

Abstract
The occurrence and progression of cerebral β-amyloid angiopathy (CAA) and β-amyloid plaques in sporadic Alzheimer's disease may be attributed to aging-related deficiencies in β-amyloid drainage along cerebral perivascular pathways. To elucidate high-definition characteristics of cerebral β-amyloid deposition, we performed immunogold silver staining for β-amyloid-40 and β-amyloid-42 on semithin LR White-embedded tissue sections from 7 Alzheimer's disease/severe CAA, 9 Alzheimer's disease/mild CAA, 5 old control and 4 young control autopsy brains. In vessel walls, β-amyloid-40 and β-amyloid-42 deposits were unevenly distributed along the adventitia and among the medial smooth muscle cells. β-Amyloid-40 immunoreactivity appeared greater than that of β-amyloid-42 in vessel walls, with β-amyloid-42 being preferentially located on their abluminal regions. In capillary walls, either β-amyloid-40 or β-amyloid-42 deposits or both were present in 6 of 7 severe CAA and 1 of 9 mild CAA cases, with a marked variation in thickness and focally abluminal excrescences. In 5 of 7 severe CAA cases, a subset of β-amyloid-laden capillaries revealed either β-amyloid-40 or β-amyloid-42 deposits or both radiating from their walls into the surrounding neuropil (“pericapillary deposits”). No vascular β-amyloid-40 or β-amyloid-42 deposits were observed in any of the controls. In conclusion, the patterns of β-amyloid-42 and β-amyloid-40 immunoreactivity in vessel walls suggest that β-amyloid deposits occur in the vascular basement membranes along cerebral perivascular drainage pathways, extending from cortical capillaries to leptomeningeal arteries. The presence of pericapillary β-amyloid deposits suggests that a subset of β-amyloid plaques originate from β-amyloid-laden capillaries, particularly in Alzheimer's disease brains that exhibit preferential capillary CAA involvement.
Keywords: Cerebral amyloid angiopathy, LR White resin, Immunogold silver staining
1. Introduction
In sporadic late-onset Alzheimer's disease (AD) the neuropathologic characteristics, β-amyloid (Aβ) plaques and hyperphosphorylated-tau neurofibrillary changes in brain parenchyma, are commonly accompanied by cerebral Aβ angiopathy (CAA) [1]. Amyloid deposits in AD brains are mainly composed of Aβ peptides 1-40 and 1-42 [2]. Aβ42 is much more fibrillogenic than Aβ40, and the Aβ40/42 ratio predicts their distribution in vessel walls and parenchymal plaques, whereby an increased Aβ40/42 ratio is associated with the development of CAA [2]. The apolipoprotein E ε4 allelic dosage was shown to correlate with Aβ40 accumulation in parenchymal plaques [3,4] and vessel walls [5] previously seeded with Aβ42 in AD brains. Nonetheless, the mechanistic cascade of cerebral Aβ deposition is still unclear. In aging and sporadic AD there is no consistent evidence for either overproduction or abnormal isoforms of Aβ, in contrast to Down's syndrome and familial forms of AD [2]. Progressive cerebral Aβ accumulation may be associated with decreased enzymatic degradation of Aβ originating from neurons, deficient efflux of soluble Aβ from the interstitial fluid, increased influx of Aβ from the blood circulation, or a combination of these and other factors [6]. In addition to receptor-mediated transcytosis of Aβ across the blood-brain barrier, Aβ elimination may be mediated by perivascular macrophages [7], via bulk flow of interstitial fluid into the ventricles [8], and through perivascular interstitial fluid drainage of soluble Aβ along the basement membranes of capillaries and arteries [6,9].
In histologic preparations, insoluble or fibrillar Aβ deposits can be visualized in correlation with tissue morphology. Previous immunohistochemical studies of cerebral Aβ deposition were performed mostly on paraffin-embedded tissue sections [5,10-13], and occasionally using immunoelectron microscopy [14-16]. Immunohistologic evaluation on paraffin sections is likely restricted by relatively low definition. While ultrastructural visualization of protein markers of interest can be achieved by immunoelectron microscopy, the field size of examination is very limited with this approach. Immunogold silver staining of semithin resin-embedded tissue sections is considered a “compromise” method that provides higher-definition immunohistologic observations compared with those of paraffin immunohistochemistry, and larger field sizes compared to those that can be visualized by immunoelectron microscopy.
The present study was aimed at elucidating high-definition characteristics of Aβ deposition in cortical blood vessels and capillaries in AD brains by using immunogold silver staining for Aβ40 and Aβ42 on semithin LR White-embedded tissue sections.
2. Materials and methods
2.1. Study sample
The brains of demented patients with a neuropathologic diagnosis of definite or probable AD (according to the Consortium to Establish a Registry for Alzheimer's Disease [17]) were selected from the Mary S. Easton Center for Alzheimer's Disease Research Brain Bank at the University of California, Los Angeles. The neuropathologic protocols used in evaluating autopsy cases have been described elsewhere [18]. A total of 16 AD cases selected (Table 1) were grouped on the basis of CAA severity in accordance with Vonsattel criteria [19] into severe CAA (Vonsattel grade III, 7 cases: SA1 to SA7) and mild CAA (Vonsattel grade I, 9 cases: MA1 to MA9). All demented patients showed definite AD neuropathologic features, with Braak & Braak stage VI [20], except for case SA2 that had “mixed” probable AD/vascular dementia, with Braak & Braak stage V neuropathologic findings. Non-demented controls consisted of 5 old controls (OC1 to OC5) and 4 young controls (YC1 to YC4), which at autopsy showed no significant neuropathologic changes. Included in our present study for immunogold silver staining were LR White-embedded tissue blocks from the occipital cortex. In case the occipital cortical blocks were not available for a given case, those from the frontal cortex were used (Table 1).
Table 1.
Summary of demographic data and immunohistologic findings in autopsy specimens
| Case | Age (yr)-Sex | Postmortem delay (hr) | Brain weight (g) | Cerebral cortex | Aβ40 artery / arteriole | Aβ42 artery / arteriole | Aβ40 capillary | Aβ42 capillary |
|---|---|---|---|---|---|---|---|---|
| SA1 | 86-F | 19 | 970 | RO | + | + | + | − |
| SA2 | 85-M | 4 | 1250 | RO | − | − | − | − |
| SA3 | 87-F | 14 | 925 | LO | + | + | + (PD) | − |
| SA4 | 83-F | 24 | 1320 | RO | + | + | + (PD) | + (PD) |
| SA5 | 88-M | 14 | 1120 | RF | + | + | + (PD) | + (PD) |
| SA6 | 79-M | 21 | 1100 | LO | + | + | + | + (PD) |
| SA7 | 96-F | 12 | 920 | RO | + | + | − | + (PD) |
| MA1 | 88-F | 6 | No record | LO | − | − | − | − |
| MA2 | 74-M | 12 | 1160 | RO | − | − | − | − |
| MA3 | 90-M | 13 | 1110 | RO | − | − | − | − |
| MA4 | 65-F | 9 | 1160 | RF | − | − | − | − |
| MA5 | 72-F | 16 | 950 | RO | − | − | − | − |
| MA6 | 91-F | 4 | 1120 | RO | + | + | − | − |
| MA7 | 63-F | 18 | 950 | LO | − | − | − | − |
| MA8 | 69-F | 16 | 910 | RO | − | + | − | + |
| MA9 | 89-M | 15 | 1165 | RO | − | − | − | − |
| OC1 | 72-F | 16 | 1150 | RO | − | − | − | − |
| OC2 | 86-M | 28 | 1170 | RO | − | − | − | − |
| OC3 | 67-M | 22 | 1670 | LO | − | − | − | − |
| OC4 | 81-M | 24 | 1470 | RO | − | − | − | − |
| OC5 | 71-M | 26 | 1270 | RO | − | − | − | − |
| YC1 | 43-F | 30 | No record | RO | − | − | − | − |
| YC2 | 35-F | 15 | 1560 | LO | − | − | − | − |
| YC3 | 23-M | 21 | 1570 | LO | − | − | − | − |
| YC4 | 13-M | 22 | 1430 | LO | − | − | − | − |
Abbreviations:Aβ, β-amyloid; SA1-SA7, brains of Alzheimer's disease patients with severe cerebral Aβ angiopathy (Vonsattel grade III); MA1-MA9, brains of Alzheimer's disease patients with mild cerebral Aβ angiopathy (Vonsattel grade I); OC1-OC5, brains of non-demented old controls; YC1-YC4, brains of non-demented young controls; F, female; M, male; RO, right occipital; LO, left occipital; RF, right frontal; “+”, presence; “−”, absence; PD, pericapillary deposits (ie, Aβ deposits radiating from Aβ-laden capillaries into the surrounding neuropil).
2.2. LR White embedment
Fresh brain tissue fragments of approximately 2 mm in greatest dimension were fixed at autopsy with 0.1 M Sorensen's phosphate buffer (pH 7.4) containing 2% paraformaldehyde, 0.5% glutaraldehyde, 2% sucrose, 0.15 M sodium chloride and 0.15 mg/ml calcium chloride for 2 hr and then washed with Sorensen's buffer (3 times, 30 min each). Following full dehydration with a graded ethanol series in Sorensen's buffer (50, 70, 70, 100 and 100%, 30 min each), the tissue fragments were gradually infiltrated with a mixture of medium-grade LR White resin (Ted Pella, Redding, CA, USA) and ethanol (1:2, 1:1 and 2:1) at room temperature for 1 hr each, and then with neat LR White resin at room temperature (2 times, 1 hr each), at 4°C (overnight) and at room temperature (1 hr). The tissue fragments were embedded with neat LR White resin in airtight gelatin capsules (size 00, Ted Pella) at 60°C for 24 hr to allow thermal polymerization.
2.3. Immunogold silver staining
LR White-embedded tissue block faces were trimmed to an approximately 2 × 3 mm size. Serial 1 micrometer-thick tissue sections were cut with a motorized ultramicrotome equipped with a diamond knife, and transferred to Fisherbrand Superfrost/Plus microscope slides (3 consecutive sections per slide). One slide from each tissue block was stained with 0.4% toluidine blue and reviewed to confirm the presence of cerebral cortical tissue, one was used for Aβ40 immunostaining, and one for Aβ42 immunostaining. Following incubation with formic acid (1:200 dilution of 90% solution in deionized water [Barnstead Nanopure Diamond, Thermo Scientific, Waltham, MA, USA]) at room temperature for 10 min, the sections were rinsed in water and in PBS. The sections were then incubated with 0.05 M glycine in PBS for 5 min to quench aldehyde groups left in tissue from fixation, rinsed in PBS, and then incubated for 10 min with blocking solution (0.5% bovine serum albumin and 0.1% cold-water-fish-skin gelatin in PBS). The sections were blotted and incubated with rabbit polyclonal anti-Aβ40 (Millipore, Temecula, CA, USA, #AB5074P) or anti-Aβ42 (Millipore, #AB5078P) primary antibody diluted 1:200 in the blocking solution (for both antibodies), at 4°C for 24 hr. After being rinsed with the blocking solution, the sections were incubated with Fab' fragments of goat anti-rabbit IgG secondary antibody conjugated with 1.4 nm-in diameter Nanogold (Nanoprobes, Yaphank, NY, USA, #2004; 1:100 dilution in 1% normal goat serum in the blocking solution) at room temperature for 3 hr. Following PBS wash and water wash, the sections were incubated with silver solution (LI Silver Enhancement Kit, Nanoprobes, #2013) at room temperature for 30 min. The sections were then rinsed in water and in PBS, and incubated with 1.25% neutrally buffered glutaraldehyde in PBS at room temperature for 3 min. After being rinsed in water, the sections were counterstained with 0.4% toluidine blue at room temperature for 1 min. Following water wash, the sections were blotted, air dried, and mounted with Cytoseal 60 (Richard-Allan Scientific, Waltham, MA, USA). Two negative reagent controls were made, one by replacing the primary antibody with the primary-antibody diluent, and the other by replacing secondary antibody with the secondary-antibody diluent.
2.4. Qualitative assessment of Aβ immunoreactivity
By light microscopy, immunostained sections were qualitatively evaluated independently by two neuropathologists (V. S. and H. V. V.) for the distribution patterns of Aβ40 and Aβ42 deposits in blood vessels and capillaries. On each tissue section, the presence of parenchymal Aβ40 or Aβ42 plaques served as a built-in positive tissue control. No immunogold silver staining signals were observed in any of the negative reagent controls. The differential distribution of Aβ40 and Aβ42 immunoreactive deposits in vessel walls was assessed with regard to their relative locations within the vascular wall (ie, the intima, media and adventitia), relative extents of mural involvement (ie, greater vs. less than half of the mural thickness), and longitudinal distribution patterns (ie, continuous vs. segmental, and uniformly vs. unevenly spaced). In capillary walls, the presence of Aβ40 or Aβ42 immunoreactive deposits radiating from their walls into the surrounding neuropil (“pericapillary deposits”) was recorded, as were the longitudinal distribution patterns. Aβ40 and Aβ42 plaques in cortical parenchyma were classified into diffuse plaques and dense-core plaques. The relative abundance of Aβ40 and Aβ42 plaques of diffuse or dense-core type was evaluated on the adjacent tissue sections on a scale of 1+ to 3+, with no attempt to systematically count the plaques. The relative immunostaining intensity of Aβ40 and Aβ42 plaques was also assessed. In addition, the presence of close spatial relationship between Aβ-laden capillaries and Aβ plaques was recorded.
3. Results
For each case, the demographic data, postmortem delay, fresh brain weight, cerebral cortical region examined, and presence or absence of Aβ40- and Aβ42-laden blood vessels and capillaries are shown in Table 1. The patients' ages at death ranged from 79 to 96 years (mean 86.1, standard deviation [SD] 5.3) in the severe CAA group (4 female and 3 male), from 63 to 91 years (mean 77.9, SD 11.5) in the mild CAA group (6 female and 3 male), from 67 to 86 years (mean 75.4, SD 7.8) in the old control group (1 female and 4 male), and from 13 to 43 years (mean 28.5, SD 13.2) in the young control group (2 female and 2 male).
3.1. Aβ deposition in blood vessel walls
In AD brains, both Aβ40 and Aβ42 deposits in vessel walls were observed in 6 of 7 severe CAA and 1 of 9 mild CAA brains. Aβ42 only-laden vessels were present in 1 of 9 mild CAA brains. Aβ40 and Aβ42 deposits were unevenly distributed along the adventitia and among the medial smooth muscle cells of leptomeningeal arteries (Figs. 1A-F) and cortical arteries and arterioles (Figs. 1G-K). In the majority of AD brains that exhibited both Aβ40 and Aβ42 deposits in vessel walls, Aβ40 immunoreactivity appeared greater than that of Aβ42 in the walls of leptomeningeal and cortical arteries on the adjacent tissue sections, with Aβ42 being preferentially located on their abluminal regions (Figs. 1E-I); the opposite immunoreactivity pattern was not observed in any of these AD brains. Nonetheless, this differential distribution of Aβ40 and Aβ42 deposits was not always obvious in the walls of arterioles on the adjacent tissue sections. Note that no Aβ40 or Aβ42 deposits were seen in any of the leptomeningeal or cortical vessels in any of the old or young non-demented controls.
Fig. 1.
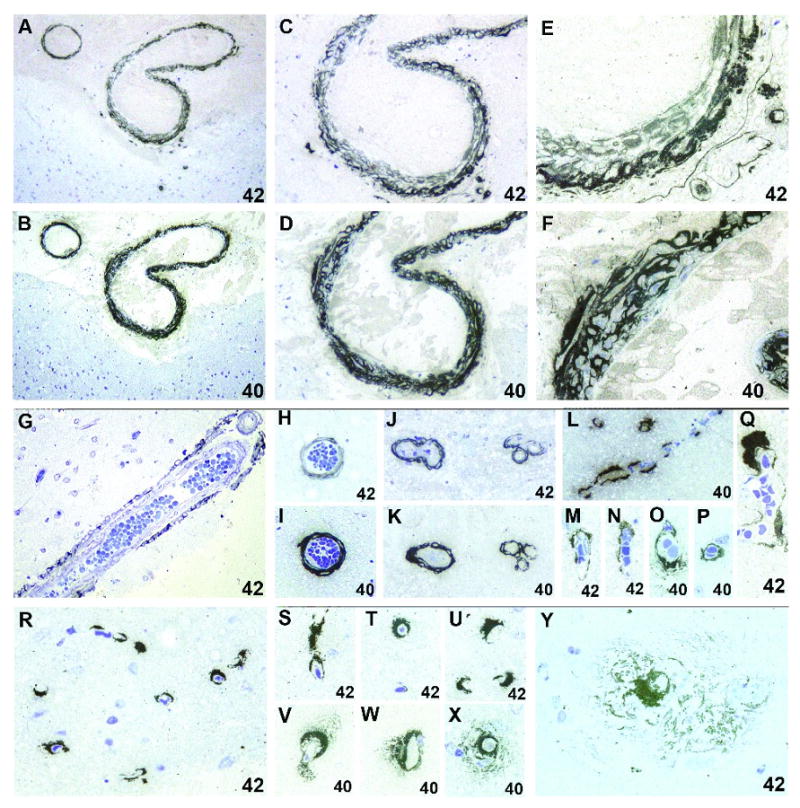
Leptomeningeal arteries show Aβ42 (A, C and E) and Aβ40 (B, D and F) deposits in the adventitia and among the vascular smooth muscle cells of the media. While Aβ40 deposits are distributed through most of the vessel wall (F), those of Aβ42 are preferentially located on their abluminal regions (E). In cortical vessels, Aβ42 (G, H and J) and Aβ40 (I and K) deposits are distributed in a pattern similar to that seen in leptomeningeal vessels. Note that pairs of Aβ42- and Aβ40-immunostained vessels shown in (A-F) are from the adjacent tissue sections, as are those in (H and I) and (J and K). In capillary walls, Aβ40 (L, O and P) and Aβ42 (M, N and Q) deposits are unevenly distributed, with a marked variation in thickness and focally abluminal excrescences. In a subset of Aβ-laden capillaries, there are Aβ42 (R-U and Y) and Aβ40 (V-X) deposits radiating from their walls into the surrounding neuropil (“pericapillary deposits”). Original magnification ×200 (A and B), ×400 (C, D and G-L), and ×1000 (E, F and M-Y). Cases SA7 (A-F), SA5 (G-P, X and Y), and SA4 (Q-W).
3.2. Aβ deposition in capillary walls
In AD brains, either Aβ40 or Aβ42 deposits or both in capillary walls were observed in 6 of 7 severe CAA and 1 of 9 mild CAA brains. These Aβ deposits were unevenly distributed, with a marked variation in thickness and focally abluminal excrescences (Figs. 1L-Q). No qualitative difference between Aβ40 and Aβ42 immunoreactivity was appreciated in capillary walls. In 5 of 7 severe CAA brains, a subset of Aβ-laden capillaries showed pericapillary deposits of either Aβ40 or Aβ42 or both (Figs. 1R-Y, Table 1). No pericapillary Aβ deposits were seen in any of the mild CAA brains. No Aβ-laden capillaries were found in any of the non-demented controls.
3.3. Aβ deposition in parenchymal plaques
In AD brains, both Aβ40 and Aβ42 plaques exhibiting diffuse or dense-core morphology were present in cortical parenchyma, Aβ42 plaques being more abundant on the adjacent tissue sections (Fig. 2A). On a scale of 1+ to 3+, the approximate extent of greater abundance of Aβ42 plaques compared to Aβ40 plaques was 1-level (ie, 2+ vs. 1+, or 3+ vs. 2+, data not shown). Aβ42 immunostaining intensity appeared in most instances greater than that of Aβ40 in parenchymal plaques on the adjacent tissue sections. Occasionally seen were Aβ plaques in close proximity to Aβ-laden arterioles (Fig. 2B) or capillaries (Fig. 2C). Diffuse Aβ42 plaques were observed in 1 of 5 old controls and none of the young controls. No Aβ40 plaques were seen in any of the non-demented controls.
Fig. 2.
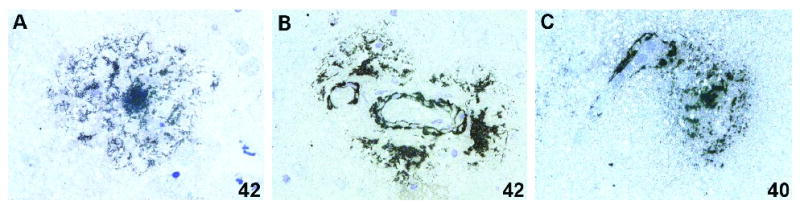
A dense-core Aβ42 plaque (A, case SA6). Aβ42 plaques are in close proximity to an Aβ42-laden arteriole (B, case MA8). An Aβ40 plaque is in close proximity to an Aβ40-laden capillary (C, case SA5). Original magnification ×1000 (A-C).
4. Discussion
By using immunogold silver staining for Aβ40 and Aβ42 on semithin LR White-embedded sections of cerebral cortical tissue from AD brains, we have demonstrated high-definition patterns of Aβ42 and Aβ40 immunoreactivity in vessel walls that are consistent with deposition within the vascular basement membranes. Our observation in resin-embedded tissue, together with the findings in other studies using paraffin immunohistochemistry [10,21] and immunoelectron microscopy [16], supports a hypothesis that Aβ deposits occur in cerebral perivascular drainage pathways. In vessel walls with advanced Aβ deposition, a predominance of Aβ40 over Aβ42 load was apparent with Aβ42 being preferentially located on their abluminal portion, a finding in agreement with those in other studies performed on paraffin sections of sporadic AD brains [5,22].
Experiments with soluble tracers injected into the brain parenchyma of live rodents and rabbits have indicated that perivascular pathways existed for interstitial fluid drainage along the basement membranes of capillaries and vascular smooth muscle cells [9,23]. In human brains, the ultrastructure of perivascular spaces has been described [10,24]. Neuronal tauopathy in the cholinergic nucleus basalis of Meynert in advancing age [25] may contribute to deficiencies in intracortical vasodilatation, arteriolar pulsation and consequently perivascular drainage of interstitial fluid. Degenerative changes such as increased perivascular collagen deposition and thickening of the vascular basement membranes [26] may also compromise cerebral perivascular interstitial fluid drainage. These aging-related functional and structural changes of intracortical vessels [26] might promote soluble Aβ to accumulate and be transformed into Aβ oligomers, insoluble Aβ intermediates and Aβ fibrils within vascular basement membranes and parenchymal plaques [27,28].
It remains controversial as to whether deposition of Aβ in capillary basement membranes occurs early on following deficiencies in Aβ elimination via cerebral perivascular drainage pathways in advancing age, and whether a subset of parenchymal Aβ plaques originate from Aβ-laden capillaries [28]. In the brains of patients carrying the Flemish amyloid precursor protein gene mutation (A692G), an abundance of vascular and parenchymal Aβ (primarily Aβ40) deposits, as well as large vasocentric dense-core Aβ40 plaques, were noted in the cerebral cortex [29]. In Tg2576 and PSAPP mouse models of AD, the majority of dense-core Aβ plaques were shown to be centered on capillaries [15]. In a study by Miyakawa et al [30] on parahippocampal tissue from 10 AD and 10 control brains, close spatial association of almost all dense-core Aβ40 plaques with capillaries was shown on double immunofluorescent staining for Aβ and collagen IV. On electron microscopy, they found that some degenerated capillaries containing amyloid fibrils in their walls were localized in close proximity to dense-core amyloid plaques [30]. An immunoelectron microscopic study of AD brains by Yamaguchi et al [16] has suggested that the capillary basement membrane was one of the predilection sites for early Aβ deposition. Also in this report, pericapillary Aβ deposits were shown extending from the outer basement membranes of Aβ-laden capillaries [16]. Preston et al [10] reported that diffuse Aβ plaques were seen surrounding capillaries, while dense-core Aβ plaques were not associated with blood vessels. In the same study, confocal microscopy revealed pericapillary deposits of amyloid on thioflavin-S staining in continuity with amyloid in the capillary walls [10]. On the other hand, by using double immunostaining for Aβ and collagen IV on the entorhinal cortex of two AD patients, Kawai et al [31] suggested that spatial association between capillaries and both diffuse and dense-core Aβ plaques occurred by chance, depending on the size of Aβ plaques. Aβ-laden capillaries originally associated with diffuse plaques may progressively be loaded with Aβ and undergo degeneration, as diffuse plaques evolve into dense-core plaques [30]. Therefore, there would be a higher probability for identifying Aβ-laden capillaries within diffuse Aβ plaques than in dense-core Aβ plaques on immunohistologic sections studied either at the light or electron microscopic level.
Pericapillary Aβ deposits found in our present study may represent a histopathologic intermediate in the course of development from capillary Aβ deposits to diffuse Aβ plaques. It has been shown that the Aβ40/42 ratio of Aβ-laden capillaries is lower than that of affected arteries but comparable to that found in Aβ plaques [32]. As a result of impaired drainage along cerebral perivascular pathways in old age, the less soluble Aβ42 is unable to drain as far along the clearance channels as the more soluble Aβ40. The Aβ42 thus accumulates, becomes insoluble, and predominantly deposits in brain parenchyma in the form of plaques. In contrast, the Aβ40 deposits preferentially in the basement membranes of cortico-leptomeningeal vessels to form CAA [28]. This working hypothesis would explain the consistent findings in sporadic AD brains of a predominance of Aβ40 over Aβ42 load in vessel walls and the opposite pattern in parenchymal plaques [2,5,22], which were also shown in our present qualitative analysis.
Regarding technical aspects, the immunogold silver staining of semithin resin-embedded tissue sections was useful as demonstrated in our present study, particularly in evaluating the cerebral vasculature, because the localization of protein markers of interest can be correlated more precisely with high-definition histology compared to paraffin immunohistochemistry. Immunoreactive substances (Aβ40 and Aβ42) can be seen in relation to vascular medial smooth muscle cells or capillary endothelium. Use of hydrophilic acrylic resins that can relatively preserve the antigenicity of tissue (eg, LR White resin) complemented by careful optimization of the fixation, dehydration, embedment and staining protocols is critical for high-definition immunohistologic studies on semithin tissue sections. We obtained consistently satisfactory results of post-embedding immunogold silver staining following full tissue dehydration in absolute ethanol and embedment in medium-grade LR White resin by thermal polymerization. Nonetheless, other investigators have proposed different modified methods shown to improve the antigenicity and ultrastructure of LR White-embedded tissue. For instance, Yamaguchi et al [16] pursued partial dehydration of tissue in 70% ethanol before embedment in hard-grade LR White resin by chemical catalytic polymerization at 0°C, while Sakai et al [33] treated tissue with 1% tannic acid prior to partial dehydration in 70% ethanol containing 2% phosphotungstic acid, followed by embedment in LR White resin by thermal polymerization. In our immunogold silver staining protocols for Aβ40 and Aβ42 on semithin LR White-embedded tissue sections we successfully applied 90% formic acid pretreatment at 1:200 dilution for 10 min to unmask antigens, in contrast to standard pretreatment with neat formic acid solution for paraffin Aβ immunohistochemistry [34]. For other protein markers, different antigen retrieval techniques may be needed on resin-embedded tissue sections.
The relatively small field size of semithin resin-embedded tissue sections that is subject to sampling errors may be considered a drawback to the study of human brains; nonetheless, examination of multiple serial sections on the same slide is feasible. When this method is applied to immunohistologic analysis of rodent brains in animal models of neurological diseases [15], sampling errors are of lesser concern. The presence of Aβ-laden blood vessels in 6 of 7 severe CAA brains but in only 2 of 9 mild CAA brains in our present study can probably be explained by lower detection sensitivity of this method compared to much larger and thicker tissue sections used in paraffin immunohistochemistry. By using 14 micrometer-thick paraffin sections of AD brains, Alonzo et al [5] found no significant difference in the proportion of Aβ40- or Aβ42-immunostained vessels between mild and severe CAA (classified according to the Vonsattel system, which qualitatively evaluates the degree of CAA involvement of individual vessels [19]). We found Aβ-laden capillaries in 6 of 7 severe CAA brains but in only 1 of 9 mild CAA brains. This observation differed somewhat from a report by Thai et al [12] showing that the degree of CAA severity did not predict the presence of capillary CAA. Nonetheless, CAA with capillary involvement may be pathogenetically distinct from that lacking capillary involvement, as only the former was found to correlate with the apolipoprotein E ε4 allele [12].
In summary, our study demonstrated high-definition characteristics of Aβ40 and Aβ42 deposition in the cerebral vasculature. The patterns of Aβ42 and Aβ40 immunoreactivity in blood vessel walls suggest that Aβ deposits occur in the vascular basement membranes along cerebral perivascular drainage pathways, extending from cortical capillaries to leptomeningeal arteries. The presence of pericapillary Aβ deposits supports the notion that a subset of Aβ plaques originate from Aβ-laden capillaries. As significant heterogeneity exists among individuals with AD diagnosis [35], this proposed mechanistic cascade of cerebral microvascular and parenchymal deposition of Aβ may be applied only to those individuals whose brains exhibit preferential capillary involvement such as type-1 CAA described by Thai et al [12]. Acknowledging that sporadic AD is pathogenetically heterogeneous, we propose that the validity of classifying CAA based on the presence or absence of capillary involvement be taken into account when studying the pathogenesis of this complex disease.
Acknowledgments
This work was supported by the United States National Institute on Aging grants P50 AG16570 and P01AG12435, and the Dalijit S. & Elaine Sarkaria Chair in Diagnostic Medicine held by H. V. Vinters, as well as an interdisciplinary research fellowship in neuroAIDS in the United States National Institute of Health grant MH81482 to V. Soontornniyomkij.
List of nonstandard abbreviations
Used in the manuscript
- Aβ
β-amyloid
- AD
Alzheimer's disease
- CAA
cerebral Aβ angiopathy
- SA
brains of AD patients with severe CAA
- MA
brains of AD patients with mild CAA
- OC
brains of non-demented old controls
- YC
brains of non-demented young controls
Used exclusively in the table
- F
female
- M
male
- RO
right occipital
- LO
left occipital
- RF
right frontal
- “+”
presence
- “−”
absence
- PD
pericapillary deposits
Footnotes
Disclosure: There are no real or potential conflicts of interest for any of the authors to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Virawudh Soontornniyomkij, Email: vsoontor@ucsd.edu.
Cecilia Choi, Email: cchoi@mednet.ucla.edu.
Justine Pomakian, Email: jpomakian@mednet.ucla.edu.
Harry V. Vinters, Email: hvinters@mednet.ucla.edu.
References
- 1.Ellis RJ, Olichney JM, Thai LJ, Mirra SS, Morris JC, Beekly D, Heyman A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology. 1996;46:1592–6. doi: 10.1212/wnl.46.6.1592. [DOI] [PubMed] [Google Scholar]
- 2.Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol. 2008;18:253–66. doi: 10.1111/j.1750-3639.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gearing M, Mori H, Mirra SS. Abeta-peptide length and apolipoprotein E genotype in Alzheimer's disease. Ann Neurol. 1996;39:395–9. doi: 10.1002/ana.410390320. [DOI] [PubMed] [Google Scholar]
- 4.Mann DM, Iwatsubo T, Pickering-Brown SM, Owen F, Saido TC, Perry RH. Preferential deposition of amyloid beta protein (Abeta) in the form Abeta40 in Alzheimer's disease is associated with a gene dosage effect of the apolipoprotein E E4 allele. Neurosci Lett. 1997;221:81–4. doi: 10.1016/s0304-3940(96)13294-8. [DOI] [PubMed] [Google Scholar]
- 5.Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol. 1998;57:353–9. doi: 10.1097/00005072-199804000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 2009;118:103–13. doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2009;106:1261–6. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. 2004;45:545–52. doi: 10.1016/j.neuint.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, Weller RO. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008;34:131–44. doi: 10.1111/j.1365-2990.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 10.Preston SD, Steart PV, Wilkinson A, Nicoll JA, Weller RO. Capillary and arterial cerebral amyloid angiopathy in Alzheimer's disease: defining the perivascular route for the elimination of amyloid beta from the human brain. Neuropathol Appl Neurobiol. 2003;29:106–17. doi: 10.1046/j.1365-2990.2003.00424.x. [DOI] [PubMed] [Google Scholar]
- 11.Attems J, Jellinger KA. Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology-a pilot study. Acta Neuropathol. 2004;107:83–90. doi: 10.1007/s00401-003-0796-9. [DOI] [PubMed] [Google Scholar]
- 12.Thal DR, Ghebremedhin E, Rüb U, Yamaguchi H, Del Tredici K, Braak H. Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2002;61:282–93. doi: 10.1093/jnen/61.3.282. [DOI] [PubMed] [Google Scholar]
- 13.Stopa EG, Butala P, Salloway S, et al. Cerebral cortical arteriolar angiopathy, vascular beta-amyloid, smooth muscle actin, Braak stage, and APOE genotype. Stroke. 2008;39:814–21. doi: 10.1161/STROKEAHA.107.493429. [DOI] [PubMed] [Google Scholar]
- 14.Singhrao S, Cole G, Henderson WJ, Newman GR. LR White embedding allows a multi-method approach to the analysis of brain tissue from patients with Alzheimer's disease. Histochem J. 1990;22:257–68. doi: 10.1007/BF01387181. [DOI] [PubMed] [Google Scholar]
- 15.Kumar-Singh S, Pirici D, McGowan E, et al. Dense-core plaques in Tg2576 and PSAPP mouse models of Alzheimer's disease are centered on vessel walls. Am J Pathol. 2005;167:527–43. doi: 10.1016/S0002-9440(10)62995-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamaguchi H, Yamazaki T, Lemere CA, Frosch MP, Selkoe DJ. Beta amyloid is focally deposited within the outer basement membrane in the amyloid angiopathy of Alzheimer's disease. An immunoelectron microscopic study. Am J Pathol. 1992;141:249–59. [PMC free article] [PubMed] [Google Scholar]
- 17.Gearing M, Mirra SS, Hedreen JC, Sumi SM, Hansen LA, Heyman A. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part X. Neuropathology confirmation of the clinical diagnosis of Alzheimer's disease. Neurology. 1995;45:461–6. doi: 10.1212/wnl.45.3.461. [DOI] [PubMed] [Google Scholar]
- 18.Soontornniyomkij V, Lynch MD, Mermash S, Pomakian J, Badkoobehi H, Clare R, Vinters HV. Cerebral microinfarcts associated with severe cerebral beta-amyloid angiopathy. Brain Pathol. 2010;20:459–67. doi: 10.1111/j.1750-3639.2009.00322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vinters HV, Vonsattel J. Neuropathologic features and grading of Alzheimer's related and sporadic CAA. In: Verbeek MM, de Waal RMW, Vinters HV, editors. Cerebral amyloid angiopathy in Alzheimer's disease and related disorders. Dordrecht, The Netherlands: Kluver Academic Publishers; 2000. pp. 137–55. [Google Scholar]
- 20.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 21.Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol. 1998;153:725–33. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haglund M, Kalaria R, Slade JY, Englund E. Differential deposition of amyloid beta peptides in cerebral amyloid angiopathy associated with Alzheimer's disease and vascular dementia. Acta Neuropathol (Berl) 2006;111:430–5. doi: 10.1007/s00401-006-0054-z. [DOI] [PubMed] [Google Scholar]
- 23.Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009;117:1–14. doi: 10.1007/s00401-008-0457-0. [DOI] [PubMed] [Google Scholar]
- 24.Zhang ET, Inman CB, Weller RO. Interrelationships of the pia mater and the perivascular (Virchow-Robin) spaces in the human cerebrum. J Anat. 1990;170:111–23. [PMC free article] [PubMed] [Google Scholar]
- 25.Mesulam M, Shaw P, Mash D, Weintraub S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol. 2004;55:815–28. doi: 10.1002/ana.20100. [DOI] [PubMed] [Google Scholar]
- 26.Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol. 2001;64:575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 27.Roher AE, Kuo YM, Potter PE, et al. Cortical cholinergic denervation elicits vascular A beta deposition. Ann N Y Acad Sci. 2000;903:366–73. doi: 10.1111/j.1749-6632.2000.tb06388.x. [DOI] [PubMed] [Google Scholar]
- 28.Nicoll JA, Yamada M, Frackowiak J, Mazur-Kolecka B, Weller RO. Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer's disease. Pro-CAA position statement. Neurobiol Aging. 2004;25:589–97. doi: 10.1016/j.neurobiolaging.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 29.Kumar-Singh S, Cras P, Wang R, et al. Dense-core senile plaques in the Flemish variant of Alzheimer's disease are vasocentric. Am J Pathol. 2002;161:507–20. doi: 10.1016/S0002-9440(10)64207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyakawa T, Kimura T, Hirata S, Fujise N, Ono T, Ishizuka K, Nakabayashi J. Role of blood vessels in producing pathological changes in the brain with Alzheimer's disease. Ann N Y Acad Sci. 2000;903:46–54. doi: 10.1111/j.1749-6632.2000.tb06349.x. [DOI] [PubMed] [Google Scholar]
- 31.Kawai M, Cras P, Perry G. Serial reconstruction of beta-protein amyloid plaques: relationship to microvessels and size distribution. Brain Res. 1992;592:278–82. doi: 10.1016/0006-8993(92)91686-9. [DOI] [PubMed] [Google Scholar]
- 32.Thal DR, Griffin WS, de Vos RA, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol. 2008;115:599–609. doi: 10.1007/s00401-008-0366-2. [DOI] [PubMed] [Google Scholar]
- 33.Sakai Y, Hosaka M, Hira Y, Watanabe T. Addition of phosphotungstic acid to ethanol for dehydration improves both the ultrastructure and antigenicity of pituitary tissue embedded in LR White acrylic resin. Arch Histol Cytol. 2005;68:337–47. doi: 10.1679/aohc.68.337. [DOI] [PubMed] [Google Scholar]
- 34.Vinters HV, Pardridge WM, Secor DL, Ishii N. Immunohistochemical study of cerebral amyloid angiopathy. II. Enhancement of immunostaining using formic acid pretreatment of tissue sections. Am J Pathol. 1988;133:150–62. [PMC free article] [PubMed] [Google Scholar]
- 35.Armstrong RA, Lantos PL, Cairns NJ. Overlap between neurodegenerative disorders. Neuropathology. 2005;25:111–24. doi: 10.1111/j.1440-1789.2005.00605.x. [DOI] [PubMed] [Google Scholar]