performed sequentially with appropriate Alexa Fluor-conjugated IgG-specific secondary antibodies (Invitrogen) as described previously[24]. Fluorescent images were captured with immunofluorescence microscopy.
2.7. Rac1 activity assay
Serum-starved THMC transfected with the plasmids indicated and or infected with AdCOX-2 for 24h were lysed in lysis buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 1% IgepalCA-630, 10 mM MgCl2, 1 mM EDTA, and 10% glycerol supplemented with protease inhibitor mixture). To pulldown the GTP-bound active form of Rac1, equal amounts of the lysates were incubated with 10μg of GST-PBD bound to glutathione-agarose for 1h at 4°C. The beads were then washed three times with lysis buffer, and the proteins bound to the beads were eluted with Laemmli sample buffer and detected for the amount of GTP-bound Rac1 in immunoblotting using a monoclonal anti-Rac1 antibody.
2.8. Statistical analysis
All data from promoter activity assay are expressed as means ± s.e.m. from at least three separate experiments, each performed in duplicate. We carried out statistical analyses using the Student's t-test. Values were considered statistically significant at P < 0.05.
3. Results
3.1. ELMO1 interacts with COX-2 in mesangial cells
We have observed that overexpression of COX-2 results in the formation of covalent adducts between COX-2 and some unknown proteins, likely to be mediated by products of Cox-2 activity. To identify the proteins that may be involved in regulation of COX-2 activity, we isolated COX-2 adducts by immunoprecipitation with COX-2 antibody and subjected them to tandem mass spectrometry. A following search against mammalian database indicated the presence of ELMO1 (data not shown). The interaction of ELMO1 with COX-2 was further confirmed by co-immunoprecipitation (co-IP). In primary cultured human mesangial cells (HMC), we showed that endogenous COX-2 co-immunoprecipitated endogenous ELMO1 (Fig. 1A). This interaction was also observed by immunoprecipitation studies of transformed human mesangial cells (THMC) expressing Flag-tagged ELMO1 and infected with adenovirus encoding wild-type COX-2 (AdCOX-2WT) (Fig. 1B). Ability of Flag-tagged ELMO1 to co-precipitate introduced COX-2 did not interfere with coimmunoprecipitation of Flag-tagged ELMO1 with endogenous Dock180 (Fig. 1B). COX-2 is a membrane protein and is associated with the nuclear membrane and the endoplastic reticulum. Immunofluorescence staining demonstrated that COX-2-ELMO1 interaction occurred in perinuclear regions (Fig. 1C). Our findings uncover a novel function of ELMO1 and suggest a novel mechanism of regulating COX-2.
Fig. 1.
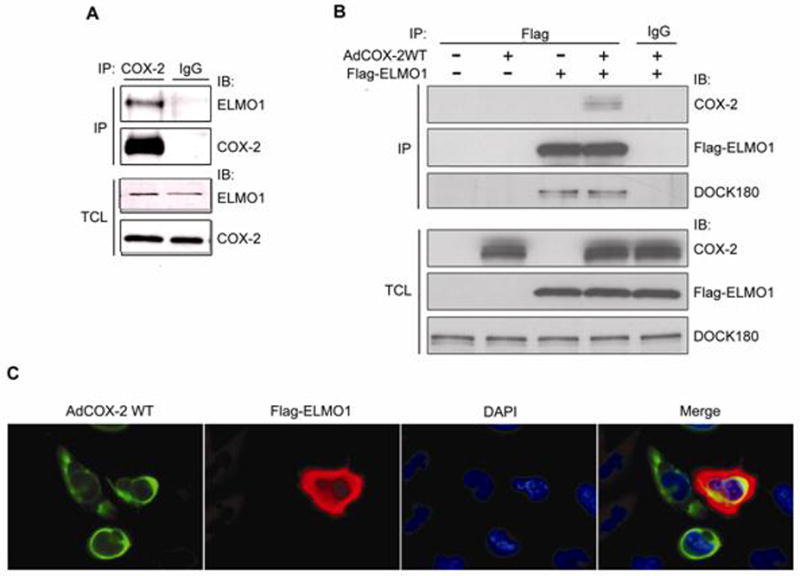
ELMO1 forms a complex with COX-2 in mesangial cells. (A) HMC lysates were immunoprecipitated with either COX-2 antibody, or control IgG, and co-precipitation of endogenous ELMO1 was determined by immunoblotting. Expression of the endogenous proteins was determined by immunoblotting of TCL. (B) Flag-ELMO1 was transfected into THMC, and co-precipitation of infected COX-2 and endogenous DOCK180 were determined by immunoblotting. Expression of the endogenous proteins or overexpressed proteins was determined by immunoblotting of TCL. (C) Flag-ELMO1 and AdWTCOX-2 expressing THMC were stained with anti-Flag (red) and anti-COX-2 (green), and their chromatin was stained with DAPI (blue). Colocalization of ELMO1 and COX-2 appears in yellow in the merge panel. Data are representative of three independent experiments.
3.2. Effect of COX-2 on fibronectin gene expression
Fibronectin is one of the major components of ECM proteins predominantly deposited in the glomerular mesangium in various renal glomerular diseases. However, little information is known about the role of COX-2 in fibronectin accumulation the process of glomerular injury. The G533 of COX-2 is critical for its cyclooxygenase activity. The G533A mutant demonstrated just 26% of wild-type total COX-2 activity, whereas the G533L mutant was unable to convert arachidonic acid to PGG2. However, both of the mutants have no effects on the peroxidase activity of COX-2[25]. To determine whether COX-2 affects fibronectin gene expression, Adenovirus encoding either wild type AdCOX-2WT or two COX-2 mutants (AdCOX-2G533A and AdCOX-2G533L) was employed to infect THMC transfected with human fibronetin promoter, Fn-luc. We demonstrated that AdCOX-2WT infection increased fibronectin promoter activity (P < 0.01), while the promoter activity was decreased to 51.4% by AdCOX-2G533A (P < 0.05) or to 32.1% by AdCOX-2G533L (P < 0.05) compared with AdCOX-2WT (Fig. 2 A, B). These results showed that COX-2 increased fibronectin gene expression, and that this effect was dependent on the cyclooxygenese activity of COX-2. To further confirm our findings, we next examined whether PGE2 participates in COX-2-mediated fibronectin gene expression as PGE2 is most abundant prostanoid detected in the kidney. As predicted, PGE2 increased fibronectin promoter activity in a concentration-dependent manner (Fig. 2C). This suggested that COX-2 increased fibronectin upregulation through its cyclooxygenase activity.
Fig. 2.
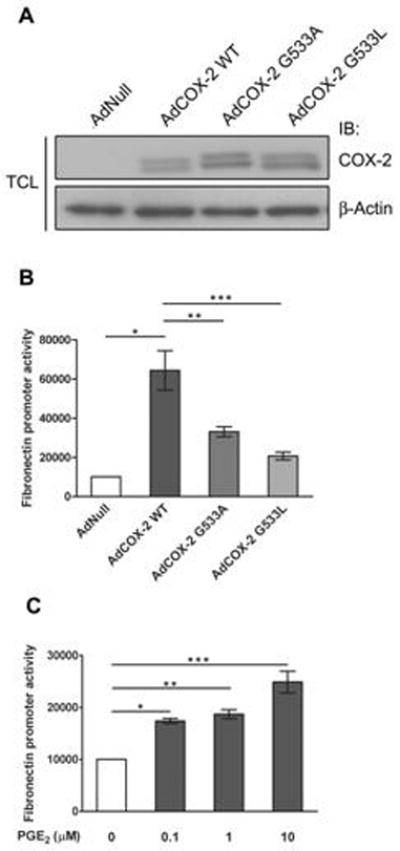
Effect of COX-2 on fibronectin gene expression. (A) Immunoblotting assay for COX-2 expression in THMC infected with AdCOX-2 WT, AdCOX-2G533A and AdCOX-2G533L. β-Actin was used for protein loading control. TCL: total cell lysates. IB: immunoblotting (B) Luciferase assay with the fibronectin promoter in THMC infected with AdCOX-2 WT, AdCOX-2G533A and AdCOX-2G533L. * P < 0.01, ** P < 0.05, *** P < 0.05 by two-tailed Student t-test. (C) Luciferase assay with the fibronectin promoter in THMC under PGE2 stimulation. * P = 0.0001, ** P < 0.001, and *** P < 0.005 by two-tailed Student t-test. All data in B and C are means ± s.e.m from at least three separate experiments, each performed in duplicate.
3.3. Effect of ELMO1 on COX-2-mediated fibronectin gene expression
3.3.1 Effect of wild-type ELMO1 and mutant ELMO1 on COX-2-mediated fibronectin promoter activity
To determine whether ELMO1 affects COX-2 activity, THMC were transfected with ELMO1 and fibronectin promoter connected with reporter gene, luciferase, followed by infection with AdCOX-2WT. We demonstrated that ELMO1 increased COX-2 activity, as ELMO1WT enhanced COX-2-mediated fibronectin promoter activity (P < 0.05) (Fig. 3A). ET625, is a dominant negative form of ELMO1 as it fails to interact with Dock180 and thus lacks Rac1 activity. Co-immunoprecipitation results showed that Flag-tagged ET625 pulled down COX-2 in THMC (Figure 3, B). We found that ET625 also increased COX-2-mediated fibronectin promoter activity (P < 0.05) (Fig. 3A).
Fig. 3.
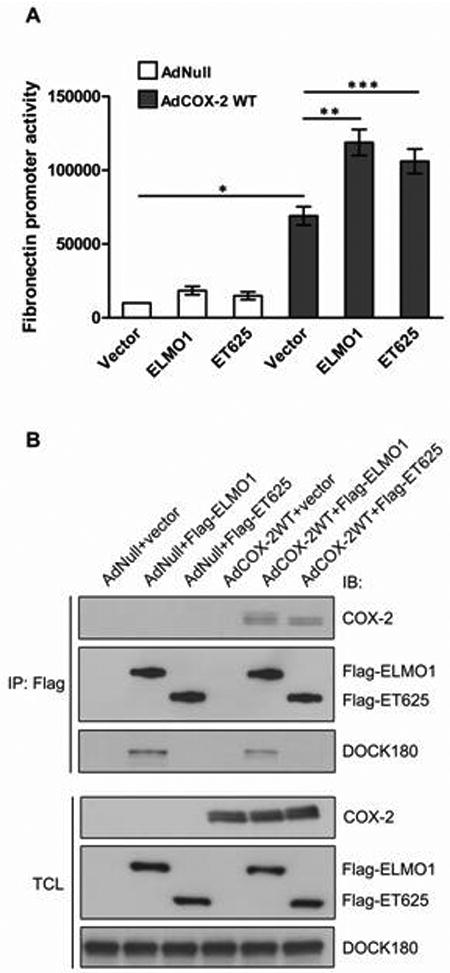
Effect of wild-type ELMO1 and mutant ELMO1 on COX-2-mediated fibronectin gene expression. (A) Luciferase assay with the fibronectin promoter in THMC co-introduced with AdCOX-2WT, ELMO1WT or ET625. * P < 0.001, ** P < 0.05, *** P < 0.05 by two-tailed Student t-test. (B) THMC transfected with the indicated plasmids were infected with AdCOX-2WT, and lysates were immunoprecipitated with anti-Flag antibody. Precipitated COX-2 and DOCK 180 were determined by immunoblotting. Expression of the endogenous proteins or overexpressed proteins was determined by immunoblotting of TCL. Data are representative of three independent experiments. All data in A are means ± s.e.m from at least three separate experiments, each performed in duplicate.
3.3.2 Effect of rac1 on COX-2-mediated fibronectin promoter activity
To further rule out the possibility that Rac1 is involved in ELMO1-stimulated COX-2 activity, we introduced the constitutive active Rac1, Rac1Q63E, into THMC infected with AdCOX-2WT, and found that Rac1Q63E had no effect on the COX-2 activity (P = 0.8170) (Fig. 4A, B). We demonstrated that both ELMO1WT and ET625 had no effects on COX-2 protein expression (Fig. 4C), and thus the possibility that ELMO1 increases COX-2 activity through inducing COX-2 gene expression could be ruled out. ELMO1 may increase fibronectin gene expression through direct modification of COX-2 activity.
Fig. 4.
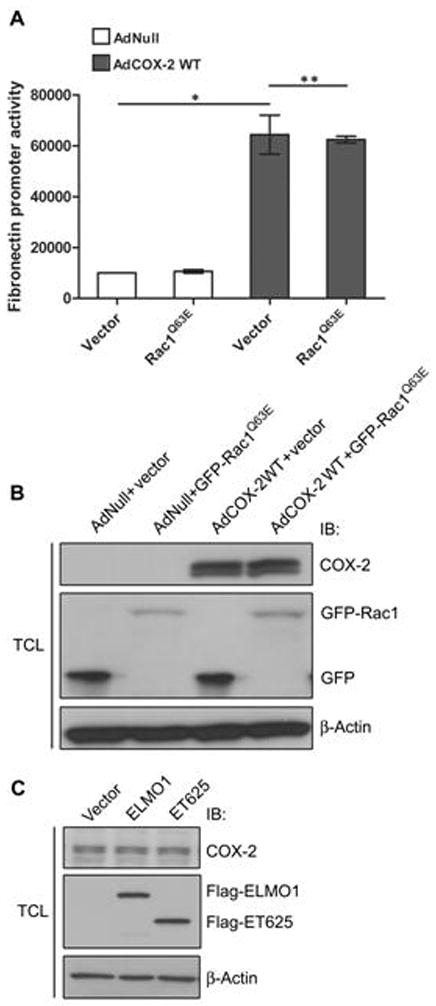
Effect of Rac1 on COX-2mediated fibronectin promoter activity. (A) Constitutive active Rac1, Rac1Q63E, has no effects on COX-2-mediated fibronectin promoter activity. * P < 0.005, ** P = 0.8170 by two-tailed Student t-test. (B) Immunoblotting assay for GFP tagged Rac1Q63E expression in THMC infected with AdCOX-2 WT. β-Actin was used for protein loading control. (C) Immunoblotting demonstrated that ELMO1WT and ET625 have no effects on endogenous COX-2 expression. Data are representative of three independent experiments. All data in A are means ± s.e.m from at least three separate experiments, each performed in duplicate.
3.4. Effect of COX-2 on ELMO1-rac1 activity
To determine whether COX-2 affected ELMO1-mediated Rac1 activity, we introduced WTELMO1 and Dock180 plasmids into THMC infected with AdCOX-2WT, as ELMO1 was reported to increase Rac1 activity through cooperating with Dock180. Here we demonstrated that ELMO1 and Dock180 co-expression increased Rac1 activity, whereas COX-2 had no effect on ELMO1/Dock180-mediated Rac1 activity (Fig. 5).
Fig. 5.
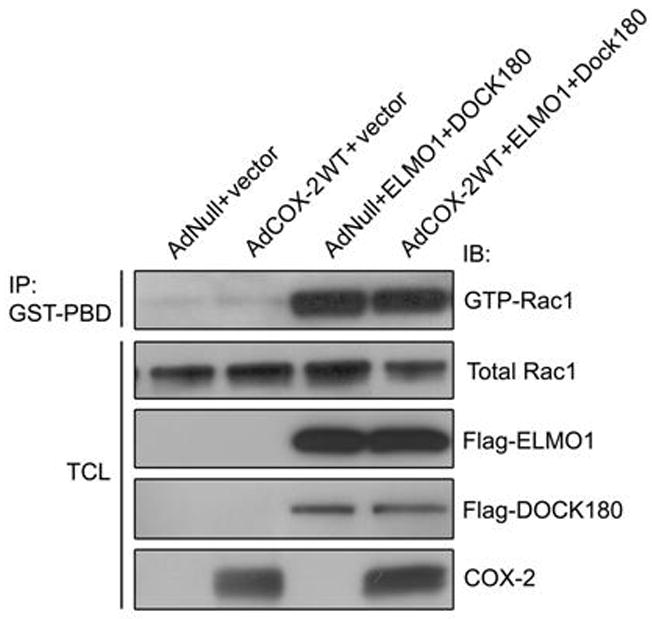
Effect of COX-2 on ELMO1-Rac1 activity. Rac1 activity assay demonstrated that ELMO1 and DOCK180 cotransfection increased the amount of GTP-bound Rac1, whereas AdCOX-2 infection has no effects on ELMO1/DOCK180-mediated Rac1 activity. Data are representative of three independent experiments.
4. Discussion
In this study, we reported that ELMO1 interacted with COX-2 and increased COX-2-mediated fibronectin upregulation in mesangial cells. Our data suggest that ELMO1 may play an important role in glomerular injury through interacting with and regulating COX-2 activity, thus resulting in accumulated deposits of ECM in the pathogenesis of glomerular disease.
Our data indicated that ELMO1 could form a complex with COX-2 in mesangial cells. Here raised an interesting question. What is the significance of the interaction of ELMO1 with COX-2? In mammalian cells, ELMO1 binds directly to Dock180 through its carboxy-terminal region, leading to Rac1 activation[19], whereas COX-2 is the rate-limiting enzyme involved in the production of prostaglandins. The interaction of ELMO1 with COX-2 may affect COX-2 activity.
The importance of COX-2 in glomerular injury has been reported in a variety of clinical and experimental studies[26, 27]. However, the precise mechanism about the role of COX-2 in ECM accumulation remains unknown. Fibronectin is one of the major components of ECM accumulated in many glomerular diseases[28]. In this study, we demonstrated that overexpression of COX-2 increased fibronectin promoter activity. We also found that PGE2, the major product of COX-2 in the kidney, stimulated fibronectin promoter activity. These data suggest that COX-2 may cause glomerular injury through stimulating fibronectin accumulation. In the process of studying effect of ELMO1 on COX-2 activity, we demonstrated that wild-type ELMO1 promoted COX-2-mediated fibronectin promoter activity, whereas, neither ET625, a dominant negative form of ELMO1 nor constitutive active Rac1, Rac1Q63E has no effect on fibronectin promoter activity. Together with the colocalization and co-immunoprecipitation data, it is suggested that ELMO1 increases COX-2 activity independent of Rac1 activity. The direct interaction between ELMO1 and COX-2 may contribute to the conformational change of COX-2 and thus increases its cyclooxygenase activity. Recent studies have established that ELMO1 is associated with susceptibility to glomerular disease. Genetic variants in intron 13, 16, 18 and 20 of the ELMO1 gene were detected in patients with diabetes[20-22]. The genetic variants of ELMO1 gene may result in elevated ELMO1 gene expression in glomeruli of patients with diabetes, as elevated ELMO1 expression appeared in glomeruli of diabetic mice[20]. Subsequent functional studies demonstrated that overexpression of ELMO1 promoted ECM genes transcription[29]. These studies suggest that ELMO1 play a role in ECM accumulation in the pathogenesis of glomerular disease. In this study, we uncovered a new function of ELMO1 serving as a regulator of COX-2 activity. ELMO1 may aggravate glomerulr injury through increasing COX-2 activity and thus stimulating COX-2-mediated fibronectin accumulation in the development of glomerulosclerosis.
In conclusion, we have shown that the interaction of ELMO1 with COX-2 was demonstrated in mesangial cells. We also found that the interaction of ELMO1 with COX-2 increased COX-2-mediated fibronectin upregulation, which is independent of Rac1 activity. These results suggest that ELMO1 serves as a regulator of COX-2 activity. ELMO1 may participate in ECM accumulation in the pathogenesis of glomerular sclerosis through modifying COX-2 activity via protein-protein interaction. The interaction of ELMO1 with COX-2 could play an important role in the development and progression of renal glomerular disease.
Acknowledgments
This work was supported by grants from National Institutes of Health to A. Sorokin (RO1DK41684) and from Biotechnology & Bioengineering Center (MCW) to A. Sorokin. We thank A. Parfenyev who participated in the early stage of this project. We also thank S. Prosser for help in preparation of the manuscript, S. Maeda (RIKEN Yokohama Institute, Yokohama, Japan), M. Matsuda (Kyoto University, Kyoto, Japan), and J. Barbieri (Department of Microbiology and Molecular Genetics, Medical College of Wisconsin) for providing Flag-tagged ELMO1, Flag-tagged Dock180 and constitutive active Rac1 (Rac1Q63E) cDNAs, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC. Am J Physiol Renal Physiol. 2003;284(2):F243–252. doi: 10.1152/ajprenal.00300.2002. [DOI] [PubMed] [Google Scholar]
- 2.Poncelet AC, Schnaper HW. Am J Physiol. 1998;275(3 Pt 2):F458–466. doi: 10.1152/ajprenal.1998.275.3.F458. [DOI] [PubMed] [Google Scholar]
- 3.Yang C, Patel K, Harding P, Sorokin A, Glass WF., 2nd Exp Cell Res. 2007;313(6):1240–1250. doi: 10.1016/j.yexcr.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith WL, DeWitt DL, Garavito RM. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 5.Harding P, Balasubramanian L, Swegan J, Stevens A, Glass WF., 2nd Kidney Int. 2006;69(9):1578–1585. doi: 10.1038/sj.ki.5000323. [DOI] [PubMed] [Google Scholar]
- 6.Pratt PF, Bokemeyer D, Foschi M, Sorokin A, Dunn MJ. J Biol Chem. 2003;278(51):51928–51936. doi: 10.1074/jbc.M309256200. [DOI] [PubMed] [Google Scholar]
- 7.Sheu ML, Chao KF, Sung YJ, Lin WW, Lin-Shiau SY, Liu SH. Cell Signal. 2005;17(8):975–984. doi: 10.1016/j.cellsig.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 8.Feng L, Xia Y, Garcia GE, Hwang D, Wilson CB. J Clin Invest. 1995;95(4):1669–1675. doi: 10.1172/JCI117842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guan Z, Buckman SY, Miller BW, Springer LD, Morrison AR. J Biol Chem. 1998;273(44):28670–28676. doi: 10.1074/jbc.273.44.28670. [DOI] [PubMed] [Google Scholar]
- 10.Tomasoni S, Noris M, Zappella S, Gotti E, Casiraghi F, Bonazzola S, Benigni A, Remuzzi G. J Am Soc Nephrol. 1998;9(7):1202–1212. doi: 10.1681/ASN.V971202. [DOI] [PubMed] [Google Scholar]
- 11.Komers R, Lindsley JN, Oyama TT, Schutzer WE, Reed JF, Mader SL, Anderson S. J Clin Invest. 2001;107(7):889–898. doi: 10.1172/JCI10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Honkanen T, Mustonen J, Kainulainen H, Myllymaki J, Collin P, Hurme M, Rantala I. Kidney Int. 2005;67(6):2187–2195. doi: 10.1111/j.1523-1755.2005.00324.x. [DOI] [PubMed] [Google Scholar]
- 13.Fujihara CK, Antunes GR, Mattar AL, Andreoli N, Malheiros DM, Noronha IL, Zatz R. Kidney Int. 2003;64(6):2172–2181. doi: 10.1046/j.1523-1755.2003.00319.x. [DOI] [PubMed] [Google Scholar]
- 14.Blume C, Heise G, Muhlfeld A, Bach D, Schror K, Gerhardz CD, Grabensee B, Heering P. Kidney Int. 1999;56(5):1770–1778. doi: 10.1046/j.1523-1755.1999.00742.x. [DOI] [PubMed] [Google Scholar]
- 15.Waldner C, Heise G, Schror K, Heering P. Eur J Clin Invest. 2003;33(11):969–975. doi: 10.1046/j.1365-2362.2003.01256.x. [DOI] [PubMed] [Google Scholar]
- 16.Cheng HF, Wang CJ, Moeckel GW, Zhang MZ, McKanna JA, Harris RC. Kidney Int. 2002;62(3):929–939. doi: 10.1046/j.1523-1755.2002.00520.x. [DOI] [PubMed] [Google Scholar]
- 17.Komers R, Lindsley JN, Oyama TT, Anderson S. Clin Exp Pharmacol Physiol. 2007;34(1-2):36–41. doi: 10.1111/j.1440-1681.2007.04534.x. [DOI] [PubMed] [Google Scholar]
- 18.Gumienny TL, Brugnera E, Tosello-Trampont AC, Kinchen JM, Haney LB, Nishiwaki K, Walk SF, Nemergut ME, Macara IG, Francis R, Schedl T, Qin Y, Van Aelst L, Hengartner MO, Ravichandran KS. Cell. 2001;107(1):27–41. doi: 10.1016/s0092-8674(01)00520-7. [DOI] [PubMed] [Google Scholar]
- 19.Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont AC, Macara IG, Madhani H, Fink GR, Ravichandran KS. Nat Cell Biol. 2002;4(8):574–582. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- 20.Shimazaki A, Kawamura Y, Kanazawa A, Sekine A, Saito S, Tsunoda T, Koya D, Babazono T, Tanaka Y, Matsuda M, Kawai K, Iiizumi T, Imanishi M, Shinosaki T, Yanagimoto T, Ikeda M, Omachi S, Kashiwagi A, Kaku K, Iwamoto Y, Kawamori R, Kikkawa R, Nakajima M, Nakamura Y, Maeda S. Diabetes. 2005;54(4):1171–1178. doi: 10.2337/diabetes.54.4.1171. [DOI] [PubMed] [Google Scholar]
- 21.Leak TS, Perlegas PS, Smith SG, Keene KL, Hicks PJ, Langefeld CD, Mychaleckyj JC, Rich SS, Kirk JK, Freedman BI, Bowden DW, Sale MM. Ann Hum Genet. 2009;73(2):152–159. doi: 10.1111/j.1469-1809.2008.00498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pezzolesi MG, Katavetin P, Kure M, Poznik GD, Skupien J, Mychaleckyj JC, Rich SS, Warram JH, Krolewski AS. Diabetes. 2009 doi: 10.2337/db09-0641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishaque A, Dunn MJ, Sorokin A. J Biol Chem. 2003;278(12):10629–10640. doi: 10.1074/jbc.M210559200. [DOI] [PubMed] [Google Scholar]
- 24.Yang C, Glass WF., 2nd Exp Biol Med (Maywood) 2008;233(6):689–693. doi: 10.3181/0710-RM-279. [DOI] [PubMed] [Google Scholar]
- 25.Rowlinson SW, Crews BC, Lanzo CA, Marnett LJ. J Biol Chem. 1999;274(33):23305–23310. doi: 10.1074/jbc.274.33.23305. [DOI] [PubMed] [Google Scholar]
- 26.Stahl RA, Thaiss F, Kahf S, Schoeppe W, Helmchen UM. Kidney Int. 1990;38(2):273–281. doi: 10.1038/ki.1990.196. [DOI] [PubMed] [Google Scholar]
- 27.Lianos EA, Bresnahan BA, Pan C. J Clin Invest. 1991;88(2):623–631. doi: 10.1172/JCI115347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi K, Osada S, Shofuda K, Horikoshi S, Shirato I, Tomino Y. J Am Soc Nephrol. 1998;9(12):2262–2271. doi: 10.1681/ASN.V9122262. [DOI] [PubMed] [Google Scholar]
- 29.Shimazaki A, Tanaka Y, Shinosaki T, Ikeda M, Watada H, Hirose T, Kawamori R, Maeda S. Kidney Int. 2006;70(10):1769–1776. doi: 10.1038/sj.ki.5001939. [DOI] [PubMed] [Google Scholar]