

Abstract
A neuropathologic study of three cases of prosaposin (pSap) deficiency (ages at death 27, 89 and 119 days), carried out in the standard autopsy tissues, revealed a neurolysosomal pathology different from that in the non-neuronal cells. Non-neuronal storage is represented by massive lysosomal accumulation of glycosphingolipids (glucosyl-, galactosyl-, lactosyl-, globotriaosylceramides, sulphatide, and ceramide). The lysosomes in the central and peripheral neurons were distended by pleomorphic non-lipid aggregates lacking specific staining and autofluorescence. Lipid storage was borderline in case 1, and at a low level in the other cases. Neurolysosomal storage was associated with massive ubiquitination, which was absent in the non-neuronal cells and which did not display any immunohistochemical aggresomal properties. Confocal microscopy and cross-correlation function analyses revealed a positive correlation between the ubiquitin signal and the late endosomal/lysosomal markers. We suppose that the neuropathology most probably reflects excessive influx of non-lipid material (either in bulk or as individual molecules) into the neurolysosomes. The cortical neurons appeared to be uniquely vulnerable to pSap deficiency. Whereas in case 1 they populated the cortex, in cases 2 and 3 they had been replaced by dense populations of both phagocytic microglia and astrocytes. We suggest that this massive neuronal loss reflects a cortical neuronal survival crisis precipitated by the lack of pSap. The results of our study may extend the knowledge of the neurotrophic function of pSap, which should be considered essential for the survival and maintenance of human cortical neurons.
Keywords: Prosaposin deficiency, Neurolysosomal disorder, Ubiquitination, Cross-correlation function, Cortical neuronal survival crisis
Introduction
Prosaposin (pSap) deficiency is a rapidly progressive fatal neurovisceral lysosomal storage disorder caused by mutations in the PSAP gene which leads to the blockage of pSap protein translation or to the premature (intranuclear) nonsense-mediated decay of pSap mRNA [13, 25, 43, 54]. The proven genotypes result in the absence of pSap and the products of pSap proteolytic processing. These polypeptide products, called saposins (Saps, sphingolipidhydrolase activating proteins), are essential for activating a set of lysosomal sphingolipid hydrolases [54]. In the absence of Saps, the sphingolipid substrates remain undegraded. The accumulated substrates, in visceral cell types, cause the lysosomes to become distended, giving them a foamy appearance and the process ultimately ends in organ failure, similar to full blown classic lipid storage disorders [3, 13, 25]. In this study, we show that in pSap deficiency the neurolysosomes of both the peripheral and central neurons express a distinct pathology, which may indicate a process resembling autophagocytosis. Given the opportunity to compare the cortical structure at the earliest stage of the disease to that in the more advanced stages, we were able to describe a unique failure of cortical neurons to survive after reaching the cerebral cortex. We would like to characterize this destructive phenomenon, precipitated by the absence of pSap, as a cortical neuronal survival crisis, which seems to be a feature of human pSap deficiency. This fits well with the concept of pSap having fundamental neurotrophic functions [47].
Materials and methods
Formaldehyde fixed paraffin embedded (FFPE) tissue samples were available from three cases of verified pSap deficiency. The life spans of the cases were: 27 days (case 1 [14]), 89 days (case 2 [25]) and 119 days (case 3 [13]). In two of the three cases (27, 119 days) we were kindly provided by colleagues from the pathology departments unembedded routinely formaldehyde fixed samples of several cortical and subcortical brain areas. The tissue samples available were as follows: case 1 (27 days)–parts of the frontal and insular cortex, striatum, pallidum, thalamus, and adjacent parts of the white matter; case 2 (89 days)–several cortical samples, thalamus, and basal ganglia; and case 3 (119 days)–cerebral cortex from all lobes, cerebellum, pons, medulla oblongata and spinal cord.
Histology, immunohistochemistry, electron microscopy and lipid biochemistry
The tissue sections (5 μm) were dewaxed, hydrated and hematoxylin-eosin stained. The primary antibodies and their respective dilutions used for immunohistochemistry are listed in Table 1. The primary antibodies (incubated overnight at 4°C) were detected with appropriate Envision TM kits (DAKO, Copenhagen, Denmark) or rabbit anti-guinea pig HRP labeled secondary antibody (DAKO).
Table 1.
Dilutions and sources of antibodies
| Antigen (Antibody clone) | Source | Dilution | |
|---|---|---|---|
| IH | IF | ||
| LAMP 1 (rabbit polyclonal) | kindly provided by Dr.S.Carlsson (University of Umea, Sweden) | 1:200 | |
| LAMP 2 (rabbit polyclonal) | kindly provided by Dr.S.Carlsson | 1:200 | 1:100 |
| Cathepsin D (rabbit polyclonal) | DAKO, Copenhagen, Denmark | 1:4000 | |
| Cathepsin D (MCA2068) | Serotec, Raleigh, NC, USA | 1:50 | |
| Ubiquitin (rabbit polyclonal) | DAKO, Copenhagen, Denmark | 1:1500 | 1:500 |
| Ubiquitin (FPM1) | Novocastra, Newcastle upon Tyne, United Kingdom | 1:100 | 1:100 |
| Poly ubiquitinylated proteins (FK1) [16] | Biomol, Plymouth Meeting, PA, USA | 1:500 | |
| Mono- and poly ubiquitinylated proteins (FK2) [16] | Biomol, Plymouth Meeting, PA, USA | 1:1000 | |
| LC3 (rabbit polyclonal) [27] | MBL, Naka-ku Nagoya, Japan | 1:500 | |
| Glial fibrillary acidic protein - GFAP (6F2) | DAKO, Copenhagen, Denmark | 1:200 | |
| GFAP (rabbit polyclonal) | DAKO, Copenhagen, Denmark | 1:500 | |
| CD68 (PGM1) | DAKO, Copenhagen, Denmark | 1:200 | 1:200 |
| Neuron specific nuclear protein – NeuN (A60) | Chemicon Int., Temecula, CA, USA | 1:500 | |
| Prohibitin (II-14–10) | Lab Vision, Westinghouse, CA, USA | 1:500 | |
| 60 kDa antigen of human mitochondria (113–1) | Biogenex, San Ramon, CA, USA | 1:100 | 1:50 |
| Subunit c of mitochondrial ATP synthase - SCMAS | kindly provided by prof. E.Kominami (Juntendo University, Tokyo, Japan) | 1:200 | |
| p62 component of aggresome (guinea pig polyclonal) [67] | Progen, Heidelberg, Germany | 1:4000 | |
| Activated caspase 3 (rabbit monoclonal) | Epitomics, Burlingame, CA, USA | 1:25 | |
| Protein disulfide isomerase (PDI) (1D3) | Stressgen, Victoria, Canada | 1:500 | |
IH immunohistochemistry, IF immunofluorescence
In conjunction, various neurolysosomal storage disorders (previously defined biochemically at the Institute of Inherited Metabolic Disorders) were studied to follow neuronal ubiquitination (for the list, see Results). The FFPE tissue samples from various parts of the brain were examined together with the samples of storage affected visceral organs.
The sections of the formaldehyde fixed unembedded samples (cases 1 and 2) were processed for electron microscopy after osmification, using standard dehydration in ethanol and embedding in an Araldite–Epon mixture. The paraffin embedded samples of the pons (case 3) were processed for electron microscopy after deparaffination, hydration, and osmification.
The residual lipids extracted from the paraffin embedded brain samples were analyzed using a procedure recently described [13].
Immunofluorescence and laser scanning confocal microscopy
The FFPE paraffin embedded tissue sample from the spinal cord (case 3) was chosen for immunofluorescence multiple labeling colocalization analysis. The tissue sections were treated as described above. The primary antibodies and their dilutions used for immunofluorescence labeling are listed in Table 1.
The combinations of the primary antibodies were as follows: anti-LAMP2/anti-ubiquitin, anti-cathepsin D/anti-ubiquitin, anti-LAMP2/anti-60 kDa antigen of human mitochondria, anti-PDI/anti-ubiquitin, anti-60 kDa antigen of human mitochondria/anti-ubiquitin. The details of the tissue section processing and staining protocol for both immunohistochemistry and immunofluorescence labeling are available from the authors upon request.
Images (xy sampled at the maximum of z fluorescence intensity) were acquired using a Nikon Eclipse E800 microscope equipped with a C1 confocal head and 488, 543 and 633 nm laser lines and appropriate 515 ± 15 nm, 590 ± 15 nm and 650LP band pass filters. The sampling density was corrected to conform to the Nyquist criterion according to the objective lens used (Nikon Plan Apo 60 × 1.4 N.A.). The microscope settings including laser intensity, pinhole size, pixel dwell times; photomultiplier gains were kept constant for all image acquisitions and the images were checked for the presence of spectral cross-talk.
Point spread functions (PSF) at appropriate excitation/detection wavelengths were measured using PS-Speck Microscope Point Source Kit (Invitrogen- Molecular Probes, Carlsbad, CA).
The cerebral cortex sample from (case 2, day 89) was double-labeled with mouse monoclonal anti-CD 68 and rabbit polyclonal anti-GFAP antibodies. Secondary detections were performed using donkey anti-mouse IgG Alexa Fluor 555 and goat anti-rabbit IgG Alexa Fluor 488 antibodies (Invitrogen- Molecular Probes, Carlsbad, CA) diluted 1:500.
Image restoration, processing and cross-correlation function (CCF) analysis
Between 25 and 30 images of neurons of the spinal cord anterior columns were acquired and further processed for each of the double-labeling experiments. The images were restored based on deconvolution using an appropriately measured PSF with a classical maximum likelihood estimate algorithm (Huygens Professional software, SVI, Hilversum, Netherlands). Only pixels included in the neurons were further evaluated without any additional thresholding or pixel intensity manipulation. Cross-correlation function analysis [65] was performed using a Tcl/Tk script employing the Huygens Scripting modality in Huygens Professional software. Single pixel shifts with parallel Pearson’s coefficient calculations were applied to the red channel of the images with respect to the green channel to the extent of −4 to +4 μm (only values −3 to +3 μm are shown, Fig. 5). The appropriate pixel shifted Pearson’s coefficient values were calculated for each individual evaluated shifted image. The median values and 5/95 percentile values and first and third quartile values of Pearson’s coefficient were plotted on the y axis for each single pixel shifted position (x axis) to get the final cross-correlation curves and to document the distribution of the values.
Fig. 5.
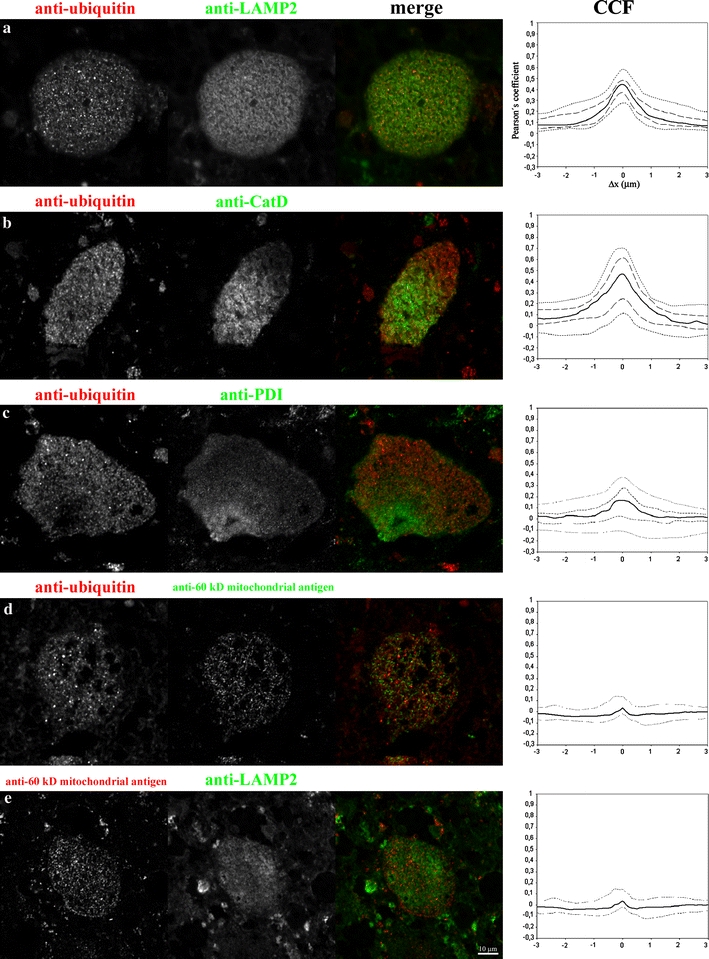
Laser scanning confocal microscopy. Figure shows isolated gray scale single channel, and merged RGB images of the double-labeled neurons for appropriate indirect immunofluorescence labels (used antibodies given in panels a–e). On the right are the corresponding CCF curves: Full line median values, dotted lines 5/95 percentile values, dashedlines 1st and 3rd quartile (for details on graph construction see Methods). CCF values were determined only for the pixels located inside the neurons, the surrounding tissue was excluded from the CCF calculations. Positive Pearson’s coefficient values demonstrate significant overlap (colocalization) of the signals, negative values demonstrate repulsion (anti-colocalization) of the signals, and values around zero demonstrate random distribution of the signals. For CCF calculation refer to Materials and methods. Scale bar (10 μm)
The Pearson’s coefficient values range from −1 to +1. Positive Pearson’s coefficient values at Δx = 0 demonstrate significant overlap (colocalization) of the signals, negative values demonstrate repulsion (anti-colocalization) of the signals, and values around zero demonstrate a random distribution of signals. The details of the Tcl/Tk script used to calculate CCF is available from the authors upon request. All the image manipulations and figure preparations were performed using Huygens Professional or ImageJ (NIH, http://www.rsb.info.nih.gov/ij/) software.
In order to avoid any ambiguity, the use of neurolysosomal in this paper refers to the lysosomal compartment of neurons.
Results
General neuropathology
The general neuropathologic findings in all the three cases have already been published [13, 14, 25]. However, to briefly summarize, the neuronal perikarya were variably distended by fine eosinophilic non-autofluorescent granules (Figs. 1a inset, 2a). Neuronal storage was present in all the regions available for examination: brain cortex (see below), basal ganglia, thalamus, pons, medulla oblongata, spinal cord, and cerebellum. The only exception was the subventricular zone (cases 1 and 2) which was cellular and devoid of histological and immunohistochemical abnormalities. There was a marked difference between the brain cortical pathology in case 1 as compared to cases 2 and 3 (see below). Storage was manifested in the peripheral nervous system in all samples available (gut, lung, liver, kidney, and urinary bladder).
Fig. 1.
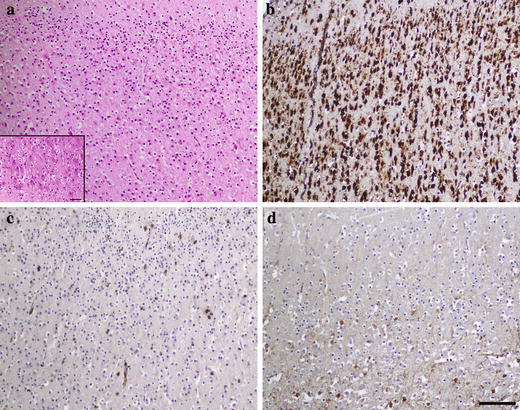
Cortex in case 1 (day 27) in pSAP deficiency. a Numerous cortical neurons with a rather normal architecture. Discrete neuronal storage accented in basal layers (inset); H&E. b Strong neuronal perikaryal staining for cathepsin D. c Paucity of microglial phagocytes demonstrated by CD 68 antibody (clone PGM1), and d of astroglia stained with GFAP antibody. a–d correspond to the identical cortical area. Scale bar 200 μm a–d. Scale bar in inset in (a) 100 μm
Fig. 2.
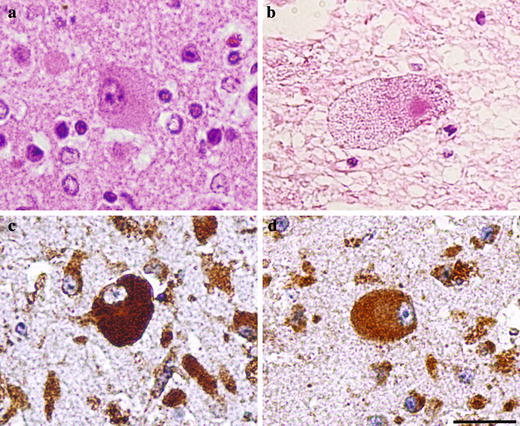
Neuronal cytology and immunodetection of lysosomal markers in pSAP deficiency. a Case 1 (day 27): a subcortical storage neuron with the perikaryal region distended by fine granules; H&E. b Case 3 (day 119): storage in spinal motor neuron with a granulovacuolar appearance of the perikaryon. c Case 1 (day 27): strong staining of the perikaryal granules for cathepsin D and for LAMP 2 d. Scale bar 50 μm
The following data represent novel and previously unreported features of the neurolysosomal storage process in pSap deficiency.
Study on the nature of neurolysosomal processes
Cytology, electron microscopy and lipid biochemistry
Contrary to the foamy appearance of the storage cells in the visceral region, the neuronal perikarya were distended even ballooned by a uniform population of fine eosinophilic granules. In case 3, their appearance changed to granulovacuolar (Fig. 2b). The granules were not autofluorescent and did not exhibit any distinct staining in routine histological stainings. Lipid histochemistry showed an absence of birefringence and no significant detectable amounts of glycolipids and sphingolipids [14].
In contrast to the neurons the astrocytes displayed significantly milder storage changes [13, 25]. Regarding the oligodendroglia, the lysosomal storage has never been demonstrated [13, 17]. Changes of microglial phagocytes are discussed below.
Electron microscopy (cases 1 and 2) showed vacuoles approximately 0.5 μm in diameter containing numerous dense granules of various densities, 45–230 nm in diameter, sometimes with amorphous or coarse membranous substructure, occasionally resembling degraded mitochondria (Fig. 3). Identical structures were observed in the dystrophic axons. Other organelles (RER, Golgi aparatus), while reduced in number, were without substantial alteration. Electron microscopy of dewaxed pontine samples (case 3) showed similar structures. The only difference was an increase of electron lucent space in the storage compartment leading to granulovacuolar neuronal cytology and an increase in the size of individual storage lysosomes, up to 1 μm in diameter on average. The ultrastructure differed from the ultrastructure of the lysosomes in the visceral storage cells [25] and from the lysosomes of the other known neurolysosomal storage disorders featured by membranous lipid deposits.
Fig. 3.
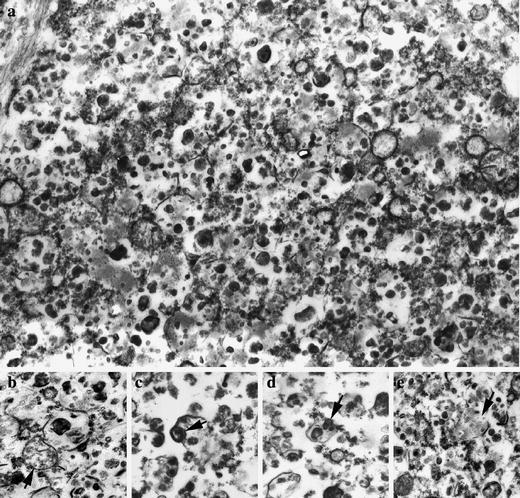
Ultrastructure of a subcortical neuron in case 1 (day 27) of prosaposin deficiency. a A dense population of lysosomes containing pleiomorphic condensed deposits. b–e Some of the deposits resemble degenerated mitochondria (arrowsc–e), for comparison refer to intact mitochondrion (b, arrow). Magnification ×28,000
Biochemical analysis of residual lipids in FFPE samples from the pons (case 3, day 119) which were rich in ballooned granulovacuolar neurons, revealed a threefold to sixfold elevation of gangliosides (GM1, GM2, GM3 types), lactosylceramide and globotriaosylceramide as compared to the controls (data not shown). At this point we must note some limitations of the analysis caused by previous dehydrations, as well as the potential contribution (although minor) to the overall lipid content of non-neuronal storage elements present in the tissue.
Immunohistochemistry, immunofluorescence, laser scanning confocal microscopy (LSCM) and cross-correlation function (CCF) analysis
In all the three cases the neuronal perikaryal storage granules displayed a uniform lysosomal immunophenotype, featured by strong staining for cathepsin D (luminal marker) and for the late endosomal/lysosomal membrane markers LAMP1, 2 (Figs. 1b, 2c, d). In the majority of storage neurons the perikaryal granules displayed variable positivity for the subunit c of mitochondrial ATP synthase (SCMAS) with the respective antibody (data not shown).
A peculiar feature of the immunophenotype was the nearly uniform, strong, granular immunostaining of ubiquitin (Fig. 4a–c), present in all analyzed samples (cortical, subcortical and extra cerebral). The cortical neurons in case 1 were ubiquitinated almost without any substantial difference between the individual layers. Axonal spheroids were ubiquitinated as well. The neurons of the peripheral nervous system (gut, lung, liver, kidney, and urinary bladder) displayed an identical lysosomal immunophenotype, including the high extent of ubiquitination (Fig. 4d). The cellular pattern of ubiquitination, seen in bright-field microscopy, was suggestive of its association with storage lysosomes (for details, see below).
Fig. 4.
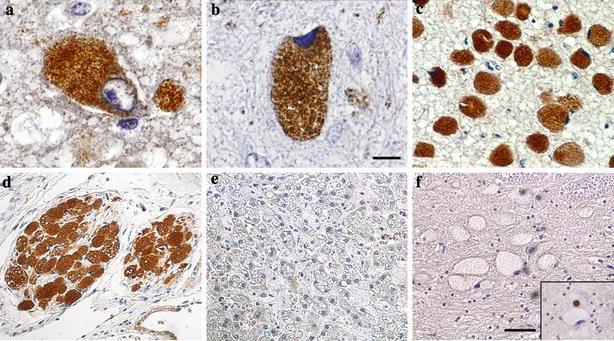
Immunodetection of ubiquitin in prosaposin deficiency (except for panel f). a Strong granular staining in perikarya of subcortical storage neurons in case 1 (day 27), and b case 2 (day 89). c Uniform strong ubiquitination in pontine storage neurons in case 3 (day 119), and d in peripheral neurons of the kidney hilus in the same case. e Absence of detectable ubiquitination in the storage cells in the liver in pSap deficiency (case 2). f Absence of ubiquitination in storage neurons in Niemann–Pick-disease type A. Inset shows intense ubiquitination of the small dystrophic axon close to another negative neuronal perikaryon. Scale bar 25 μm (a,b) and 100 μm (c–f)
To evaluate the proportion of ubiquitin protein conjugates (mono- and polyubiquitinated) in the overall ubiquitin signal, the antibodies (clone FK1 and FK2, Table 1) against these types of epitopes [16] were used. We were able to detect significant amounts of mono- and polyubiquitinated moieties (FK2 antibody, data not shown) with the same staining patterns as the above-described ubiquitin staining. Unfortunately, the FK1 antibody (polyubiquitinated protein conjugates) did not provide reliable staining in the control samples (Mallory bodies rich liver biopsy) and as such was not included in the study. The cortical neurons in age-matched controls were immunonegative for ubiquitin. The immunophenotype of storage lysosomes in all the affected non-neuronal cells, from the brain (astrocytes, microglial phagocytes) and the visceral region was identical to the neurolysosomes, except for the absence of any detectable ubiquitination (Fig. 4e).
The immunohistochemical detection of cytoplasmic aggresomal sequestered proteins using p62 antibody did not show any positivity in any of the evaluated cortical and subcortical brain samples of all the three cases (data not shown).
In contrast to pSap deficiency, neurolysosomal ubiquitination was undetectable in the following neurolysosomal storage disorders: Niemann–Pick disease type C1, type A (Fig. 4f), infantile sialic storage disorder, mucolipidosis type I, aspartylglucosaminuria, alpha-mannosidosis, polysulphatase deficiency, and glycogen storage disease type II. Only the axonal spheroids and the neuronal tangles, particularly numerous in protracted neuronopathic Niemann–Pick type C cases, displayed strong ubiquitination. In neuronal ceroid lipofuscinoses, types 2, 3, 4 and 8, the main population of storage granules was devoid of ubiquitination. The only exception was strong ubiquitination seen in a small population of transformed storage material present as globules or spheroids in the neuronal perikarya [12]. Moderate ubiquitination of storage neurons has been seen in the group of mucopolysaccharidoses (types I–III). Extra cerebral non-neuronal storage cells in all the above-mentioned disorders lacked any detectable ubiquitination. We could not confirm improved ubiquitin immunostaining with the recommended pretreatment [34].
Multiple indirect immunofluorescence labeling experiments were performed on sections of the spinal cord (case 3) in order to evaluate the relationship between the detectable ubiquitin signals and the potentially involved, cellular compartments. Prior to the evaluation of the signals in confocal images, we performed image restoration based on deconvolution using the microscope’s measured PSF, a method known to increase the effective image resolution [35, 57, 64]. The characteristics of the signals in the evaluated images allowed us to apply the cross-correlation function (CCF) estimation [65] for the analysis of their spatial (colocalization) relationship. The CCF method of colocalization evaluation of complex staining patterns is advantageous because it does not require image segmentation and can discriminate positive, negative and non-correlating signals.
In general, the signals obtained by immunofluorescence labeling followed the patterns observed by immunohistochemical staining. The intensity of the ubiquitin signal was inversely correlated to the endoplasmic reticulum (ER) and mitochondrial signals among the evaluated neurons. This resulted in a population of neurons strongly positive for ubiquitin and almost negative for ER and mitochondrial signals. This feature could be documented by the distribution of Pearson’s coefficient values in the CCF plots (Fig. 5c, d). While this loss of epitope detectability in the presence of strong ubiquitin positivity was clear for the ER and mitochondrial epitopes, a similar phenomenon was not observed for the two late endosomal/lysosomal markers (LAMP2, CatD).
In addition, the double labeling of the mitochondrial epitopes and of the late endosomal/lysosomal membrane epitope (LAMP2) in search for lysosomes with entrapped mitochondria or for mitochondrial remnants did not provide clear evidence of such events. This was, most probably, due to the combination of diffraction limited resolution of the microscope and/or advanced degradation of the mitochondrial epitopes.
Figure 5 shows images of the selected neurons with the corresponding CCFs and their distribution for the above-mentioned immunofluorescence labeling. From the five evaluated labeling combinations, only the signals of ubiquitin and LAMP 2/cathepsin D showed a non-random distribution (colocalization) according to the CCF in the population of evaluated neurons (Fig. 5a, b). The full-width of the half maximum (FWHM) of the CCF curves should correspond to the diameter of the evaluated objects [65]. This criterion stands for the median value of both the CCF curves representing the ubiquitin/LAMP2-CatD signals and corresponds to the diameter values obtained by electron microscopy. The characteristics of ubiquitin/LAMP2 and ubiquitin/cathepsin D CCF curves (FWHM or maximum Pearson’s coefficient values) did not allow unambiguous discrimination between the prevalence of the two phenomena i.e. ubiquitination of the late endosomal/lysosomal membranes or ubiquitination of the late endosomal/lysosomal contents. Despite this ambiguity, which is most likely caused by the resolution limit of the microscope, we were able to demonstrate the colocalization of the ubiquitin signal with the late endosomal/lysosomal cellular compartment in pSap deficient neurons. Analysis of the other staining combinations (anti-ubiquitin/anti-PDI, anti-ubiquitin/anti-60 kDa antigen of human mitochondria and anti-60 kDa antigen of human mitochondria/anti-LAMP2) yielded a wide distribution of values, ranging from random distributions to mild exclusion of the signals (negative values of the Pearson’s coefficient) (Fig. 5c–e). CCF found that for the anti-60 kDa antigen of human mitochondria/anti-LAMP2 (mitochondrial/lysosomal), staining could be regarded, in this case, as a positive control for a random/exclusion relationship of the signals.
Limited survival potential of cortical neurons
A comparison of brain cortical neuropathology showed pronounced differences between case 1 and cases 2 and 3. The histology in case 1 revealed a dense population of cortical neurons without substantial numerical and structural deviation from age-matched controls. The cortical neurons were moderately affected by storage, with a gradual increase toward the deeper layers (Fig. 1a). The neuronal nuclei were mostly vesicular with well-discernible nucleoli. The process culminated in the neurons of the subcortical regions (thalamus and basal ganglia). The degree of astrocytosis and the number of microglial phagocytes were proportional to the degree of neuronal storage, i.e. it was minimal in the cortex (Fig. 1c, d). In cases 2 and 3 the cortical areas displayed a profound depletion of the neurons [13, 25] and the residual neurons were identified as finely granulated single cells or small groups of cells that were detectable by using the NeuN antibody. Instead of the normal neuronal architecture there was a dense cellular population composed of equal proportions of large, coarsely vacuolated, CD 68 positive phagocytic microglia and hyperplastic, GFAP positive astrocytes (Fig. 6). In the rest of the central nervous system, the neurons were not substantially reduced in amount. No signs of caspase 3 activation or other cytological features of apoptosis were present either in the cortical or the subcortical neurons in case 1 or in the neurons in cases 2 and 3 (data not shown).
Fig. 6.
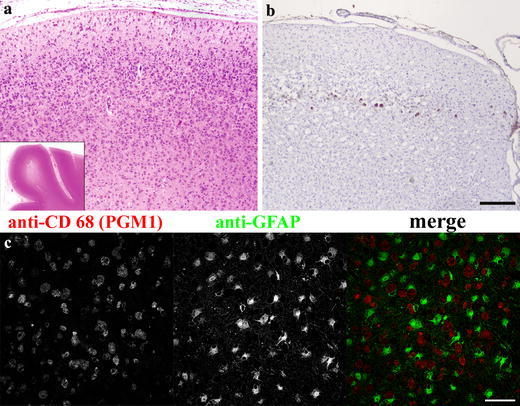
Neuronal cortical crisis in prosaposin deficiency. Case 2 (day 89). a Low power magnification (H&E) showing cellular cortex with well preserved shape and well delineated from the white matter (inset). b Very few residual neurons stained with NeuN antibody. c Dense cellularity of cerebral cortex in case 2 (day 89) formed by two distinct cell populations (CD 68 positive microglial phagocytes and GFAP positive astrocytes), LSCM acquired image. Note the intimate contact of the two cell types. Scale bar 500 μm a, b and 50 μm c. Compare with Fig. 1
Discussion
In all the three cases there was a complete absence of pSap protein caused either by the nonsense-mediated decay of the gene transcript [25] or by the failure of the mRNA translation [13, 54]. The discussion is focused on the nature of neurolysosomal storage and the remarkable sensitivity of the cortical neurons to pSap deficiency.
Nature of the neurolysosomal process
Whereas lysosomes in non-neuronal cells were distended by a massive accumulation of a set of sphingolipids due to multiple sphingolipid hydrolase insufficiency, due to pSap deficiency, leading to the foamy appearance of the affected cells, the neurons expressed an appreciably different pathology. The neuronal pathology featured finely granular lysosomes containing condensed pleiomorphic inclusions. Besides the standard membrane (LAMP 1 and LAMP 2) and luminal (cathepsin D) markers, their distinct feature was an association with strong ubiquitination including a significant proportion of ubiquitin protein conjugates detected by specific antibodies. They were devoid of ceroidlipofuscin and the lipid storage was borderline or of a low degree. We consider it important that ubiquitination has been proved to be absent in non-neuronal storage cells and even in numerous microglial phagocytes. It was also absent in the bulk of other neurolysosomal storage disorders featured by storage of defined lipid or glycoconjugate substrates due to deficient enzyme catalytic activity or due to altered lipid trafficking (see also [68]). Thus, in contrast to the non-neuronal cells, the absence of pSap leads to a different pathology in the neurolysosomes.
It should be compared to processes featured by the lysosomally associated ubiquitination. Ubiquitination associated with neurolysosomes has been described in the granulovacuolar lesions seen in Alzheimer’s disease and in scrapie infected mice, both appear to be autophagic vacuoles [39]. The ultrastructure of neurolysosomes in neuronal perikarya (Fig. 3) and axons in our cases of pSap deficiency resembled ultrastructural changes in the neuroaxonal dystrophy classified as a macroautophagy process [46] that also featured a high degree of ubiquitination [11, 42] combined with lysosomal enzyme activities [63].
In the cases of pSap deficiency we were able to document a positive correlation (colocalization) to ubiquitination and late endosomal/lysosomal markers as well as signs attributable to mitochondrial degradation (SCMAS detectability).
Ubiquitination has also been described as a part of the protein conjugation system of autophagosomal compartment [18, 38, 58, 59], a process dominated, however, by the homologs of yeast Atg ubiquitin-like proteins including mammalian LC3 protein [27, 28]. Unfortunately, the antibody against the LC3 autophagosomal membrane component provided ambiguous results in our FFPE tissues, partly due to the lack of appropriate reference samples; therefore we decided not to include these results. Ubiquitination of lysosomal contents has been repeatedly described [10, 36, 37, 41, 58] and explained by lysosomal sequestration of ubiquitin-tagged proteins.
We thus propose the existence of a similar mechanism in pSap deficiency, i.e. excessive lysosomal sequestration of the ubiquitin-tagged proteins released most probably from damaged neuronal membranes or resulting from impaired downstream pSap signaling [2, 23]. The previously demonstrated relationship of the ubiquitin/proteasome pathway and cytoplasmic vacuoles with non-lysosomal characteristics [61] does not correspond with our findings in pSap deficiency. The absence of immunodetectable p62 does not support the aggresomal-based ubiquitination [67] in pSap deficiency neuropathology, nevertheless, ubiquitination has been suggested to trigger an autophagocytosis process under certain conditions [30]. In summary, our findings can be interpreted as an excessive influx of ubiquitinated biological material (either in bulk or as individual molecules) into the neurolysosomes. It represents features compatible with the process of autophagy, encompassing a range of both bulk (macro and micro types) and of its molecular variants, [8, 49, 53] which are difficult to distinguish in routinely processed FFPE samples. It is worth mentioning that the absence of neurotrophic factors was found to activate autophagocytosis, which may culminate in a specific type of programmed cell death (PCD II) [4, 59] or persist as an ongoing process [40]. This supports our hypothesis that the specific neurolysosomal pathology in human pSap deficiency may be closely related to the absence of pSap´s trophic function.
Selective vulnerability of the cortical neurons–the cortical neuronal survival crisis
The vulnerability of the cortical neurons to cell death in pSap deficiency is immense. The findings in the three presented cases suggest a critical inability of nearly the whole cortical neuronal population to survive after a certain postnatal time point. The subependymal germinal zone as well as the neuronal migration does not seem to be affected. This massive, stage dependant neuronal loss, most probably triggered by pSap absence, occurs within a time period of 60–80 days. With respect to the extent and relative abruptness of this phenomenon we would like to designate it as a cortical neuronal survival crisis. A similar phenomenon has never been described in any of the known neurolysosomal storage disorders, with the exception of infantile NCL1 and the recently described congenital NCL [60]. In infantile NCL1, the profound neuronal cortical depopulation develops over several years [56], strongly contrasting with the early cortical neuronal survival crisis in pSap deficiency. The congenital NCL, caused by profound cathepsin D deficiency, is featured by extensive prenatal neuronal loss.
Unfortunately, brains from other cases of pSap deficiency [19, 66] were not available for study. The loss of neuronal survival capacity as described has been repeatedly reported as a consequence of neurotrophic factor deprivation leading to programmed cell death, mostly from the caspase independent (non-apoptopic) type [1, 9, 26, 29, 52]. The absence of detectable activated caspase 3 in our cases is in accord with it. It should be stressed that an amino acid sequence has been recognized in pSap [33, 48, 51] as being responsible for the neurotrophic activities in a variety of neural cells in vitro and in vivo, including differentiation, cell death prevention, regeneration [5, 24, 32, 33, 47, 55], and stimulation of myelin synthesis [7, 21, 22]. Cell death prevention by pSap has also been described in non-neuronal U937 monocytic cells [7, 44]. It has been postulated that unprocessed pSap, localized to both fetal and adult human neurons [15, 31], exerts its neurotrophic effect partly through a putative Go protein coupled receptor [20] and MAP kinase [6], and partly due to its unique affinity for glycosphingolipids, particularly gangliosides [45].
The above-mentioned data support the concept of pSap as having a crucial role in the survival of human cortical neurons and indicate that its absence may be critical for their survival in early postnatal life.
In this context it is worth noting that in the mouse pSap KO model [17, 50], the neurolysosomal storage starts around day 10 and 30 in spinal and cerebral neurons, respectively, and progresses rapidly to full blown glycolipid storage. As in human cases, storage is accompanied, by a high degree of neuronal ubiquitination. The neurological symptoms start on day 20. The animals do not survive beyond day 40. No signs of neuronal cortical depopulation have been described. These findings might point to the absence of a significant trophic effect of pSap on normal mouse neurons despite the fact that there is a strong postnatal increase in its mRNA [62].
Conclusions and perspectives
Our results indicate that pSap deficiency in humans has different consequences with respect to the cell types affected. In non-neuronal cells, the disorder does not lead to any demonstrable trophic defect and is dominated by the biochemical sequels linked to the absence of Saps i.e. inactivation of multiple sphingolipid hydrolases. The resulting storage of undigested lipids markedly distends the lysosomal compartment. However, in the peripheral and central neurons, the consequences of pSap deficiency seem to be determined by the unique sensitivity of the neurons to the lack of a trophic effect of secreted unprocessed pSap. We speculate that the initial phase of the neurolysosomal process (in both brain and peripheral neurons) strongly resembles autophagocytosis, while lipid storage plays only a minor role. In the cortical neurons the process seems to be terminated after several weeks of life by massive cell death, which by exclusion corresponds most probably to the programmed cell death type II, followed by removal by microglial phagocytes. It appears likely that pSap is one of the trophic factors essential for survival, differentiation and maintenance of human neurons particularly in the early phases of brain development.
Acknowledgments
This work was supported by the research project MSM 0021620806 of the Ministry of Education, Youth and Sports of the Czech Republic.
References
- 1.Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, Thadani A, Squinto SP, Yancopoulos GD, Lindsay RM. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- 2.Bonifacino JS, Weissman AM. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol. 1998;14:19–57. doi: 10.1146/annurev.cellbio.14.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradova V, Smid F, Ulrich-Bott B, Roggendorf W, Paton BC, Harzer K. Prosaposin deficiency: further characterization of the sphingolipid activator protein-deficient sibs. Multiple glycolipid elevations (including lactosylceramidosis), partial enzyme deficiencies and ultrastructure of the skin in this generalized sphingolipid storage disease. Hum Genet. 1993;92:143–152. doi: 10.1007/BF00219682. [DOI] [PubMed] [Google Scholar]
- 4.Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8:569–581. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- 5.Calcutt NA, Campana WM, Eskeland NL, Mohiuddin L, Dines KC, Mizisin AP, O’Brien JS. Prosaposin gene expression and the efficacy of a prosaposin-derived peptide in preventing structural and functional disorders of peripheral nerve in diabetic rats. J Neuropathol Exp Neurol. 1999;58:628–636. doi: 10.1097/00005072-199906000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Campana WM, Hiraiwa M, Addison KC, O’Brien JS. Induction of MAPK phosphorylation by prosaposin and prosaptide in PC12 cells. Biochem Biophys Res Commun. 1996;229:706–712. doi: 10.1006/bbrc.1996.1869. [DOI] [PubMed] [Google Scholar]
- 7.Campana WM, Hiraiwa M, O’Brien JS. Prosaptide activates the MAPK pathway by a G-protein-dependent mechanism essential for enhanced sulfatide synthesis by Schwann cells. Faseb J. 1998;12:307–314. doi: 10.1096/fasebj.12.3.307. [DOI] [PubMed] [Google Scholar]
- 8.Cuervo AM, Dice JF. Lysosomes, a meeting point of proteins, chaperones, and proteases. J Mol Med. 1998;76:6–12. doi: 10.1007/s109-1998-8099-y. [DOI] [PubMed] [Google Scholar]
- 9.Davies AM. Promoting motor neuron survival. Curr Biol. 1993;3:879–881. doi: 10.1016/0960-9822(93)90224-C. [DOI] [PubMed] [Google Scholar]
- 10.Davis WL, Jacoby BH, Goodman DB. Immunolocalization of ubiquitin in degenerating insect flight muscle. Histochem J. 1994;26:298–305. doi: 10.1007/BF00157762. [DOI] [PubMed] [Google Scholar]
- 11.Dickson DW, Wertkin A, Kress Y, Ksiezak-Reding H, Yen SH. Ubiquitin immunoreactive structures in normal human brains. Distribution and developmental aspects. Lab Invest. 1990;63:87–99. [PubMed] [Google Scholar]
- 12.Elleder M. A histochemical and ultrastructural study of stored material in neuronal ceroid lipofuscinosis. Virchows Arch B Cell Pathol. 1978;28:167–178. doi: 10.1007/BF02889067. [DOI] [PubMed] [Google Scholar]
- 13.Elleder M, Jerabkova M, Befekadu A, Hrebicek M, Berna L, Ledvinova J, Hulkova H, Rosewich H, Schymik N, Paton BC, Harzer K. Prosaposin deficiency–a rarely diagnosed, rapidly progressing, neonatal neurovisceral lipid storage disease. Report of a further patient. Neuropediatrics. 2005;36:171–180. doi: 10.1055/s-2005-865608. [DOI] [PubMed] [Google Scholar]
- 14.Elleder M, Jirasek A, Smid F, Ledvinova J, Besley GT, Stopekova M. Niemann–Pick disease type C with enhanced glycolipid storage. Report on further case of so-called lactosylceramidosis. Virchows Arch A Pathol Anat Histopathol. 1984;402:307–317. doi: 10.1007/BF00695084. [DOI] [PubMed] [Google Scholar]
- 15.Fu Q, Carson GS, Hiraiwa M, Grafe M, Kishimoto Y, O’Brien JS. Occurrence of prosaposin as a neuronal surface membrane component. J Mol Neurosci. 1994;5:59–67. doi: 10.1007/BF02736694. [DOI] [PubMed] [Google Scholar]
- 16.Fujimuro M, Sawada H, Yokosawa H. Production and characterization of monoclonal antibodies specific to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Lett. 1994;349:173–180. doi: 10.1016/0014-5793(94)00647-4. [DOI] [PubMed] [Google Scholar]
- 17.Fujita N, Suzuki K, Vanier MT, Popko B, Maeda N, Klein A, Henseler M, Sandhoff K, Nakayasu H. Targeted disruption of the mouse sphingolipid activator protein gene: a complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum Mol Genet. 1996;5:711–725. doi: 10.1093/hmg/5.6.711. [DOI] [PubMed] [Google Scholar]
- 18.Gropper R, Brandt RA, Elias S, Bearer CF, Mayer A, Schwartz AL, Ciechanover A. The ubiquitin-activating enzyme, E1, is required for stress-induced lysosomal degradation of cellular proteins. J Biol Chem. 1991;266:3602–3610. [PubMed] [Google Scholar]
- 19.Harzer K, Paton BC, Poulos A, Kustermann-Kuhn B, Roggendorf W, Grisar T, Popp M. Sphingolipid activator protein deficiency in a 16-week-old atypical Gaucher disease patient and his fetal sibling: biochemical signs of combined sphingolipidoses. Eur J Pediatr. 1989;149:31–39. doi: 10.1007/BF02024331. [DOI] [PubMed] [Google Scholar]
- 20.Hiraiwa M, Campana WM, Martin BM, O’Brien JS. Prosaposin receptor: evidence for a G-protein-associated receptor. Biochem Biophys Res Commun. 1997;240:415–418. doi: 10.1006/bbrc.1997.7673. [DOI] [PubMed] [Google Scholar]
- 21.Hiraiwa M, Campana WM, Mizisin AP, Mohiuddin L, O’Brien JS. Prosaposin: a myelinotrophic protein that promotes expression of myelin constituents and is secreted after nerve injury. Glia. 1999;26:353–360. doi: 10.1002/(SICI)1098-1136(199906)26:4<353::AID-GLIA9>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 22.Hiraiwa M, Taylor EM, Campana WM, Darin SJ, O’Brien JS. Cell death prevention, mitogen-activated protein kinase stimulation, and increased sulfatide concentrations in Schwann cells and oligodendrocytes by prosaposin and prosaptides. Proc Natl Acad Sci U S A. 1997;94:4778–4781. doi: 10.1073/pnas.94.9.4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horak J. The role of ubiquitin in down-regulation and intracellular sorting of membrane proteins: insights from yeast. Biochim Biophys Acta. 2003;1614:139–155. doi: 10.1016/S0005-2736(03)00195-0. [DOI] [PubMed] [Google Scholar]
- 24.Hozumi I, Hiraiwa M, Inuzuka T, Yoneoka Y, Akiyama K, Tanaka R, Kikugawa K, Nakano R, Tsuji S, O’Brien JS. Administration of prosaposin ameliorates spatial learning disturbance and reduces cavity formation following stab wounds in rat brain. Neurosci Lett. 1999;267:73–76. doi: 10.1016/S0304-3940(99)00325-0. [DOI] [PubMed] [Google Scholar]
- 25.Hulkova H, Cervenkova M, Ledvinova J, Tochackova M, Hrebicek M, Poupetova H, Befekadu A, Berna L, Paton BC, Harzer K, Boor A, Smid F, Elleder M. A novel mutation in the coding region of the prosaposin gene leads to a complete deficiency of prosaposin and saposins, and is associated with a complex sphingolipidosis dominated by lactosylceramide accumulation. Hum Mol Genet. 2001;10:927–940. doi: 10.1093/hmg/10.9.927. [DOI] [PubMed] [Google Scholar]
- 26.Hutchins JB, Barger SW. Why neurons die: cell death in the nervous system. Anat Rec. 1998;253:79–90. doi: 10.1002/(SICI)1097-0185(199806)253:3<79::AID-AR4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 27.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, Noda T, Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147:435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirkland RA, Adibhatla RM, Hatcher JF, Franklin JL. Loss of cardiolipin and mitochondria during programmed neuronal death: evidence of a role for lipid peroxidation and autophagy. Neuroscience. 2002;115:587–602. doi: 10.1016/S0306-4522(02)00512-2. [DOI] [PubMed] [Google Scholar]
- 30.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kondoh K, Sano A, Kakimoto Y, Matsuda S, Sakanaka M. Distribution of prosaposin-like immunoreactivity in rat brain. J Comp Neurol. 1993;334:590–602. doi: 10.1002/cne.903340407. [DOI] [PubMed] [Google Scholar]
- 32.Kotani Y, Matsuda S, Sakanaka M, Kondoh K, Ueno S, Sano A. Prosaposin facilitates sciatic nerve regeneration in vivo. J Neurochem. 1996;66:2019–2025. doi: 10.1046/j.1471-4159.1996.66052019.x. [DOI] [PubMed] [Google Scholar]
- 33.Kotani Y, Matsuda S, Wen TC, Sakanaka M, Tanaka J, Maeda N, Kondoh K, Ueno S, Sano A. A hydrophilic peptide comprising 18 amino acid residues of the prosaposin sequence has neurotrophic activity in vitro and in vivo. J Neurochem. 1996;66:2197–2200. doi: 10.1046/j.1471-4159.1996.66052197.x. [DOI] [PubMed] [Google Scholar]
- 34.Kovacs GG, Flicker H, Budka H. Immunostaining for ubiquitin: efficient pretreatment. Neuropathol Appl Neurobiol. 2003;29:174–177. doi: 10.1046/j.1365-2990.2003.00433.x. [DOI] [PubMed] [Google Scholar]
- 35.Landmann L. Deconvolution improves colocalization analysis of multiple fluorochromes in 3D confocal data sets more than filtering techniques. J Microsc. 2002;208:134–147. doi: 10.1046/j.1365-2818.2002.01068.x. [DOI] [PubMed] [Google Scholar]
- 36.Laszlo L, Doherty FJ, Osborn NU, Mayer RJ. Ubiquitinated protein conjugates are specifically enriched in the lysosomal system of fibroblasts. FEBS Lett. 1990;261:365–368. doi: 10.1016/0014-5793(90)80593-8. [DOI] [PubMed] [Google Scholar]
- 37.Laszlo L, Doherty FJ, Watson A, Self T, Landon M, Lowe J, Mayer RJ. Immunogold localisation of ubiquitin-protein conjugates in primary (azurophilic) granules of polymorphonuclear neutrophils. FEBS Lett. 1991;279:175–178. doi: 10.1016/0014-5793(91)80142-P. [DOI] [PubMed] [Google Scholar]
- 38.Lenk SE, Dunn WA, Jr, Trausch JS, Ciechanover A, Schwartz AL. Ubiquitin-activating enzyme, E1, is associated with maturation of autophagic vacuoles. J Cell Biol. 1992;118:301–308. doi: 10.1083/jcb.118.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lowe J, McDermott H, Kenward N, Landon M, Mayer RJ, Bruce M, McBride P, Somerville RA, Hope J. Ubiquitin conjugate immunoreactivity in the brains of scrapie infected mice. J Pathol. 1990;162:61–66. doi: 10.1002/path.1711620112. [DOI] [PubMed] [Google Scholar]
- 40.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 41.Mayer RJ, Lowe J, Landon M, McDermott H, Laszlo L. The role of protein ubiquitination in neurodegenerative disease. Acta Biol Hung. 1991;42:21–26. [PubMed] [Google Scholar]
- 42.Migheli A, Attanasio A, Pezzulo T, Gullotta F, Giordana MT, Schiffer D. Age-related ubiquitin deposits in dystrophic neurites: an immunoelectron microscopic study. Neuropathol Appl Neurobiol. 1992;18:3–11. doi: 10.1111/j.1365-2990.1992.tb00760.x. [DOI] [PubMed] [Google Scholar]
- 43.Millat G, Verot L, Rodriguez-Lafrasse C, Di Marco JN, Rimet Y, Poujol A, Girard N, Monges G, Livet MO, Vanier MT (2003) Fourth reported family with prosaposin deficiency. 14th ESGLD Workshop (September 18th-21st, 2003). Podebrady/Prague, Czech Republic
- 44.Misasi R, Garofalo T, Di Marzio L, Mattei V, Gizzi C, Hiraiwa M, Pavan A, Grazia Cifone M, Sorice M. Prosaposin: a new player in cell death prevention of U937 monocytic cells. Exp Cell Res. 2004;298:38–47. doi: 10.1016/j.yexcr.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 45.Misasi R, Sorice M, Garofalo T, Griggi T, Campana WM, Giammatteo M, Pavan A, Hiraiwa M, Pontieri GM, O’Brien JS. Colocalization and complex formation between prosaposin and monosialoganglioside GM3 in neural cells. J Neurochem. 1998;71:2313–2321. doi: 10.1046/j.1471-4159.1998.71062313.x. [DOI] [PubMed] [Google Scholar]
- 46.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 47.O’Brien JS, Carson GS, Seo HC, Hiraiwa M, Kishimoto Y. Identification of prosaposin as a neurotrophic factor. Proc Natl Acad Sci U S A. 1994;91:9593–9596. doi: 10.1073/pnas.91.20.9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Brien JS, Carson GS, Seo HC, Hiraiwa M, Weiler S, Tomich JM, Barranger JA, Kahn M, Azuma N, Kishimoto Y. Identification of the neurotrophic factor sequence of prosaposin. Faseb J. 1995;9:681–685. doi: 10.1096/fasebj.9.8.7768361. [DOI] [PubMed] [Google Scholar]
- 49.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 50.Oya Y, Nakayasu H, Fujita N, Suzuki K. Pathological study of mice with total deficiency of sphingolipid activator proteins (SAP knockout mice) Acta Neuropathol (Berl) 1998;96:29–40. doi: 10.1007/s004010050857. [DOI] [PubMed] [Google Scholar]
- 51.Qi X, Qin W, Sun Y, Kondoh K, Grabowski GA. Functional organization of saposin C. Definition of the neurotrophic and acid beta-glucosidase activation regions. J Biol Chem. 1996;271:6874–6880. doi: 10.1074/jbc.271.12.6874. [DOI] [PubMed] [Google Scholar]
- 52.Rabizadeh S, Bredesen DE. Ten years on: mediation of cell death by the common neurotrophin receptor p75(NTR) Cytokine Growth Factor Rev. 2003;14:225–239. doi: 10.1016/S1359-6101(03)00018-2. [DOI] [PubMed] [Google Scholar]
- 53.Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem. 2000;275:27447–27456. doi: 10.1074/jbc.M001394200. [DOI] [PubMed] [Google Scholar]
- 54.Sandhoff K, Kolter T, Harzer K. Sphingolipid activator proteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3371–3388. [Google Scholar]
- 55.Sano A, Matsuda S, Wen TC, Kotani Y, Kondoh K, Ueno S, Kakimoto Y, Yoshimura H, Sakanaka M. Protection by prosaposin against ischemia-induced learning disability and neuronal loss. Biochem Biophys Res Commun. 1994;204:994–1000. doi: 10.1006/bbrc.1994.2558. [DOI] [PubMed] [Google Scholar]
- 56.Santavuori P, Gottlob I, Haltia M, Rapola J, Lake BD, Tyynelä J, Peltonen L. CLN1. Infantile and other types of NCL with GROD. In: Goebel HH, Mole SE, Lake BD, editors. The Neuronal Ceroid Lipofuscinoses (Batten Disease) Amsterdam: IOS Press; 1999. pp. 16–36. [Google Scholar]
- 57.Shaw PJ, Rawlins DJ. Three-dimensional fluorescence microscopy. Prog Biophys Mol Biol. 1991;56:187–213. doi: 10.1016/0079-6107(91)90013-I. [DOI] [PubMed] [Google Scholar]
- 58.Schwartz AL, Ciechanover A, Brandt RA, Geuze HJ. Immunoelectron microscopic localization of ubiquitin in hepatoma cells. Embo J. 1988;7:2961–2966. doi: 10.1002/j.1460-2075.1988.tb03158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwartz LM, Smith SW, Jones ME, Osborne BA. Do all programmed cell deaths occur via apoptosis? Proc Natl Acad Sci U S A. 1993;90:980–984. doi: 10.1073/pnas.90.3.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Siintola E, Partanen S, Stromme P, Haapanen A, Haltia M, Maehlen J, Lehesjoki AE, Tyynela J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006;129:1438–1445. doi: 10.1093/brain/awl107. [DOI] [PubMed] [Google Scholar]
- 61.Skinner PJ, Vierra-Green CA, Clark HB, Zoghbi HY, Orr HT. Altered trafficking of membrane proteins in purkinje cells of SCA1 transgenic mice. Am J Pathol. 2001;159:905–913. doi: 10.1016/S0002-9440(10)61766-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun Y, Witte DP, Grabowski GA. Developmental and tissue-specific expression of prosaposin mRNA in murine tissues. Am J Pathol. 1994;145:1390–1398. [PMC free article] [PubMed] [Google Scholar]
- 63.Suzuki K, Terry RD. Fine structural localization of acid phosphatase in senile plaques in Alzheimer’s presenile dementia. Acta Neuropathol (Berl) 1967;8:276–284. doi: 10.1007/BF00688828. [DOI] [PubMed] [Google Scholar]
- 64.Voort HT, Strasters KC. Restoration of Confocal Images for Quantitative Image Analysis. J Micr. 1995;178:165–181. [Google Scholar]
- 65.Steensel B, Binnendijk EP, Hornsby CD, Voort HT, Krozowski ZS, Kloet ER, Driel R. Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J Cell Sci. 1996;109:787–792. doi: 10.1242/jcs.109.4.787. [DOI] [PubMed] [Google Scholar]
- 66.Vanier MT, Millat G, Verot L, Di Marco JN, Rimet Y, Poujol A, Girard N, Monges G, Livet MO (2003) Fourth reported family with prosaposin deficiency. In: Proceedings of international symposium on lysosomal storage diseases, Santiago de Compostela, Spain
- 67.Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H. p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160:255–263. doi: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhan SS, Beyreuther K, Schmitt HP. Neuronal ubiquitin and neurofilament expression in different lysosomal storage disorders. Clin Neuropathol. 1992;11:251–255. [PubMed] [Google Scholar]