

Summary
Background
Innovative prevention strategies for HIV-1 transmission are urgently needed. PRO2000 vaginal gel was efficacious against HIV-1 transmission in studies in macaques; we aimed to assess efficacy and safety of 2% and 0·5% PRO2000 gels against vaginal HIV-1 transmission in women in sub-Saharan Africa.
Methods
Microbicides Development Programme 301 was a phase 3, randomised, double-blind, parallel-group trial, undertaken at 13 clinics in South Africa, Tanzania, Uganda, and Zambia. We randomly assigned sexually active women, aged 18 years or older (≥16 years in Tanzania and Uganda) without HIV-1 infection in a 1:1:1 ratio to 2% PRO2000, 0·5% PRO2000, or placebo gel groups for 52 weeks (up to 104 weeks in Uganda). Randomisation was done by computerised random number generator. Investigators and participants were masked to group assignment. The primary efficacy outcome was incidence of HIV-1 infection before week 52, which was censored for pregnancy and excluded participants without HIV-1 follow-up data or with HIV-1 infection at enrolment. HIV-1 status was established by rapid tests or ELISA at screening at 12 weeks, 24 weeks, 40 weeks, and 52 weeks, and confirmed in a central reference laboratory. The primary safety endpoint was an adverse event of grade 3 or worse. Use of 2% PRO2000 gel was discontinued on Feb 14, 2008, on the recommendation of the Independent Data Monitoring Committee because of low probability of benefit. This trial is registered at http://isrctn.org, number ISRCTN 64716212.
Findings
We enrolled 9385 of 15 818 women screened. 2591 (95%) of 2734 participants enrolled to the 2% PRO2000 group, 3156 (95%) of 3326 in the 0·5% PRO2000 group, and 3112 (94%) of 3325 in the placebo group were included in the primary efficacy analysis. Mean reported gel use at last sex act was 89% (95% CI 86–91). HIV-1 incidence was much the same between groups at study end (incidence per 100 woman-years was 4·5 [95% CI 3·8–5·4] for 0·5% PRO2000 vs 4·3 [3·6–5·2] for placebo, hazard ratio 1·05 [0·82–1·34], p=0·71), and at discontinuation (4·7 [3·8–5·8] for 2% PRO2000 gel, 3·9 [3·0–4·9] for 0·5% PRO2000 gel, and 3·9 [3·1–5·0] for placebo gel). Incidence of the primary safety endpoint at study end was 4·6 per 100 woman-years (95% CI 3·9–5·4) in the 0·5% PRO2000 group and 3·9 (3·2–4·6) in the placebo group; and was 4·5 (3·7–5·5) in the 2% PRO2000 group at discontinuation.
Interpretation
Although safe, 0·5% PRO2000 and 2% PRO2000 are not efficacious against vaginal HIV-1 transmission and are not indicated for this use.
Funding
UK Department for International Development, UK Medical Research Council, European and Developing Countries Clinical Trials Partnership, International Partnership for Microbicides, and Endo Pharmaceuticals Solutions.
Introduction
With an estimated 1·9 million new HIV-1 infections in sub-Saharan Africa in 2008, innovative strategies for prevention are urgently needed.1 Women in Africa are disproportionately affected by HIV-1 and many are unable or unwilling to negotiate condom use, want to conceive, or both. Vaginal microbicides are a potential method for prevention of HIV-1 transmission that is controlled by the woman. After several disappointing trials,2–5 the phase 2/2b trial, HPTN035,6 reported that the vaginal microbicide 0·5% PRO2000 led to a non-significant reduction in HIV-1 incidence of 30% (hazard ratio 0·7, 95% CI 0·46–1·08, p=0·10) compared with placebo gel.
We report the Microbicides Development Programme (MDP) 301 trial, which assessed the efficacy and safety of 0·5% and 2% PRO2000 gels compared with hydroxyethylcellulose placebo gel7 for prevention of vaginally acquired HIV-1 infection. This trial was done by the MDP, a collaborative partnership of institutions in Africa and Europe. PRO2000 is a synthetic naphthalene sulphonate polymer of around 5 kDa molecular weight, with antiviral activity against HIV-1 and other sexually transmitted infections as shown in laboratory studies and in studies in animals, including macaque challenge experiments with X4 and R5 simian–human immunodeficiency virus,8–13 and a favourable safety profile in phase 1/2 clinical trials.14–17 We aimed to assess the efficacy and safety of PRO2000 gels for prevention of vaginal HIV-1 transmission in sub-Saharan Africa.
Methods
Participants and design
MDP301 was a phase 3, randomised, double-blind, parallel-group trial. Full details of trial design, sample size, research sites, study populations, study conduct including the randomisation and masking, and data underpinning the sample size assumptions, have been reported.18 We did a social science substudy to assess accuracy of behavioural and adherence data, which is described in detail elsewhere.19,20
Participants were enrolled at 13 clinics, which were managed by six research centres, in Africa (three in South Africa, one in Tanzania, one in Uganda, and one in Zambia). Eligible women were 18 years or older (≥16 years in Tanzania and Uganda); did not have HIV-1 infection at screening and were willing to be tested for HIV-1 infection and receive the result; were willing to have regular speculum examinations and urinary pregnancy tests; were willing to use gel as instructed; were likely to be sexually active; were willing to receive health education about condoms; and were willing and able to give informed consent. Women were not eligible if they were unable or unwilling to provide a reliable method of contact; were likely to move out of the area within 12 months; were likely to have sex more than 14 times a week on a regular basis (a regulatory requirement was that no more than 60 applicators were to be dispensed at every 4 weekly visit); used spermicides regularly; were pregnant or within 6 weeks post partum; had a severe clinical or laboratory abnormality; needed referral for assessment of a suspicious cervical lesion; had received treatment to the cervix or other gynaecological procedure within 30 days of enrolment; were allergic to latex; or were participating or had participated in another clinical trial that was likely to affect the primary efficacy endpoint within 30 days before enrolment.
The protocol was approved by local and national ethics committees, in all participating countries and in the UK. Authorisation was obtained from the national regulatory authority in all participating countries and the US Food and Drug Administration. Participants indicated their consent by signature or witnessed thumbprint.
Randomisation and masking
Participants were randomly assigned in a 1:1:1 ratio to 2% PRO2000, 0·5% PRO2000, or placebo groups. Participants were randomly assigned on the basis of lists that were created with randomised permuted blocks of varying size, for each of the 13 clinics, by an independent statistician with a computerised random number generator, containing unique trial numbers matched to nine sets of study product codes. Site pharmacists dispensed gel in identical applicators on the basis of the trial number and the assigned study product codes on the clinic randomisation list. No other site personnel had access to the list. Success of masking was not formally addressed. At enrolment, women were assigned a unique trial number selected sequentially from the clinic trial register. Only statisticians responsible for preparation of the Independent Data Monitoring Committee reports and essential manufacturing and distribution staff had access to the list matching study product codes to gel.
Procedures
Visits were scheduled every 4 weeks for 52 weeks (up to 104 weeks in Uganda to provide long-term safety data). Gel was dispensed in packs of ten prefilled single dose applicators (maximum of 60 applicators in ten packs per visit), after a negative urinary pregnancy test. Women were instructed to apply gel within an hour before sexual intercourse. They were counselled to use condoms during all sex acts and received unrestricted supplies of free condoms at the research clinics. Gel supply was interrupted if a participant had a positive pregnancy test and could be resumed after a negative pregnancy test. At every 4 week visit, women were asked about gel and condom use at the most recent sex act and returned used and unused applicators, which were were counted and recorded for assessments of adherence.
HIV-1 status was assessed at 12 weeks, 24 weeks, 40 weeks, and 52 weeks (up to 104 weeks in Uganda) and when gel supply was interrupted or discontinued because of a positive pregnancy test. A clinical interview and pelvic examination to report genital and non-genital adverse events were also done at these visits, at week 4, and if a pregnancy was diagnosed. Adverse events were defined by ICH guidelines and MedDRA coded. Solicited genital symptoms and signs were non-menstrual bleeding, genital sores or ulcers, genital discomfort (itching, burning, or dryness), and external or internal epithelial disruption, genital erythema, and genital oedema.
Routine haematology and biochemistry tests were done for the first 500 participants enrolled in centres in Durban and Johannesburg, South Africa, and all 840 participants in Uganda at screening and at 12 weeks, 24 weeks, and 52 weeks (and 104 weeks or at final visit in Uganda). Additionally, a plasma sample was obtained from these participants at the final visit for PRO2000 analysis.
To confirm HIV-1 status, serum was obtained up to 6 weeks before enrolment, at enrolment, and then at weeks 4, 12, 24, 40, and 52 (and week 72 and 104 in Uganda); buffy coat was obtained at enrolment, and at weeks 24, 40, and 52 (and week 104 in Uganda). The HIV-1 testing algorithm comprised parallel HIV-1 rapid tests at the clinics, with discordant or positive tests after enrolment triggering ELISA testing at local laboratories and confirmation at a central laboratory in South Africa. A second serum sample was obtained at the subsequent visit after a first positive rapid test result. The central laboratory analysed samples from the visit at which a positive rapid test result was obtained and from all previous visits at which samples were obtained. This analysis allowed detection of seroconversion to be established as at or before enrolment.
The algorithm was designed to confirm HIV-1 infection on the basis of two separate samples, with two different methods of diagnosis. Serum samples were tested for HIV-1 antibodies with Abbott AxSYM HIV Ag/Ab Combo (fourth-generation ELISA; Wiesbaden, Germany), Bio-Rad Genetic Systems HIV-1 ELISA (third-generation ELISA; Redmond, WA, USA), and Genetic Systems HIV-1 Western Blot (Redmond, WA, USA) assays. We used Biomerieux Vironostika HIV-1 antigen ELISA (Boxtel, Netherlands) as a confirmatory assay for p24 testing. Buffy coat samples were tested with the Roche qualitative DNA PCR Version 1.5 assay (Roche Diagnostic Systems, Branchburg, NJ, USA). The Roche COBAS Amplicor HIV-1 Monitor (Roche Diagnostic Systems) was used for the detection of HIV-1 RNA if the buffy coat specimen was not satisfactory.
Endpoints
The primary efficacy endpoint was HIV-1 infection, confirmed by the central reference laboratory, in participants who were confirmed without HIV-1 infection at enrolment. Secondary efficacy endpoints were acquisition of herpes simplex virus type 2 (HSV-2) infection by participants who were HSV-2 seronegative at enrolment, which was established serologically and confirmed by the central laboratory at 40 weeks and 52 weeks; or presence of Neisseria gonorrhoeae or Chlamydia trachomatis, which was established by a positive nucleic acid amplification assay at 24 weeks.
The primary safety endpoint was a grade 3 (severe) or worse clinical or laboratory adverse event, irrespective of relation to trial gel. Secondary safety endpoints were reported local toxic effects (any grade of genital itching, burning, internal epithelial disruption, internal erythema, or internal oedema) and systemic toxic effects (any increase in grade from baseline in routine laboratory variables). We systematically assessed these outcomes because of a probable or known association with PRO2000 when administered vaginally or systemically in previous trials.21,22
The primary efficacy outcome was originally designed to be measured at 40 weeks, which changed to 52 weeks after commencement of the trial.18
Discontinuation
The MDP301 trial protocol stated that the Independent Data Monitoring Committee could recommend early termination of the trial “if there was proof beyond reasonable doubt that one of the trial interventions is clearly indicated or clearly contraindicated in terms of a net difference in seroincidence or adverse events”. The 2% PRO2000 gel was discontinued on Feb 14, 2008, after a review by the committee on Feb 8, 2008 of available data up to Jan 15, 2008. The committee advised there was little chance of 2% PRO2000 gel showing benefit given the planned sample size and postulated effect size. However, the conditional power for significant benefit from the 0·5% PRO2000 dose, based on the original sample size assumptions, was sufficiently high to warrant trial continuation. We report data for the 0·5% PRO2000 and placebo groups to study end, and data for the 0·5% PRO2000, 2% PRO2000, and placebo groups with data up until discontinuation on Feb 14, 2008.
Statistical analysis
MDP301 was designed with 80% power to detect a 35% reduction in HIV-1 incidence (90% for a 40% reduction), assuming an incidence in the placebo group of 4·0 per 100 woman-years. The assumptions made for incidence and loss to follow-up (we predicted 3300 participants would be needed per group, assuming 20% loss of woman-years of follow-up) were derived from data in previous cohort studies.18
The primary efficacy analysis comprised all enrolled participants, excluding those with HIV-1 infection at enrolment, those without follow-up data for HIV infection, and with censoring at 52 weeks (plus 6 week window for final visit) and while gel use was discontinued because of pregnancy. For women who did not have HIV-1 infection when gel use was resumed after pregnancy, additional time was added to the woman-years of follow-up for the primary efficacy analysis.
A second efficacy analysis was done with the same criteria as the primary efficacy analysis, but without censoring for pregnancy and using all follow-up data. We assessed secondary efficacy endpoints with two further analyses, censoring at 24 and 40 weeks (plus 4-week window) from enrolment, excluding those with HIV-1 infection at enrolment and censoring for pregnancy.
We did two planned subgroup analyses of patients, including tests for interaction. One subgroup analysis was stratified by research centre and one was done with postrandomisation data and categorised women according to the consistency of gel use, with the expectation that efficacy against HIV-1 transmission would be greater in consistent users than it was in sporadic users. Gel use was predefined as consistent if women reported use during the last sex act at 12 (92%) or more of 13 visits, or at least 92% of visits attended if fewer than 13; returned at least one used applicator to support their answer when appropriate; and attended at least seven of the expected 13 visits (unless they became pregnant or were infected with HIV-1 during follow-up).
In patients with HIV-1 seroconversion, the date of detection of HIV-1 infection was established by an endpoint committee. Woman-years of observation were censored at the date of seroconversion, estimated by the midpoint between the last negative test and date of detection, or at the last HIV-1-negative test for patients who did not become infected.
We analysed the primary efficacy endpoint as time-to-event, and groups were compared by use of hazard ratios (HRs) relative to the placebo group, 95% CI, and p values, which were obtained by Cox proportional hazards regression and stratified by clinic. We analysed secondary efficacy endpoints as binary outcomes by logistic regression, the proportion infected with HSV-2 at week 40 and week 52 for those HSV-2 negative at baseline; and cross-sectional prevalence of N gonorrhoeae and C trachomatis for all patients tested at week 24.
All women with follow-up clinical data were included in the safety analyses. Safety endpoints were analysed as time-to-first-event and groups compared as for the efficacy analysis. All analyses for the 2% PRO2000 group were undertaken with all three groups, and were censored when the 2% PRO2000 gel was discontinued on Feb 14, 2008. All statistical tests were two-sided and all analyses were done with Stata version 10.1.
This trial is registered at http://isrctn.org, number ISRCTN 64716212.
Role of the funding source
MDP301 was supported by the UK Department for International Development (DFID), the UK Medical Research Council (MRC), the European and Developing Countries Clinical Trials Partnership, and the International Partnership for Microbicides. The sponsors of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. Endo Pharmaceuticals Solutions donated the study gels, provided regulatory support, and participated in design and management of the study. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Figure 1 shows the trial profile and table 1 shows patient characteristics at baseline. We screened women for HIV-1 infection between September, 2005 and August, 2008, and enrolled 9404 participants; 19 of these participants enrolled twice, and data for their second enrolment were excluded from all analyses, including one seroconversion during the participant's second enrolment. 2591 (95%) of 2734 participants randomly assigned to the 2% PRO2000 gel group were eligible for inclusion in the primary efficacy analysis, 3156 (95%) of 3326 were eligible in the 0·5% PRO2000 gel group, and 3112 (94%) of 3325 were eligible in the placebo gel group.
Figure 1.
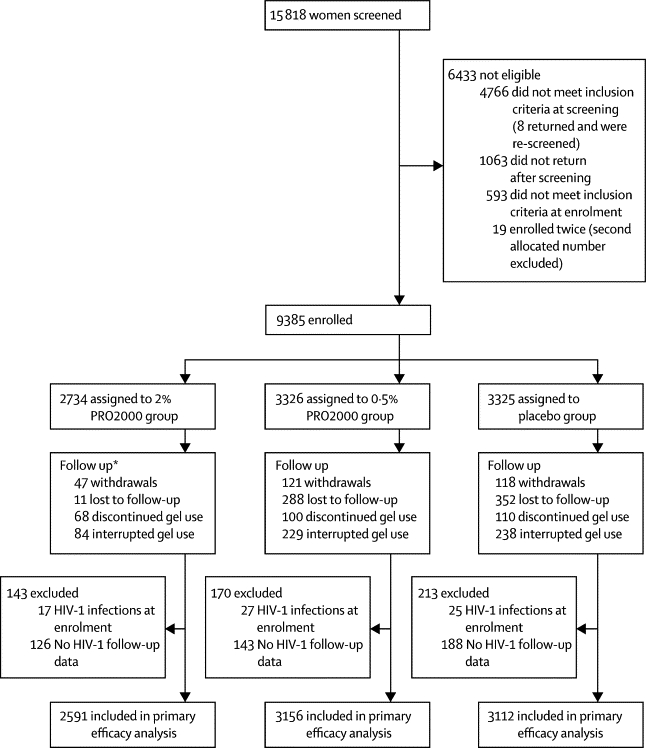
Trial profile
*1564 participants were in follow-up at Feb 14, 2008.
Table 1.
Baseline characteristics
| 2% PRO2000*(n=2734) | 0·5% PRO2000 (n=3326) | Placebo (n=3325) | ||
|---|---|---|---|---|
| Demographic | ||||
| Age (years) | ||||
| 15–24 | 1022/2734 (37%) | 1249/3326 (38%) | 1267/3324 (38%) | |
| 25–34 | 955/2734 (35%) | 1076/3326 (32%) | 1096/3324 (33%) | |
| 35–44 | 532/2734 (19%) | 723/3326 (22%) | 702/3324 (21%) | |
| ≥45 | 225/2734 (8%) | 278/3326 (8%) | 259/3324 (8%) | |
| Education | ||||
| None | 177/2734 (6%) | 240/3326 (7%) | 233/3324 (7%) | |
| Primary | 1927/2734 (70%) | 2309/3326 (69%) | 2271/3324 (68%) | |
| Secondary or higher | 630/2734 (23%) | 777/3326 (23%) | 820/3324 (25%) | |
| Medical history (ever) | ||||
| Non-menstrual bleeding | 266/2734 (10%) | 339/3326 (10%) | 307/3325 (9%) | |
| Sores or ulcers | 294/2734 (11%) | 357/3326 (11%) | 342/3325 (10%) | |
| Unusual genital discomfort | 656/2734 (24%) | 786/3326 (24%) | 820/3325 (25%) | |
| Unusual genital discharge | 686/2734 (25%) | 796/3326 (24%) | 820/3325 (25%) | |
| Pain during sex | 140/2734 (5%) | 181/3326 (5%) | 163/3325 (5%) | |
| Other genital disorders | 22/2734 (1%) | 21/3326 (1%) | 28/3325 (1%) | |
| Positive laboratory results | ||||
| Chlamydia trachomatis | 202/2698 (7%) | 256/3293 (8%) | 266/3295 (8%) | |
| Neisseria gonorrhoeae | 93/2698 (3%) | 110/3293 (3%) | 119/3295 (4%) | |
| Herpes simplex virus type 2 seropositive | 1622/2725 (60%) | 2029/3312 (61%) | 1982/3311 (60%) | |
| Syphilis† | 106/2726 (4%) | 116/3300 (4%) | 137/3304 (4%) | |
| Trichomonas vaginalis | 268/2723 (10%) | 312/3314 (9%) | 314/3311 (9%) | |
| Behavioural | ||||
| Effective contraception‡ | 1514/2732 (55%) | 1850/3326 (56%) | 1873/3325 (56%) | |
| Sex acts in previous week | ||||
| 0 | 542/2732 (20%) | 627/3326 (19%) | 610/3325 (19%) | |
| 1 | 433/2732 (16%) | 573/3326 (17%) | 534/3325 (17%) | |
| ≥2 | 1754/2732 (64%) | 2124/3326 (64%) | 2178/3325 (64%) | |
| Missing/unknown | 3/2732 (<1%) | 2/3326 (<1%) | 3/3325 (<1%) | |
| Partners in previous week (median [IQR]) | 1 (1–1) | 1 (1–1) | 1 (1–1) | |
| Condom use at last sex act | 1500/2732 (55%) | 1894/3326 (57%) | 1826/3325 (55%) | |
| Anal sex in previous 4 weeks | 28/2732 (1%) | 42/3326 (1%) | 31/3325 (1%) | |
Data are n/n of participants with reported data (%) unless otherwise stated.
2% PRO2000 gel was discontinued Feb 14, 2008.
Active syphilis was defined on the basis of rapid plasma reagin titre.
Sterilisation, intrauterine contraceptive device, or use of injected, implanted, or oral contraception.
Table 2 shows HIV-1 incidence in the 0·5% PRO2000 gel and placebo gel groups at study end, and for all gel groups at censoring on Feb 14, 2008. In the primary efficacy analysis, incidence of HIV-1 did not differ between groups at discontinuation of 2% PRO2000 or study end (table 2). Equally, incidence of HIV-1 did not differ between groups in the second analysis that did not censor for pregnancy (table 2).
Table 2.
Primary efficacy outcome: HIV-1 incidence in the primary and secondary efficacy analyses
|
End of study |
Censored at 2% PRO2000 gel discontinuation, Feb 14, 2008 |
||||
|---|---|---|---|---|---|
| 0·5% PRO2000 (n=3326) | Placebo (n=3325) | 2% PRO2000 (n=2734) | 0·5% PRO2000 (n=2732) | Placebo (n=2722) | |
| Primary efficacy analysis* | |||||
| Participants | 3156 | 3112 | 2591 | 2587 | 2543 |
| Woman-years of follow-up | 2873 | 2836 | 1741 | 1732 | 1717 |
| Seroconversions | 130 | 123 | 82 | 67 | 67 |
| Incidence† | 4·5 (3·8–5·4) | 4·3 (3·6–5·2) | 4·7 (3·8–5·8) | 3·9 (3·0–4·9) | 3·9 (3·1–5·0) |
| Hazard ratio | 1·05 (0·82–1·34) | 1 | 1·21 (0·88–1·68) | 0·99 (0·70–1·39) | 1 |
| p value | 0·71 | .. | 0·24 | 0·94 | .. |
| Second efficacy analysis‡ | |||||
| Participants | 3156 | 3112 | 2591 | 2587 | 2543 |
| Woman-years of follow-up | 3133 | 3099 | 1847 | 1846 | 1832 |
| Seroconversions | 145 | 143 | 86 | 70 | 77 |
| Incidence† | 4·6 (3·9–5·4) | 4·6 (3·9–5·4) | 4·7 (3·8–5·8) | 3·8 (3·0–4·8) | 4·2 (3·4–5·3) |
| Hazard ratio | 1·00 (0·79–1·26) | 1 | 1·11 (0·82–1·51) | 0·90 (0·65–1·24) | 1 |
| p value | 0·99 | .. | 0·50 | 0·53 | .. |
Data are n or n (95% CI) unless otherwise stated. ..=not applicable.
All enrolled participants, excluding those with HIV-1 infection at enrolment, those without follow-up data for HIV-1 infection, and censored at 52 weeks (plus 6 week window for final visit) or pregnancy.
Equivalent to primary efficacy analysis, but not censored for pregnancy or at week 52.
Per 100 woman-years.
Gel efficacy against HIV-1 transmission did not differ between centres (p=0·19 for interaction), or between consistent (HR 0·97, 95% CI 0·87–1·35, p=0·87) and inconsistent (1·17, 0·42–1·72, p=0·42) gel users (p=0·47 for interaction). HIV-1 incidence did not differ between gel groups at weeks 24 or 40 (data not shown).
Table 3 shows analyses for secondary efficacy endpoints. No differences between groups were noted for data obtained before discontinuation of 2% PRO2000 gel or at study end. For participants who were HSV-2-negative at randomisation, around 7% of the 0·5% PRO2000 and placebo groups were HSV-2 seropositive at week 40, and 11–13% were HSV-2 seropositive at week 52. At week 24, around 3% of participants in each group were positive for N gonorrhoeae and around 6% were positive for C trachomatis (table 3). No differences were reported between the three gel groups for these secondary efficacy endpoints up to discontinuation of 2% PRO2000 (table 3).
Table 3.
Secondary efficacy outcomes
|
End of study |
Censored at 2% PRO2000 gel discontinuation, Feb 14, 2008 |
|||||
|---|---|---|---|---|---|---|
| 0·5% PRO2000 (n=3326) | Placebo (n=3325) | 2% PRO2000 (n=2734) | 0·5% PRO2000 (n=2732) | Placebo (n=2722) | ||
| Herpes simplex virus type 2 | ||||||
| Infection by week 40 | 59/890 (6·6%) | 66/907 (7·3%) | 18/395 (4·6%) | 22/374 (5·9%) | 22/380 (5·8%) | |
| Odds ratio | 0·90 (0·63–1·30) | 1 | 0·78 (0·41–1·47) | 1·01 (0·55–1·87) | 1 | |
| p value | 0·59 | .. | 0·44 | 0·55 | .. | |
| Infection by week 52 | 109/919 (11·9%) | 115/888 (13·0%) | 34/297 (11·5%) | 34/292 (11·6%) | 32/284 (11·3%) | |
| Odds ratio | 0·90 (0·68–1·20) | 1 | 1·01 (0·61–1·70) | 1·04 (1·62–1·73) | 1 | |
| p value | 0·48 | .. | 0·95 | 0·89 | .. | |
| Neisseria gonorrhoeae | ||||||
| Infection at week 24 (±4 weeks) | 77/2674 (2·9%) | 74/2597 (2·9%) | 42/1613 (2·6%) | 44/1632 (2·7%) | 48/1550 (3·1%) | |
| Odds ratio | 1·01 (0·73–1·40) | 1 | 0·85 (0·56–1·29) | 0·88 (0·56–1·29) | 1 | |
| p value | 0·95 | .. | 0·44 | 0·54 | .. | |
| Chlamydia trachomatis | ||||||
| Infection at week 24 (±4 weeks) | 159/2674 (6·0%) | 164/2597 (6·3%) | 79/1611 (4·9%) | 95/1632 (5·9%) | 90/1570 (5·8%) | |
| Odds ratio | 0·94 (0·75–1·17) | 1 | 0·82 (0·60–1·11) | 0·98 (0·73–1·31) | 1 | |
| p value | 0·58 | .. | 0·20 | 0·90 | .. | |
Data are n/n (%) or n (95% CI) unless otherwise stated. ..=not applicable.
Our main results are for the comparison of 0·5% PRO2000 and placebo. Of 6651 women allocated to 0·5% PRO2000 and placebo groups, 5418 (81%) attended the week 52 visit, providing data for 2816 (85%) of the maximum possible woman-years for the 0·5% PRO2000 group, and 5591 (83%) for the placebo group (data not shown).
Reported adherence did not differ between the three gel groups. Reported condom use at last sex act increased gradually with time, and was similar across gel groups but varied between centres (figure 2). The mean percentage reported gel use at last sex act was 89% (95% CI 86–91) after enrolment. This percentage changed little during the trial, and did not differ between centres or between participants who used or did not use a condom (figure 2). Reported gel use decreased during the study for participants reporting sex acts without a condom in Durban and Johannesburg centres (South Africa), but this analysis was done on the basis of only 78 and 198 sex acts, respectively, at week 52.
Figure 2.

Adherence to condom and gel use, overall and by research centre
(A) Reported condom use, with or without gel use. (B) Reported gel use, with or without condom use. (C) Reported gel use in unprotected sex (no condom used). 2391 participants were enrolled in Durban, South Africa; 2499 in Johannesburg, South Africa; 840 in Masaka, Uganda; 1146 in Mwanza, Tanzania; 1177 in Africa Centre, South Africa; and 1332 in Mazabuka, Zambia.
181 (2%) women reported having anal sex in the previous 4 weeks at one or more of four visits; most (>90%) of these reports were from the South African sites, where HIV-1 incidence was 5·4 per 100 woman-years. Of these 181 women, 11 seroconverted, giving an incidence of 6·3 per 100 woman-years.
Table 4 summarises adverse events. No deaths or serious adverse events were regarded as related to study gels. Rates of primary safety events did not differ between the 0·5% PRO2000, 2% PRO2000, and placebo groups (table 4). 1450 (16%) of 9154 participants with follow-up clinical data had at least one solicited local toxic event after enrolment; genital itching was most commonly reported. 595 (33%) of 1796 participants with routine laboratory data experienced at least one systemic toxic event (most commonly high concentrations of aspartate aminotransferase or bilirubin). Rates for local and systemic toxic effects were similar between all gel groups (data not shown). PRO2000 concentrations were below the lower limit of quantification in all 1789 plasma specimens analysed.
Table 4.
Reported serious adverse events, selected genital adverse events, and primary safety events
|
End of study |
Censored at 2% PRO2000 gel discontinuation Feb 14, 2008 |
|||||
|---|---|---|---|---|---|---|
| 0·5% PRO2000 (n=3326) | Placebo (n=3325) | 2% PRO2000 (n=2734) | 0·5% PRO2000 (n=2732) | Placebo (n=2722) | ||
| Attended at least one visit after enrolment | 3258 | 3223 | 2571 | 2524 | 2511 | |
| Primary safety events* | 163 | 137 | 92 | 99 | 85 | |
| Woman-years of follow-up | 3349 | 3317 | 1956 | 1929 | 1911 | |
| Primary safety event (first) | 154 | 128 | 88 | 96 | 80 | |
| Incidence† | 4·6 (3·9–5·4) | 3·9 (3·2–4·6) | 4·5 (3·7–5·5) | 5·0 (4·1–6·1) | 4·2 (3·4–5·2) | |
| Hazard ratio | 1·18 (0·93–1·49) | 1 | 1·05 (0·78–1·43) | 1·19 (0·88–1·60) | 1 | |
| p value | 0·17 | .. | 0·74 | 0·26 | .. | |
| Adverse events | ||||||
| Non-menstrual bleeding | 551 (17%) | 527 (16%) | 320 (13%) | 339 (13%) | 318 (13%) | |
| Ulcers (internal) | 32 (1%) | 38 (1%) | 20 (1%) | 22 (1%) | 25 (1%) | |
| Ulcers (external) | 161 (5%) | 157 (5%) | 121 (5%) | 111 (4%) | 116 (5%) | |
| Oedema (internal) | 11 (<1%) | 15 (<1%) | 5 (<1%) | 5 (<1%) | 9 (<1%) | |
| Oedema (external) | 8 (<1%) | 5 (<1%) | 6 (<1%) | 3 (<1%) | 5 (<1%) | |
| Erythema (internal) | 201 (6%) | 201 (6%) | 134 (5%) | 117 (5%) | 122 (5%) | |
| Erythema (external) | 54 (2%) | 35 (1%) | 39 (2%) | 32 (1%) | 29 (1%) | |
| Itching | 349 (11%) | 310 (10%) | 214 (9%) | 247 (10%) | 232 (9%) | |
| Burning | 72 (2%) | 56 (2%) | 52 (2%) | 53 (2%) | 46 (2%) | |
| Other genital events | 379 (12%) | 356 (11%) | 232 (9%) | 241 (10%) | 239 (10%) | |
| Other non-genital events | 685 (21%) | 631 (20%) | 442 (18%) | 435 (17%) | 424 (17%) | |
| Serious adverse events‡ | ||||||
| Deaths | 9 (<1%) | 5 (<1%) | 7 (<1%)§ | 4 (<1%) | 0 | |
| Other serious adverse events | 142 (4%) | 119 (4%) | 86 (3%) | 95 (3%) | 75 (3%) | |
Data are n (%) or n (95% CI). ..=not applicable.
Defined as adverse events of grade 3 or more reported any time after enrolment. For women with more than one event, the time-to-event analysis uses the first event only.
Per 100 woman-years.
Serious adverse events were death, an immediate threat to life, admission to hospital, disability, congenital abnormality, vaginal oedema with sloughing, profuse non-menstrual bleeding, and cervical or gynaecological cancer.
Two additional deaths were reported after Feb 14, 2008.
For all participants enrolled, incidence of pregnancy was 11·6 per 100 woman-years (95% CI 10·9–12·4), ranging from 8·2 to 17·2 between centres but much the same between the three gel groups (11·9 per 100 woman-years in 0·5% PRO2000 and placebo groups at study end; and 10·1 in 2% PRO2000 group, 11·3 in 0·5% PRO2000 group, and 10·9 in placebo group at discontinuation).
Discussion
Despite high rates of reported adherence, 0·5% PRO2000 and 2% PRO2000 were not effective for prevention of vaginally acquired HIV-1 infection or other sexually transmitted infections. MDP301 was done to the highest international standard to support a licence application, if indicated. Alongside the clinical, laboratory, and data management procedures for which international guidelines exist, we integrated social science into the trial protocol to a high degree,19 and a commitment to active community engagement and liaison was made at all sites. Completion of this multidisciplinary and multicountry trial in resource restricted settings is testimony to the success of the MDP partnership.
Accurate incidence data for the target population are essential to ensure a phase 3 HIV-1 prevention trial is adequately powered.4 In cohort studies18 done before MDP301, weighted HIV-1 incidence rates were estimated to be 6·2 per 100 woman-years. Our power calculations were based on a conservative estimate of four per 100 woman-years because HIV-1 incidence in the target populations was assumed to fall during the study, and might have become even lower than the estimate because of the more frequent visits with HIV-1 counselling and testing. This strategy was justified as the reported overall HIV-1 incidence in the placebo group was 4·5 per 100 woman-years. Our power calculations allowed for a 20% loss of woman-years by 52 weeks, but only 12% were lost, which could translate to approximately 22 unobserved seroconversions, and is a potential limitation of this study. However, even in the extreme case that all 22 were attributed to either PRO2000 or to placebo, this would not alter the overall result of the trial. Diligent promotion of effective methods of contraception resulted in a loss of only 4% of woman-years from the primary efficacy analysis because of pregnancy. Incidence of pregnancy was similar across the gel groups, justifying the modification to the protocol analysis to censor patients while pregnant. In October 2007, after a Trial Steering Committee review of masked data, recruitment targets were revised to ensure that there was 75–80% power to detect efficacy at a lower rate (30–35% reduction).
Self-reported adherence has been high in all microbicide trials so far,2–6 but scepticism remains about reporting of adherence in the absence of a reliable biomarker. In MDP301, self-reported adherence was corroborated by the return of used applicators, and participants were asked at every visit, which provided more opportunity to report non-adherence, increasing the stringency of the definition of consistent. Information was also obtained from 725 women randomly selected to take part in a social science substudy. As well as the clinic interviews and gel returns, these women completed coital diaries and were interviewed in-depth about sexual behaviour.
Triangulation of data from all sources suggested that adherence was high,20 and qualitative data suggested that women and their partners enjoyed using the gel.23 There were no differences in reported adherence between the three gel groups, justifying use of these data to define consistency for the prespecified subgroup analysis.
We suggest several reasons why PRO2000 did not provide protection in women despite promising results in vitro and in challenge studies in animals, with 10 of 14 macaques protected in one study.13 In early studies,24,25 active drug was recoverable from cervicovaginal lavage several hours after insertion, suggesting that it was released from the formulation and not overly diluted by vaginal secretions. However, in a study26 in which cervicovaginal lavage samples were obtained from ten women who inserted 0·5% PRO2000 gel, significantly lower concentrations of PRO2000 were recovered after sex without a condom (median 14 μg/mL, IQR 3–27) than were reported in the absence of sex (28 μg/mL, 22–110, p=0·04). This difference could result from drug redistribution, binding to semen, loss from leakage, or difficulty in assaying drug because of physical changes after interaction with semen.
Several HIV-1 transmissions could have been the result of unprotected anal intercourse, although reported anal sex on the case report form was very uncommon, 1% at enrolment and 2% ever in the trial. Most reports of anal sex were from the South African sites (marginally lower than 2%), where the overall HIV-1 incidence was high. Although qualitative data from interviews and focus groups suggest that the frequency of anal sex might have been under-reported to the clinics, we do not attribute the absence of efficacy noted in this trial to it.
The rationale for including two concentrations was that the 2% concentration of PRO2000 might have resulted in local inflammation, which might have inadvertently facilitated HIV-1 transmission, offsetting any potential gain in biological potency against HIV-1. Although there was no evidence of this effect in histology or colposcopy investigations in the phase 1 and 2 studies, we postulated that symptoms and signs might emerge in a long phase 3 trial with cumulative exposure in a large population. The absence of local toxic effects with PRO2000 was reassuring; and the absence of systemic toxic effects was expected and consistent with the failure to detect PRO2000 in plasma after extended use.
Seven different molecular candidates have been assessed in 13 phase 2b/3 microbicide trials. One trial6 was designed to inform a decision algorithm, but only four of the remaining 12 had adequate statistical power to detect the prespecified effect of interest, which ranged from 35% to 50% for various reasons, including early termination3 and lower-than-expected incidence.4 One of the four trials with sufficient statistical power, COL-1492,27 reported a significant increase in HIV infections in women who used nonoynol-9 compared with placebo; two trials, including MDP301, reported no effect.2 In July, 2010, CAPRISA 00428 reported a 39% (95% CI 6–60%) reduction in HIV incidence in women who used the antiretroviral tenofovir 1% formulated as a vaginal gel, compared with women who used placebo gel. Women were advised to administer gel before and after sex, and to use no more than two applicators every 24 h. Tenofovir acts specifically to block HIV replication at the intracellular level and is much more potent in vitro than any of the non-antiretroviral candidate gels. CAPRISA provided the proof of concept for antiretroviral prophylaxis and for microbicides.
Five trials assessing effectiveness of antiretroviral prophylaxis are in progress and will report in the next 4 years. These five trials will assess daily tenofovir-based regimens taken oral, although one, the VOICE trial, will also assess tenofovir 1% as a microbicide, administered daily. Investigators need to establish whether coitally dependent dosing is effective, ideally in non-South African populations, as there are hypothetical advantages associated with the lower systemic drug concentrations in a coitally dependent dosing regimen (eg, fewer toxic effects or lower risk of drug resistance emerging during breakthrough infections).
MDP301 shows that licensing trials can be done in resource-scarce settings, and the self-reported quantitative data obtained in MDP301, supported by the return of the used applicators, and the quantitative and qualitative data obtained in the social-science substudy, reinforce the acceptability of microbicides as potential products for prevention of HIV-1 transmission. CAPRISA 00428 showed that microbicides can prevent HIV infection.
Acknowledgments
Acknowledgments
We thank the women who participated in the Microbicides Development Programme (MDP) 301 trial for their commitment, and the male partners who supported them; the MDP staff; the Trial Steering Committee (Anna Glasier [Chair], Mike Chirenje, Anne Johnson, Ade Lucas, Alwyn Mwinga, Angelina Wapakabulo, and Christopher Smangaliso Ntshele); the Independent Data Monitoring Committee (Alasdair Breckenridge [Chair], Catherine Hill, Isaac Malonza, and Florence Mirembe); the HIV Endpoint Committee (CJL, JW, and Adrian Purens); and Alan Stone (Chair, International Working Group on Microbicides) who facilitated international links. MDP is a partnership of African and European academic and governmental institutions with commercial organisations, and is funded by the UK Department for International Development and UK Medical Research Council. All employee institutions listed by the investigators, apart from the University of Amsterdam, contributed through provision of facilities or salary contributions. Study gels were provided by Endo Pharmaceutical Solutions.
Contributors
All authors contributed to study design and conduct and revision of the final report. SM prepared the first draft, and AMC did the statistical analysis and the initial draft of the tables. All authors read and approved the final report.
Conflicts of interest
AP was an employee of Endo Pharmaceuticals Solutions (formerly Indevus Pharmaceuticals), the owner of PRO2000, and held an equity interest in the company. All other authors declare that they have no conflicts of interest.
References
- 1.UNAIDS/WHO AIDS epidemic update, Joint United Nations Programme on HIV/AIDS (UNAIDS) 2009. http://data.unaids.org/pub/Report/2009/JC1700_Epi_Update_2009_en.pdf (accessed Aug 11, 2010).
- 2.Skoler-Karpoff S, Ramjee G, Ahmed K. Efficacy of Carraguard for prevention of HIV infection in women in South Africa: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1977–1987. doi: 10.1016/S0140-6736(08)61842-5. [DOI] [PubMed] [Google Scholar]
- 3.Van Damme L, Govinden R, Mirembe FM. Lack of effectiveness of cellulose sulfate gel for the prevention of vaginal HIV transmission. N Engl J Med. 2008;359:463–472. doi: 10.1056/NEJMoa0707957. [DOI] [PubMed] [Google Scholar]
- 4.Feldblum PJ, Adeiga A, Bakare R. SAVVY vaginal gel (C31G) for prevention of HIV infection: a randomized controlled trial in Nigeria. PLoS One. 2008;3:e1474. doi: 10.1371/journal.pone.0001474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halpern V, Ogunsola F, Obunge O. Effectiveness of cellulose sulfate vaginal gel for the prevention of HIV infection: results of a phase III trial in Nigeria. PLoS One. 2008;3:e3784. doi: 10.1371/journal.pone.0003784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abdool Karim SS, Coletti A, Richardson B, et al. Safety and effectiveness of vaginal microbicides buffer gel and 0.5% PRO 2000/5 gel for the prevention of HIV infection in women: results of the HPTN 035 trial. 16th Conference on Retroviruses and Opportunistic Infections; Montreal, Canada; Feb 8–11, 2009. (abstr 48LB).
- 7.Indevus Pharmaceuticals I. Investigator's brochure: universal placebo gel. 2007 (Version 2.0). Lexington: MA, USA.
- 8.Bourne N, Bernstein DI, Ireland J, Sonderfan AJ, Profy AT, Stanberry LR. The topical microbicide PRO 2000 protects against genital herpes infection in a mouse model. J Infect Dis. 1999;180:203–205. doi: 10.1086/314853. [DOI] [PubMed] [Google Scholar]
- 9.Cheshenko N, Keller MJ, MasCasullo V. Candidate topical microbicides bind herpes simplex virus glycoprotein B and prevent viral entry and cell-to-cell spread. Antimicrob Agents Chemother. 2004;48:2025–2036. doi: 10.1128/AAC.48.6.2025-2036.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenhead P, Hayes P, Watts PS, Laing KG, Griffin GE, Shattock RJ. Parameters of human immunodeficiency virus infection of human cervical tissue and inhibition by vaginal virucides. J Virol. 2000;74:5577–5586. doi: 10.1128/jvi.74.12.5577-5586.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rusconi S, Moonis M, Merrill DP. Naphthalene sulfonate polymers with CD4-blocking and anti-human immunodeficiency virus type 1 activities. Antimicrob Agents Chemother. 1996;40:234–236. doi: 10.1128/aac.40.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spencer SE, Valentin-Bon IE, Whaley K, Jerse AE. Inhibition of Neisseria gonorrhoeae genital tract infection by leading-candidate topical microbicides in a mouse model. J Infect Dis. 2004;189:410–419. doi: 10.1086/381125. [DOI] [PubMed] [Google Scholar]
- 13.Weber J, Nunn A, O'Connor T. ‘Chemical condoms’ for the prevention of HIV infection: evaluation of novel agents against SHIV(89.6PD) in vitro and in vivo. AIDS. 2001;15:1563–1568. doi: 10.1097/00002030-200108170-00014. [DOI] [PubMed] [Google Scholar]
- 14.Tabet SR, Callahan MM, Mauck CK. Safety and acceptability of penile application of 2 candidate topical microbicides: BufferGel and PRO 2000 Gel: 3 randomized trials in healthy low-risk men and HIV-positive men. J Acquir Immune Defic Syndr. 2003;33:476–483. doi: 10.1097/00126334-200308010-00008. [DOI] [PubMed] [Google Scholar]
- 15.Mayer KH, Karim SA, Kelly C. Safety and tolerability of vaginal PRO 2000 gel in sexually active HIV-uninfected and abstinent HIV-infected women. AIDS. 2003;17:321–329. doi: 10.1097/00002030-200302140-00005. [DOI] [PubMed] [Google Scholar]
- 16.Van Damme L, Wright A, Depraetere K. A phase I study of a novel potential intravaginal microbicide, PRO 2000, in healthy sexually inactive women. Sex Transm Infect. 2000;76:126–130. doi: 10.1136/sti.76.2.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamali A, Byomire H, Muwonge C. A randomised placebo-controlled safety and acceptability trial of PRO 2000 vaginal microbicide gel in sexually active women in Uganda. Sex Transm Infect. 2010;86:222–226. doi: 10.1136/sti.2009.038372. [DOI] [PubMed] [Google Scholar]
- 18.Nunn A, McCormack S, Crook AM, Pool R, Rutterford C, Hayes R. Microbicides Development Programme: design of a phase III trial to measure the efficacy of the vaginal microbicide PRO 2000/5 for HIV prevention. Trials. 2009;10:99. doi: 10.1186/1745-6215-10-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pool R, Montgomery CM, Morar NS. A mixed methods and triangulation model for increasing the accuracy of adherence and sexual behaviour data: the Microbicides Development Programme. PLoS One. 2010;5:e11600. doi: 10.1371/journal.pone.0011600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pool R, Montgomery CM, Morar NS. Assessing the accuracy of adherence and sexual behaviour data in the MDP301 vaginal microbicides trial using a mixed methods and triangulation model. PLoS One. 2010;5:e11632. doi: 10.1371/journal.pone.0011632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mauck C, Rosenberg Z, Van Damme L, for the International Working Group on Microbicides Recommendations for the clinical development of topical microbicides: an update. AIDS. 2001;15:857–868. doi: 10.1097/00002030-200105040-00006. [DOI] [PubMed] [Google Scholar]
- 22.Indevus Pharmaceuticals I. Investigator's Brochure: PRO2000/5 Gel (P). June 16, 2008. Lexington, MA, USA.
- 23.Montgomery CM, Lees S, Stadler J. The role of partnership dynamics in determining the acceptability of condoms and microbicides. AIDS Care. 2008;20:733–740. doi: 10.1080/09540120701693974. [DOI] [PubMed] [Google Scholar]
- 24.Lacey CJ, Wright A, Weber JN, Profy AT. Direct measurement of in-vivo vaginal microbicide levels of PRO 2000 achieved in a human safety study. AIDS. 2006;20:1027–1030. doi: 10.1097/01.aids.0000222075.83490.ca. [DOI] [PubMed] [Google Scholar]
- 25.Keller MJ, Zerhouni-Layachi B, Cheshenko N. PRO 2000 gel inhibits HIV and herpes simplex virus infection following vaginal application: a double-blind placebo-controlled trial. J Infect Dis. 2006;193:27–35. doi: 10.1086/498533. [DOI] [PubMed] [Google Scholar]
- 26.Keller MJ, Mesquita PMM, Torres NM. Postcoital bioavailability and antiviral activity of 0.5% PRO 2000 gel: implications for future microbicide clinical trials. PLoS One. 2010;5:e8781. doi: 10.1371/journal.pone.0008781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Damme L, Ramjee G, Alary M. Effectiveness of COL-1492, a nonoxynol-9 vaginal gel, on HIV-1 transmission in female sex workers: a randomised controlled trial. Lancet. 2002;360:971–977. doi: 10.1016/s0140-6736(02)11079-8. [DOI] [PubMed] [Google Scholar]
- 28.Karim QA, Karim SS, Frohlich JA. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science. 2010 doi: 10.1126/science.1193748. published online July 19. [DOI] [PMC free article] [PubMed] [Google Scholar]