

Abstract
Thalamocortical afferents innervate both excitatory and inhibitory cells, the latter in turn producing disynaptic feedforward inhibition, thus creating fast excitation–inhibition sequences in the cortical cells. Since this inhibition is disynaptic, the time lag of the excitation–inhibition sequence could be ∼2–3 ms, while it is often as short as only slightly above 1 ms; the mechanism and function of such fast IPSPs are not fully understood. Here we show that thalamic activation of inhibitory neurons precedes that of excitatory neurons, due to increased conduction velocity of thalamic axons innervating inhibitory cells. Developmentally, such latency differences were seen only after the end of the second postnatal week, prior to the completion of myelination of the thalamocortical afferent. Furthermore, destroying myelination failed to extinguish the latency difference. Instead, axons innervating inhibitory cells had consistently lower threshold, indicating they had larger diameter, which is likely to underlie the differential conduction velocity. Since faster activation of GABAergic neurons from the thalamus can not only curtail monosynaptic EPSPs but also make disynaptic ISPSs precede disynaptic EPSPs, such suppression theoretically enables a temporal separation of thalamically driven mono- and disynaptic EPSPs, resulting in spike sequences of ‘L4 leading L2/3’. By recording L4 and L2/3 cells simultaneously, we found that suppression of IPSPs could lead to deterioration of spike sequences. Thus, from the end of the second postnatal week, by activating GABAergic neurons prior to excitatory neurons from the thalamus, fast feedforward disynaptic suppression on postsynaptic cells may play a role in establishing the spike sequences of ‘L4 leading L2/3 cells’.
Introduction
The neocortex receives its fundamental input from the thalamus. In the rodent somatosensory cortex, thalamocortical afferents make synaptic contacts not only with excitatory relay cells but also with inhibitory cells, the latter of which then make synapses with neighbouring cells, thus forming feedforward inhibition (Agmon & Connors, 1992; Gil & Amitai, 1996; Beierlein et al. 2003; Gabernet et al. 2005; Inoue & Imoto, 2006; Sun et al. 2006). This feedforward inhibition creates a short temporal window of excitation during which action potentials are allowed to pass through for further processing. Thus, it controls spike timing, or works as a ‘coincidence detector’ (Gabernet et al. 2005). To create a narrower window, disynaptic inhibition is expected to occur in a short delay from the onset of monosynaptic EPSPs, which is, in fact, the case; the onset of IPSPs is delayed by little more than 1 ms in most cases from that of EPSPs (Gabernet et al. 2005; Cruikshank et al. 2007). This is remarkably short, considering that it includes the time for (1) spike generation in the inhibitory cell, (2) conduction of an action potential from soma to the axon terminal and (3) synaptic delay to the postsynaptic excitatory cell, whereas spike generation alone takes not less than 1 ms.
In the current study, we have found a mechanism that directly accounts for this timing challenge. Our observations indicate that thalamocortical latency is shorter to inhibitory cells than to excitatory cells. Such differential latency results from differential conduction velocity of axons, due most likely to the differences in axon diameter. We also show theoretical and experimental evidence that such earlier activation of feedforward inhibition could possibly create temporal separation of monosynaptic and disynaptic excitation from thalamus by precisely timed disynaptic inhibition. Thus, we revealed a precise network mechanism of regulating spike sequences from thalamic input in the neocortex.
Methods
Ethical information
All procedures comply with the policies and regulations of The Journal of Physiology (Drummond, 2009) and the rules of the Animal Experiment Committee of Osaka University.
Strain and maintenance of mice
We used GAD67-GFP (Δneo) mice expressing an enhanced green fluorescent protein (EGFP) under the control of the endogenous promoter for glutamate decarboxylase 67 (GAD67), as described in detail previously (Tamamaki et al. 2003). We crossed these transgenic mice with wild-type C57BL/6 mice and used the resultant heterozygous transgenic mice, which are here referred to as GAD67-GFP mice for simplicity.
Whole-cell patch recording
A total of 76 mice were used in the present study. The animals were housed with ad libitum access to food and water in a room air-conditioned at 22–23°C with a standard 12 h light–dark cycle. Mice, aged 5–32 postnatal day, were deeply anaesthetized with isoflurane (>2%, inhalation); their brains were removed and thalamocortical slices were cut (Agmon & Connors, 1991), as detailed elsewhere with some modification (Itami et al. 2001; Yanagisawa et al. 2004). In brief, slices were cut in a ‘slicing solution’ which contained (mm): sucrose 240, KCl 5, NaHCO3 26, glucose 10, MgCl2 1. Slices were subsequently transferred to artificial cerebrospinal fluid (ACSF) containing (mm): NaCl 124, KCl 3, KH2PO4 1.2, MgSO4 1.3, NaHCO3 26, CaCl2 2, and glucose 10, then bubbled with 95% O2–5% CO2. Whole-cell patch pipettes (5–8 MΩ) were used to record membrane voltages or currents from GFP-positive or negative cells in layer 4, and in some cases layer 2/3 as described in Results, using one or two Multiclamp 700A amplifiers (Molecular Devices, Sunnyvale, CA, USA), low-pass filtered at 5 kHz, digitally sampled at 10–20 kHz, and monitored with pCLAMP software (Molecular Devices). Micropipettes were pulled from borosilicate glass capillary (Sutter Instrument Co., Novato, CA, USA). Recordings were performed at room temperature maintained at 27–29°C. Upon inserting the electrode into the bath, stray pipette capacitance was compensated, and so was the bridge balance through a built-in circuit of the amplifier. Bridge balance was checked repeatedly and readjusted if necessary.
Recordings were obtained from visually identified neurons using infrared differential interference contrast optics. Only cells showing resting potentials deeper than –65 mV with overshooting action potentials were accepted. When whole-cell recordings were established, the spike frequency adaptation ratio (SR) was examined for identification of cell types; it was defined as the ratio of the first interspike interval to the average of the last three interspike intervals during 500 ms long spike trains caused by depolarizing current injections. GFP-positive cells with high SR (>0.7), spike half-height width <0.6, having large monophasic afterhyperpolarization (AHP) were classified as fast spiking (FS) cells (Porter et al. 2001; Beierlein et al. 2003; Itami et al. 2007; Tan et al. 2008; Yazaki-Sugiyama et al. 2009), while others were classified as regular spiking non pyramidal (RSNP) cells (Fig. 3G). GFP-positive cells without clear identification of cell types were referred to as GABA cells. When wild-type were used, excitatory cells were identified by characteristic sudden decrease in the number of spikes in response to increasing current steps, as we previously reported (Itami et al. 2007). Postsynaptic responses were recorded simultaneously from multiple (up to four) cells located within the same barrel, in which at least one of the cells was GFP-positive. Each barrel was clearly identified under an upright microscope. In some experiments, L2/3 cells located immediately above the L4 barrel were simultaneously recorded, as described in Results. To avoid responses resulting from antidromic activation of cortico-thalamic axons, only those with short latency (<4 ms), paired pulse depression, and displaying no supernormality were adopted in the present analysis (Beierlein & Connors, 2002). The internal electrode solution consisted of (mm): potassium methanesulfonate 130, KCl 10, Hepes 10, K-EGTA 0.5, MgATP 5, NaGTP 1, spermine 0.1, and phosphocreatinine 10 (pH 7.3, 285–290 mosmol l−1). In some experiments, biocytin or neurobiotin was added at a concentration of 0.3–0.5%. Concentric bipolar stimulating electrodes (Inter Medical, Nagoya, Japan or Frederic Haer & Co, Bowdoin, ME, USA), through which electrical stimuli consisting of square pulses for 100 μs, 1–50 V were applied at 0.1–0.16 Hz, were placed on the ventrobasal (VB) nucleus of the thalamus or on the thalamic fibres issuing from the thalamus to the cortex. When necessary, an additional stimulating electrode was placed on the white matter (WM), as described in Results.
Figure 3. Inhibitory neurons had shorter latencies than excitatory neurons from the thalamus in the same barrel.
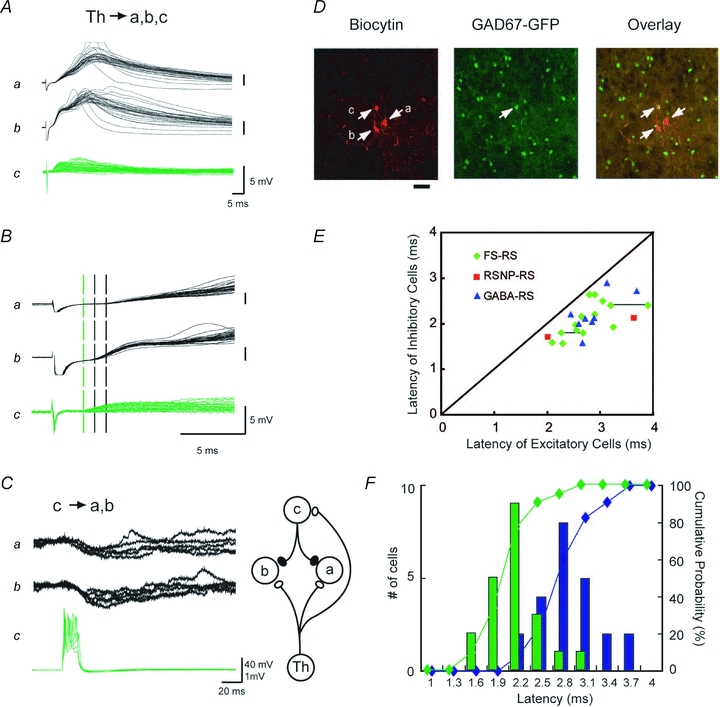
A–D, an example of three cells located in the same barrel, one inhibitory (c) and two excitatory (a and b). The shortest latency following thalamic stimulation was obtained from the inhibitory cell (c). A, EPSPs elicited by thalamic stimulation. B, EPSP onsets in A are expanded. Three broken vertical lines (one green and two black) indicate the onset of each EPSP (one GABA and two excitatory cells, respectively). C, action potentials in cell c reliably produced IPSPs in both cells a and b, confirming electrophysiologically that cell c was a functionally active inhibitory cell. D, fluorescence confocal photomicrographs of biocytin labelling (left, red), GFP (middle, green), and overlay (right, yellow), showing that only cell c was GFP-positive. Note that, as the pial surface is toward the top, c was located above the excitatory cells, and thus, was the most distant from the thalamus of the three, as illustrated in C right. Scale; 20 μm. E, for simultaneously recorded pairs and triplets, latencies for thalamically evoked EPSP(C)s from GFP-positive (ordinate) and -negative (abscissa) cells were plotted against each other. Symbols represent the types of GABAergic cells in each pair, as determined by characteristic firing properties in response to depolarizing current pulses as illustrated in Fig. 2E. Those pairs with unknown cell types of GFP-positive cells are labelled GABA (blue triangle). Symbols connected by a line are from triple recordings, consisting of one excitatory and two GABAergic cells. F, distribution histogram of latencies for thalamically evoked EPSP(C)s from GFP-positive GABAergic (green) and GFP-negative excitatory cells (blue), together with cumulative plots for GABA cells (green) and excitatory cells (blue). All the data collected across the different barrels and mice were pooled.
Measurement of the onset latency of PSP(C)s and action potentials
Onset latency of EPSP(C)s was determined by measuring the time interval between the beginning of the stimulation pulse and the onset of EPSP(C)s in each trace, then the mean of 10 values was calculated for a given connection. Onset of EPSP(C)s was defined as the point that exceeded 3 ×s.d. of the noise of the baseline region (2 ms) prior to stimulation. In principle, voltage recording is a filtered version of synaptic conductance; as far as the onset latency was concerned, current and voltage recordings provided no significant differences in the measurement. In 12 cells from young mice (age 5–10 days postnatal, n= 6), we performed both voltage and current recordings and compared the values, and found that there was no significant differences (4.65 ± 0.18 ms and 4.61 ± 0.18 ms, for voltage and current recordings, respectively, P > 0.01, two-tailed paired t-test). Onset latency of IPSPs was determined in the same way with depolarized membrane potential (−45 to −50 mV) by current injection. When IPSPs appeared over EPSPs, onset of IPSPs was determined in the following way. First derivatives of voltage changes (dV/dt) were calculated from recordings of a period of several milliseconds including the IPSP onset, then the time giving the derivative of 0 was defined as the onset of IPSPs (Fig. 1Ab). This method provides the time where the inhibitory and excitatory currents are equal, which is slightly longer than the exact inhibitory latency. Latencies for the peak of action potentials were measured from the onset of EPSPs, as defined above, to the peak of action potentials. In cases where averages of PSP amplitudes are given, only the averages of successes were calculated.
Figure 1. Fast IPSPs elicited by thalamic stimulation appeared in a short delay from monosynaptic thalamic EPSPs.
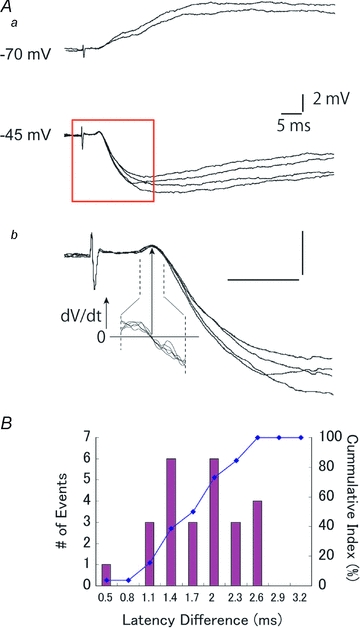
Aa, an example of EPSP–IPSP sequences in response to thalamic stimulations recorded at −70 and −45 mV. Ab, recordings in the red square in Aa are shown expanded. To identify the onset of IPSP over EPSP, the first derivative (dV/dt) of the membrane voltage at the onset region was calculated, then the zero crossing of the dV/dt curve was defined as the onset of IPSP, as shown by an arrow. B, distribution for the temporal difference between the onset of thalamic EPSPs and fast IPSPs.
Calculation of conduction velocity of thalamic axons
In some experiments, another stimulating electrode was placed at the white matter, and EPSCs were recorded by white matter stimulation in addition to thalamic stimulation. Then, conduction velocities of thalamic axons were calculated by the differential distances divided by the differential latencies from these two stimulations. The measurement of distance was performed on digital images taken after the experiment including two stimulation sites and a recording site, by estimating the axon trajectory along the tract, as carried out previously (Salami et al. 2003).
Histology
After recordings, slices were fixed with 4% paraformaldehyde with 10% sucrose in 0.1 m phosphate buffered saline (PBS) for at least 1 h, washed in PBS with 0.2% Triton X-100 (PBS-TX), and then incubated with streptavidin conjugated with Alexa Fluor 546 (Molecular Probes, diluted 1:400) in PBS-TX overnight at 4°C. Subsequently, the slices were washed twice in PBS-TX, and then observed under a confocal microscope.
Lysolecithin injection
To examine the involvement of myelination in producing differences in conduction velocity, we attempted to destroy the myelination of the axons in the cortex by introducing lysophosphatidyl choline, or lysolecithin (Sigma). A total of three mice were anaesthetized by a mixture of ketamine and xylazine (0.05 g and 4.5 mg kg−1, i.p.). Heart rate was continuously monitored during the operation. The depth of anaesthesia was consistently checked by light pinches applied to the tail, and when animals responded by an increase of respiration and/or heart rate, additional doses (0.02 g and 2 mg kg−1, i.p.) were given. After administrating local xylocaine anaesthesia (∼1 ml, subcutaneous), a small hole (less than 1 mm in diameter) was made over the right S1 area. We first injected 2 μl of lysolecithin solution (∼1–2% in saline) using a Hamilton syringe. However, because the drug diffusion was quite restricted to the injection site, we then used a micro-osmotic pump for slow but wider perfusion of lysolecithin solution. A 30G stainless steel cannula connected to a micro-osmotic pump (Alzet 1003D, Alza, Cupertino, USA) was stereotaxically implanted into one hemisphere (from bregma, 3.2 mm lateral, 1.6 mm caudal, 0.7 mm depth) of the barrel cortex in three mice at 22, 27 and 29 postnatal days on the day of electrophysiology, and lysolecithin solution was infused continuously for 3–5 days (1 μl h−1, total perfusion, approximately 90 μl). Buprenorphine (0.05 mg kg−1s.c., b.i.d.) and procaine penicillin (4000 u kg−1, s.c., u.i.d.) were applied for analgesic and antibiotic purpose, respectively. On the day of electrophysiology, the implanted pump and cannula were removed under anaesthesia and special care was taken not to induce physical damage to the injected area of the cortex. Thalamocortical slices were then prepared as described above.
All data are expressed as means ± standard error of the mean (s.e.m.), unless otherwise stated in the text.
Results
Fast disynaptic IPSPs following thalamic activation
Thalamocortical afferents make excitatory synapses onto inhibitory as well as excitatory relay cells, and the inhibitory cells make feedforward connections onto neighbouring cells (Agmon & Connors, 1992; Gil & Amitai, 1996; Beierlein et al. 2003; Gabernet et al. 2005; Inoue & Imoto, 2006; Sun et al. 2006). The resultant disynaptic inhibitory responses often appear after a considerably short delay from the preceding monosynaptic EPSPs (Agmon & Connors, 1992; Porter et al. 2001; Gabernet et al. 2005; Cruikshank et al. 2007; Hull & Scanziani, 2007) (Fig. 1A). To quantify this, we measured the latency differences between the onset of IPSPs and that of monosynaptic EPSPs, as illustrated in Fig. 1B. On average, the onset of IPSPs was delayed from EPSPs by 1.7 ± 0.1 ms (range: 0.5–2.6 ms, mode: 1.4 ms and 2 ms, n= 26). Values of some IPSPs are remarkably short, considering that the delay includes time for (1) spike generation in the inhibitory cell that is monosynaptically activated from the thalamus, (2) conduction of an action potential from the soma to the axon terminal, and (3) synaptic delay to the postsynaptic cells. To obtain precise evaluation of each process, we performed a series of experiments with multiple patch clamp recordings from neighbouring cells within a single barrel, then measured monosynaptic latencies for excitatory and inhibitory connections and the time for spike generations. We used GAD67-GFP mice in which GABAergic cells express GFP (Tamamaki et al. 2003) to facilitate the discrimination of GABAergic neurons in slices.
We first confirmed that all GFP-labelled cells were indeed GABAergic neurons (Supplemental Fig. 1); thus, we could reasonably trust the GFP labelling as an index for a GABAergic cell in the present study. As summarized in Fig. 2, it took 0.96 ± 0.09 ms (n= 36) for monosynaptic excitatory connections; similarly, 0.87 ± 0.08 ms (n= 16) for monosynaptic inhibitory connections. To generate an action potential, it took 1.43 ± 0.41 ms (n= 12) for fast spiking inhibitory cells, as measured from the onset of EPSPs to the peak of action potentials elicited by stimulation strength moderately above an action potential threshold (typically, less than 1.5 × threshold).
Figure 2. Properties of monosynaptic intracortical connections obtained by dual whole cell recordings from neurons located in a single barrel.
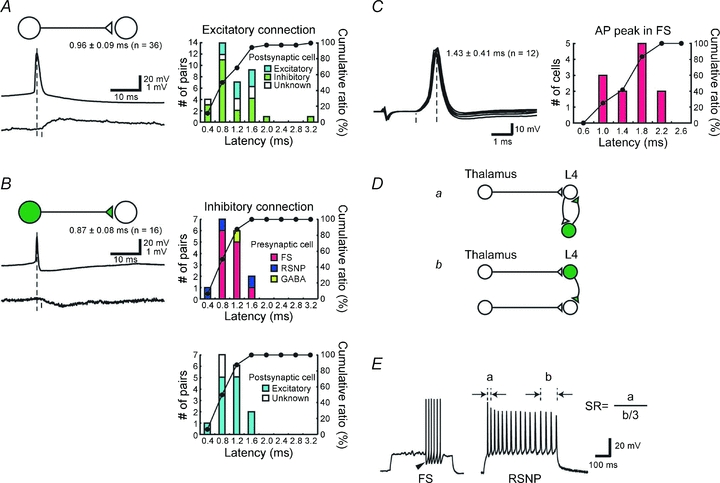
A and B, latency analysis of monosynaptic excitatory (A) and inhibitory (B) connections. Distribution histograms and cumulative plots are shown on the right. Postsynaptic cell types, either excitatory or inhibitory, are indicated by colour in A. In B, the same histograms are presented with different breakdown of presynaptic (upper) and postsynaptic (lower) cell types. C, histogram showing latencies for action potentials measured from the onsets of EPSPs to the action potential peaks in FS cells. D, schematic drawings for generating fast IPSPs. a, thalamic activation of excitatory cells produces feedback IPSPs via neighbouring inhibitory cells, and thus IPSPs are trisynaptic from the thalamic cell. b, direct activations of inhibitory cells from the thalamus that are fed forward to excitatory cells. Thus IPSPs are disynaptic from the thalamus, and therefore faster by one synapse than the trisynaptic IPSP shown in a. E, example responses of inhibitory neurons to depolarizing current injections. GFP-positive neurons were divided into fast spiking (FS) or regular spiking non-pyramidal (RSNP) by the spike frequency adaptation ratio (SR) and large monophasic AHP (arrowhead) and half-height spike width. SR was defined as the ratio of the first interspike interval divided by the average of the last three interspike intervals, as shown here.
Based on these statistics, we estimated how much the fastest IPSP could be delayed from the thalamocortical EPSPs in the cortical cells. Figure 2D shows two possible example circuits that could potentially produce fast inhibition. Figure 2Da represents an indirect activation of GABAergic cells through an excitatory cell; feedback IPSPs are generated trisynaptically from the thalamus. In contrast, Fig. 2Db represents direct activation of GABAergic cells from the thalamus, which are fed forward to produce IPSPs disynaptically from the thalamus in nearby cells. Obviously, feedforward inhibition is faster by one excitatory synapse than feedback inhibition. On the basis of the results from dual recordings (Fig. 2A–C), the delay of the feedforward IPSP from the thalamic EPSP would be calculated to be approximately 2.3 ms (1.43 + 0.87 ms for spike generation, conduction and transmission from a GABA cell to an excitatory cell) in disynaptic feedforward inhibition (Fig. 2Db). This is substantially longer than the observed latency differences (1.7 ± 0.1 ms Fig. 1B). In cases of trisynaptic inhibition (Fig. 2Da), the IPSP would be delayed by 3.3 ms (0.96 + 1.43 + 0.87 ms) without considering the spike generation time for excitatory cells. This calculation assumes spike initiation latency in GABA cells is the same between thalamocortical and intracortical excitatory afferents. This is because it is impossible to directly measure the EPSP to spike latency by intracortical excitatory afferents in GABAergic cells, for an activation of a single intracortical excitatory cell never produced action potentials in postsynaptic GABAergic cells. This assumption, however, may well be an underestimation, because thalamocortical afferents produce stronger responses compared with intracortical inputs (Gil & Amitai, 1996; Gil et al. 1999). Thus there is no possibility that this circuit (Fig. 2Da) is involved, and it can be excluded from consideration. How could a feedforward circuit as simple as the one shown in Fig. 2Db shorten the time for the appearance of IPSPs? Since we have studied the intracortical inhibitory connections, the only possibility that might account for this discrepancy is the thalamocortical connection. If the latency to inhibitory cells was shorter than that to excitatory cells, feedforward IPSPs could appear with a shorter latency. Thus, we tested this possibility directly.
Faster activation of inhibitory cells than excitatory cells from thalamic inputs
We examined whether the thalamocortical latency to a postsynaptic inhibitory cell was shorter than that to an excitatory cell using thalamocortical slices. From simultaneous recordings of inhibitory and excitatory cells, we found that, in a given barrel, latencies for GFP-positive GABAergic neurons were shorter than those for GFP-negative excitatory cells. An example is illustrated in Fig. 3A–D, where EPSPs of a GFP-positive neuron (cell c), exhibited the shortest latency of the three in a simultaneous recording (a: 3.7 ms, b: 3.0 ms, c: 2.2 ms). Cell c was monosynaptically connected to both cells a and b, and action potentials in cell c produced IPSPs in postsynaptic cells a and b, indicating that this cell was indeed a functional GABAergic neuron (Fig. 3C). Subsequent histological staining with biocytin, injected through the recording pipettes revealed that c was the sole GFP-positive cell among the three (Fig. 3D). Results from similar experiments are summarized in Fig. 3E and F. When the latencies of inhibitory and excitatory cells recorded simultaneously from a single barrel (18 pairs and 3 triplets) were plotted against each other (Fig. 3E), all points fell below the diagonal line, indicating that GABAergic cells had shorter onset latencies than excitatory cells.
In a cumulative histogram from all barrels (Fig. 3F), the mean latencies for inhibitory and excitatory cells were 2.1 ± 0.1 (n= 21) and 2.8 ± 0.1 ms (n= 23), respectively, demonstrating that the curve for inhibitory cells was shifted towards the left by 0.7 ms with a substantial overlap, compared with that for excitatory cells. Thus, although there were some excitatory cells with shorter latencies than inhibitory cells across the barrels, in a given barrel, inhibitory cells were activated earlier.
Such a displacement of the two distributions could result from an anatomically heterogeneous distribution of the cells or a biased sampling of the cells recorded, although GABAergic cells are rather evenly distributed within a barrel (Staiger et al. 1996). In fact, we intentionally avoided such a biased sampling, and rather tried to record inhibitory cells from the upper and excitatory cells from the lower part of the barrels, as shown in Fig. 3D. However, we further examined this possibility in detail by recording still more neurons from a single barrel, as shown in Fig. 4A. In this experiment, we recorded thalamic EPSPs from four neurons simultaneously in the first session. Subsequently, we replaced three electrodes while holding one neuron still recording, then we succeeded in obtaining EPSPs from three more neurons, and thus in total we recorded EPSPs from seven neighbouring cells quasi-simultaneously. Of these seven neurons, only two were GFP-positive inhibitory cells (b and c in Fig. 4A–C). In later analysis, these two inhibitory neurons turned out to have shorter latencies than the other five excitatory cells (Fig. 4B and C). It appeared that the greater the distance of a cell from the bottom cell, the larger the latency tended to be in the three excitatory cells (Fig. 4C, d, a′ and c′), indicating that these neurons may have been innervated by the same afferent. Although there is no such linear tendency between distance and latency, inhibitory neurons had shorter latencies than any of the excitatory neurons. Similar tendencies were seen in three independent experiments (Fig. 4D–F). These data might indicate that the latencies for excitatory and inhibitory cells from the thalamus were independently controlled. From these experiments, we concluded that GABAergic neurons had shorter latencies than excitatory cells irrespective of their locations in a barrel.
Figure 4. GABAergic cells had shorter latencies than excitatory cells irrespective of their locations in a barrel.
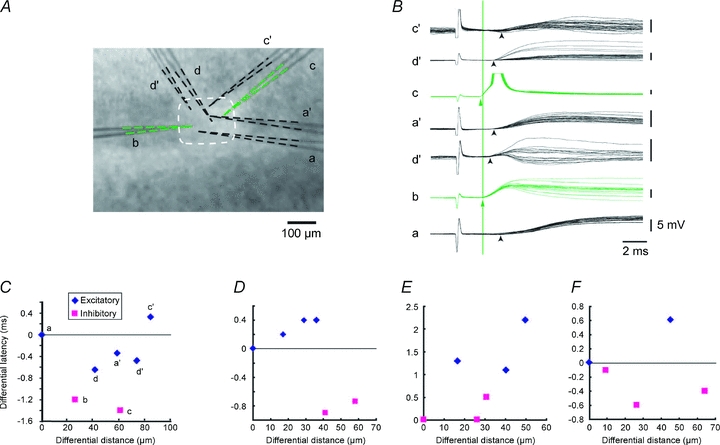
A, photomicrograph showing the locations of the cells by electrodes used for recordings. Two photomicrographs were superimposed. Electrodes a, b, c and d were first used to record thalamic EPSPs simultaneously, then all the electrodes but b were replaced with a′, c′ and d′, which were used in turn to record from the other 3 cells. Electrode b remained recording throughout the experiment without touching at all. At the end of each recording session, photomicrographs were taken, then two photomicrographs were superimposed so that the electrode b matched completely. Of these, b and c were patched on GFP-positive GABAergic cells (B). Tips of each electrode were delineated to clarify the location of the recorded cells. B, EPSPs recorded from the seven electrodes as labelled in response to thalamic stimulation. Arrowheads indicate the onsets of EPSPs. Green vertical line indicates the onset of EPSP in the GABA cell recorded with electrode b. C, differential distances in a vertical direction were measured between the recorded cells and latency differences were measured from respective EPSP recordings. Measurements were carried out from the anatomically deepest cell a to each cell, then differential latencies were plotted against differential distances. D–F, results from three independent groups of cells, in which similar analyses were carried out.
To better delineate the possible causes of the differences in onset latencies between inhibitory and excitatory cells, we next examined whether similar latency differences could be seen in response to WM stimulation. We placed an additional stimulating electrode in the WM and compared the responses elicited by activations of the two sites so that we could determine whether the latency difference was generated in axons between the VB and the WM, or between the WM and layer IV cells (Fig. 5A). As exemplified in this case, latencies in the inhibitory cell were shorter than those in excitatory cells from both the VB (1.6 ms vs. 3.2 ms, inhibitory and excitatory, respectively) and WM (1.3 ms vs. 2.6 ms, inhibitory and excitatory, respectively) stimulation (Fig. 5B). Similar results were obtained in a total of seven pairs (Fig. 5C). Thus, the activation of an inhibitory neuron prior to that of an excitatory cell was confirmed not only for VB stimulation but also for WM stimulation. Also, as illustrated in Fig. 5B, we observed that the EPSCs in GABAergic cells are larger than in excitatory cells at the same stimulus strength in all seven pairs used in voltage clamp experiments (182.8 ± 87.5 vs. 38.3 ± 13.9 pA, holding potential =−70 mV). The difference was significant (paired t test, P < 0.05, n= 7), and consistent with recent reports (Gabernet et al. 2005; Cruikshank et al. 2007; Hull et al. 2009).
Figure 5. White matter stimulation also caused latency differences.
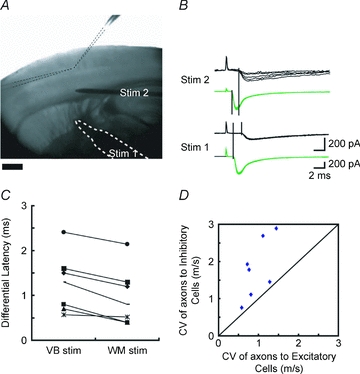
A, photomicrograph showing the experimental set-up. Two recording electrodes (delineated by dotted lines) were inserted into a single barrel, a stimulating electrode (bordered by white dotted line, Stim. 1) was placed on the thalamic fibres issuing from ventrobasal nucleus of the thalamus, and an additional stimulating electrode was placed a little above the white matter (Stim. 2). B, sample EPSCs recorded from GFP-positive (green) and -negative (black) cells in response to the stimulation to Stim. 1 and Stim. 2. Holding potential =−70 mV. No bicuculline was added. C, differences in latencies of EPSCs between excitatory and inhibitory cells were plotted at two stimulation sites, showing that there were still latency differences even when the stimulation was applied to the white matter (Stim. 2), but the differences became smaller. D, from the differences in latencies and the distances between two stimulation sites, conduction velocities (CV) for thalamic axons innervating excitatory and inhibitory cells could be calculated. CVs of axons to inhibitory cells were plotted against those of axons to excitatory cells in simultaneous recordings of 7 pairs. All of the points fell above the diagonal line, indicating that CVs for axons innervating inhibitory cells were faster than those for excitatory cells between VB and white matter.
By placing two independent stimulating electrodes, we could estimate conduction velocities (CV) for thalamocortical axons innervating inhibitory and excitatory cells. The average CV was faster for axons innervating inhibitory cells (1.80 ± 0.78 m s−1, n= 7) than that for axons innervating excitatory cells (0.96 ± 0.32 m s−1, P= 0.005, one-tailed paired t test). Importantly, for individual pairs recorded in a slice, CVs for axons innervating inhibitory cells were always faster than those for excitatory cells (Fig. 5D). We conclude that differences in CVs were the basis for the latency differences between the thalamocortical fibres innervating excitatory and inhibitory cells.
Developmental profile of the latency differences
What is responsible for the observed differences in CV? There are at least two factors that influence the CV of axons, namely, their myelination and diameter. Myelination of thalamocortical fibres occurs rather gradually during development in mice, starting after the second postnatal week and finishing around 4 weeks after birth (Jacobson, 1963). We questioned whether the latency difference between inhibitory and excitatory cells developed in parallel with the myelination process. Figure 6A and B summarizes the results of a developmental analysis of latency differences in neonatal to juvenile mice from pre-myelination stage to matured myelination phase. As expected, latencies for thalamically evoked EPSCs were longer in younger animals with immature myelination as reported previously (Jacobson, 1963; Salami et al. 2003). We found that differences in latencies from simultaneously recorded pairs of excitatory and inhibitory cells were consistently observed after P12. In other words, the onset latency difference between inhibitory and excitatory neurons appeared rather abruptly within a few days after around P12 and was seen consistently thereafter, unlike the gradual myelination of thalamocortical fibres. Thus, latency difference and completion of myelination do not progress in parallel in the mouse thalamocortical system, indicating that myelination is unlikely to be responsible for the observed differences in the CV of thalamocortical axons.
Figure 6. CV difference was not created by the differences in myelination.
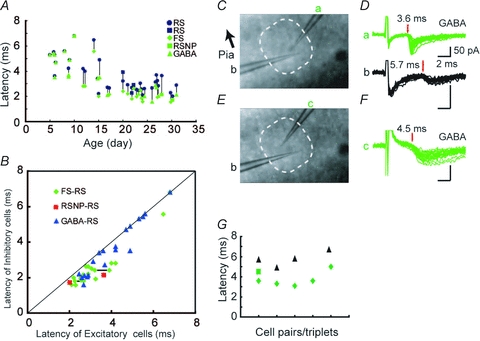
A, developmental changes in the latencies of thalamically evoked EPSP(C)s from GFP-positive and -negative cells. Data for mice older than P20 are the same as those shown in Fig. 3E. Simultaneously recorded cells from a single barrel are connected by lines. Blue symbols indicate regular spiking excitatory cells, green symbols indicate GABAergic cells. B, the data from A are plotted in the same way as in Fig. 3E, showing several data points from young animals on the diagonal line, whose latencies are, without exception, longer than for older animals. C–G, demyelination prolonged the latency of thalamically induced EPSCs in both excitatory and inhibitory cells to a similar extent, but failed to eliminate the latency differences, indicating that myelination is not responsible for generating the latency differences. C, photomicrograph showing an arrangement of two simultaneously recorded cells. D, EPSCs from electrodes a (GFP-positive) and b are shown. E and F, electrode a was replaced with electrode c, then EPSCs were recorded from another GFP-positive cell, as shown in F. G, summary of demyelination experiments. Latencies for simultaneously recorded cells are plotted in a vertical column. In one case, recording was obtained from a GFP-positive cell only (2nd column from right). Black triangles represent excitatory cells and green diamonds and a square represent inhibitory cells.
Effect of myelin destruction
By demyelinating fibres with an infusion of lysolecithin, we directly tested the role of myelination of thalamocortical fibres in creating latency differences. We examined whether the differences in latency persisted after demyelinating the thalamocortical fibres in matured mice by injecting lysolecithin (1–2%) (Hall, 1972; Blakemore, 1978) into the barrel cortex using an osmotic mini pump. Lysolecithin has been used widely for a long time, and it is known to have minimal axonal damage (Woodruff & Franklin, 1999a,b;). There is even a report that lysolecithin infusion to visual cortex did not change the visual response (M. Fagiolini, 2006, FENS Forum). After a survival period of 3–5 days, we conducted electrophysiological experiments. In an experiment shown in Fig. 6C–F, two cells were recorded simultaneously in a single barrel from a P27 mouse following 3 days of lysolecithin infusion. EPSCs from a GFP-positive GABAergic neuron (cell a) had longer onset latency (3.6 ms) when compared to intact mice at this age, but these were within the range of young mice before myelin formation (see Fig. 6A). Simultaneously recorded EPSCs from an excitatory cell (cell b) had an even longer latency (5.7 ms) (Fig. 6D). Subsequently recorded EPSCs from another GABAergic neuron (c, Fig. 6E and F) had again shorter onset latency (4.5 ms) than the excitatory cell (b). Recordings from three other pairs of neurons from independent experiments yielded similar results (Fig. 6G). The mean latency for the inhibitory cells was 3.9 ± 0.3 ms (n= 6 including one cell without a simultaneously recorded excitatory cell) and that for the excitatory cells was 5.8 ± 0.4 ms (n= 4). The latency of EPSCs in lysolecithin-infused mice was thus longer than that in intact mice of the same age and was comparable to that in mice before the second postnatal week, indicating that thalamocortical axons were successfully demyelinated. However, the latency differences between excitatory and inhibitory cells persisted. These results indicated that myelination was at least not a primary cause for the latency difference in thalamocortical activation of inhibitory and excitatory cells.
Lower threshold for axons innervating GABA cells
Exclusion of myelination in accounting for the observed differences in CV leaves the other possibility as a strong candidate, that is, a difference in the diameters of axons depending on the type of target neurons. The activation threshold of an axon is directly related to its diameter; the larger the diameter, the lower the threshold. This is because axons with larger diameters have lower internal resistances in the longitudinal direction, thus allowing a larger current to flow at a given voltage difference (BeMent & Ranck, 1969; Koester & Siegelbaum, 2000). We investigated whether axons innervating inhibitory neurons had lower thresholds than those innervating excitatory neurons by gradually increasing stimulus strength. As shown in Fig. 7A and B, thalamic fibre(s) innervating an inhibitory neuron had a lower threshold than those innervating an excitatory neuron. Of 12 pairs tested, the same tendency was consistently observed except for a single case (Fig. 7C, 11.2 ± 1.9 V vs. 16.9 ± 4.0 V, for inhibitory vs. excitatory cells, P= 0.02, one-tailed paired t test, n= 12). Similarly, in the lysolecithin experiments, thalamic axons innervating GABA cells had significantly lower thresholds (17 ± 4.9 V, n= 4) than those innervating excitatory cells (27.5 ± 10.5 V, P= 0.04, one-tailed paired t test, n= 4), explaining the observed difference of latency. These results strongly indicated that differences in axon diameters were likely to underlie the observed differences in CVs in the thalamocortical fibres.
Figure 7. Thresholds for axons innervating GABAergic cells were consistently lower than those for axons innervating excitatory cells in the same barrel.
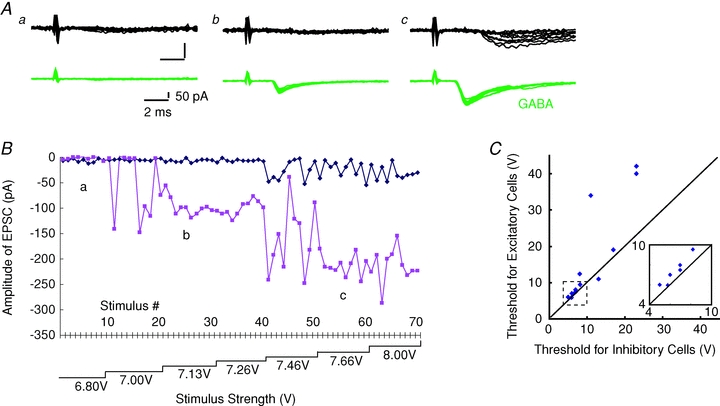
A, simultaneously recorded EPSCs from GFP-negative (top, black) and -positive (bottom, green) cells at three different stimulus intensities (a–c) as indicated in B. B, amplitudes of EPSCs recorded from pairs of GFP-negative (blue) and -positive (red) cells were plotted against stimulus number. Stimulus intensities were increased in a stepwise manner as indicated below. C, summary of the similar experiments from 12 pairs of cells. Each point represents thresholds for the excitatory (ordinate) and inhibitory (abscissa) cells in the recoded pairs. All but one fell above the diagonal line, indicating that the threshold for inhibitory cells was lower than for excitatory cells. Inset shows the same graph expanded for the region of 4–10 V as shown as a dotted square.
Cell type-dependent IPSP delay from thalamic EPSPs
As we have shown so far, if fast disynaptic IPSPs were made possible by differential activation of inhibitory and excitatory neurons, the predicted appearance of fast disynaptic IPSP would depend on the type of postsynaptic cells since there is less latency difference among inhibitory neurons than between excitatory and inhibitory neurons. To examine this, the histogram of latency differences between thalamic EPSPs and disynaptic IPSPs as shown in Fig. 1B was reconstructed according to cell type. Determination of cell type was done by GFP fluorescence and/or electrophysiological characteristics. Of the 26 cells examined, 14 turned out to be excitatory and nine were inhibitory; the remaining three cells were not classified. The resultant histograms are shown in Fig. 8, which clearly demonstrates that the two distributions were significantly different (1.5 ± 0.2 vs. 1.9 ± 0.1 ms for excitatory and inhibitory cells, respectively, P < 0.05, unpaired t test) and, as predicted, faster disynaptic IPSPs were selectively seen in excitatory cells. These results further support our findings that latency differences in thalamocortical connection depending on the target cell types underlies the generation of fast disynaptic IPSPs.
Figure 8. Delays of disynaptic IPSPs from thalamic EPSPs are shorter in excitatory cells.
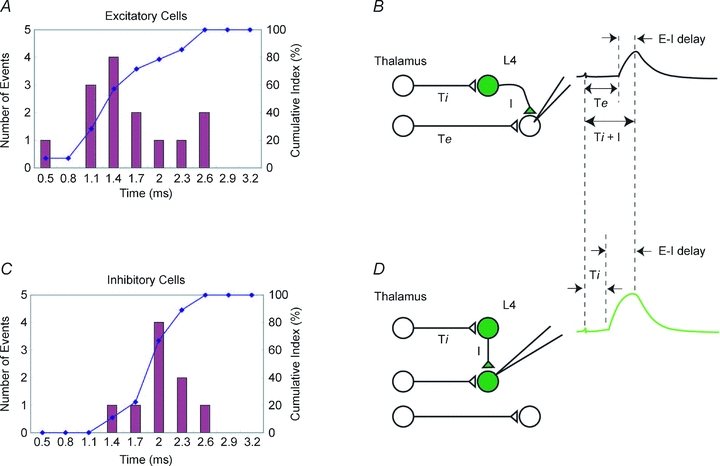
The histogram of latency difference between thalamic EPSP and disynaptic IPSP shown in Fig. 1B was reconstructed depending on each cell type. A and C, distributions of latency differences obtained from excitatory cells (A) and inhibitory cells (C). In C, all but one were FS cells; the cell type of the remaining cell was an unidentified GFP-positive GABAergic cell. B and D, schematic drawing for thalamic innervation to L4 cells. Thalamic axons to inhibitory cells are shorter than to excitatory cells, reflecting thalamic latency to each cell. Delays of disynaptic IPSPs recorded from excitatory cells (E–I delay in B) should be shorter than those from inhibitory cells (E–I delay in D) because there are latency differences in monosynaptic EPSPs from the thalamus between excitatory and inhibitory cells in B, while there is little latency difference in monosynaptic EPSPs from the thalamus between the cells in the same class (inhibitory) in D, as demonstrated in A and C. Ti and Te: thalamic latencies to inhibitory and excitatory cells, respectively. I: intracortical monosynaptic latency for inhibitory connection.
Possible relevance to the regulation of spike timing: faster activation of GABA cells might be necessary to produce fast disynaptic IPSPs in L4 and L2/3
As we have seen so far, thalamocortical axons innervating inhibitory neurons in L4 became slightly thicker, resulting in the faster activation of inhibitory neurons and the generation of fast feedforward IPSPs especially in excitatory cells. What, then, is the role of such a fast suppression on excitatory cells? Fast feedforward suppression is advantageous in rapidly curtailing the thalamocortical EPSPs by suppressing possible delayed firing of L4 cells. In fact, previous studies showed that whisker responses are suppressed by fast GABA-mediated inhibition. An iontophoretic application of GABAA antagonist revealed strong suppression right after the beginning of or during the whisker responses in L4 cells (Kyriazi et al. 1998). An in vivo intracellular study in the cat visual cortex revealed that thalamic stimulation elicited disynaptic IPSPs following monosynaptic EPSPs in all L4 cells (Ferster & Lindstrom, 1983). Interestingly, this study revealed that thalamic stimulation produced disynaptic IPSPs before disynaptic EPSPs in L3 cells in the cat visual cortex (Ferster & Lindstrom, 1983). Consistent with this finding, iontophoresis of GABAA antagonist revealed hidden GABAergic inhibition prior to whisker responses in L2/3 cells (see Fig. 2 of Foeller et al. 2005). These studies indicate that strong GABAergic inhibitions are exerted not only right after the L4 cells’ firings, but also immediately before L2/3 cells’ activities in response to sensory stimulation. Conversely, if there were some suppressing inputs after L4 cells’ as well as before L2/3 cells’ firings following sensory activation, those would be a highly desirable inhibition in order to generate ‘L4 leading L2/3’ spike sequences. Previous studies indeed demonstrated that whisker stimulation produces spike activities occurring first in L4 cells, followed by L3, then by L2 cells (Armstrong-James et al. 1992; Brumberg et al. 1999; Celikel et al. 2004), as consistent with this. Thus, we considered a possibility that fast feedforward inhibition based on the preceding activation of GABAergic to excitatory cells from thalamus may serve for the regulation of spike sequences occurring into ‘L4 leading L2/3’, by producing inhibitions after L4 and before L2/3 cells’ firings. Anatomically, feedforward inhibitory cells in L4 do indeed send axonal arbours not only within L4 but also to L2/3 (Ferster & Lindstrom, 1983; Agmon & Connors, 1992; Porter et al. 2001; Itami et al. 2007). One previous study reported that the onset of whisker responses in L3 and L2 cells was delayed by only 1.6 ms and 4.4 ms, respectively, from that of L4 cells (Celikel et al. 2004). If spike sequence of ‘L4 leading L2/3’ is to be achieved by disynaptic IPSPs, inhibitory inputs are required to reach both L4 and L3 (or L2) cells no later than 1.6 ms (or 4.4 ms in L2 cells) from the onset of the thalamic excitation of L4 cells in order to suppress L4 cells after L2/3 cells’ firing, and similarly to suppress L2/3 cells until incoming spikes of L4 cells reach L2/3 cells. What neuronal circuits enable such fast inhibition? We have shown that in excitatory cells, disynaptic IPSPs appear at 1.5 ± 0.2 ms from the onset of thalamic EPSPs in L4 cells (Fig. 8A). We have discussed that such fast IPSPs could only be possible by disynaptic inhibition through prior activation of GABA cells to excitatory cells from the thalamus. Thus, fast IPSPs that inhibit L4 cells immediately after their firings could only be achieved by thalamically driven fast feedforward GABA cells based on their preceding activation to excitatory cells. The same argument could be applied to IPSPs in L2/3, as long as the IPSPs are disynaptic from the thalamus. In other words, in order to produce disynaptic IPSPs within 1.6 ms from the onset of thalamic EPSPs, regardless of whether the target cells are L4 or L2/3, it takes 2.3 ms (1.43 + 0.87, Fig. 2B, C) for spike generation in a GABA cell and monosynaptic inhibitory connection, thus, thalamic activation of the GABA cell must be earlier than that of an excitatory cell by at least 0.7 ms (2.3–1.6), which is indeed satisfied as we have seen (Fig. 3F, the actual difference was 0.7 ms). In addition to these, from a qualitative point of view, the following argument holds true. To generate disynaptic IPSPs prior to disynaptic EPSPs in L2/3 cells (Ferster & Lindstrom, 1983; Foeller et al. 2005) following the thalamic input, feedforward GABAergic cells that generate disynaptic IPSPs must be activated earlier than excitatory cells by the thalamic input in order to outrun the excitatory connection from L4 to L2/3 (Fig. 9A left, thick lines), under the condition that both the excitation and inhibition are to be provided by monosynaptically activated cells from thalamus, and monosynaptic excitatory and inhibitory connections have comparable conduction time (Fig. 2A and B). Thus, fast disynaptic IPSPs preceding disynaptic EPSPs in L2/3 could again be achieved only by a prior (more than 0.7 ms) activation of GABA cells to excitatory cells from thalamic inputs.
Figure 9. Schematic drawings of the present results showing a faster activation of GABA cells from thalamus and fast feedforward suppression to L4 and L2/3 cells.
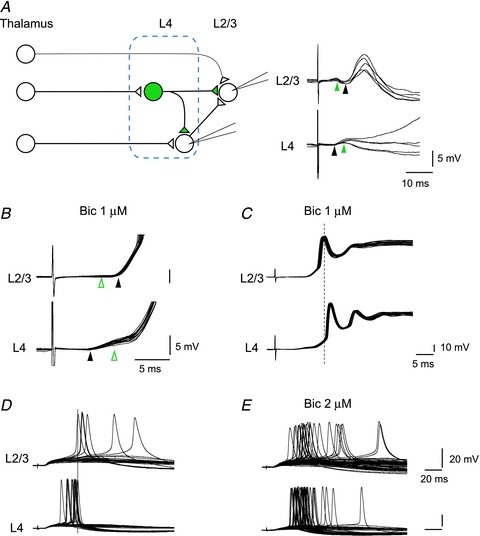
The distances from thalamus to L4 excitatory and inhibitory cells are different reflecting the latency difference. On the right, examples are shown of such simultaneous recordings. In the L4 excitatory cell, thalamic input produced EPSPs followed by IPSPs. In L2/3, the same thalamic stimulation caused clear IPSP followed by EPSPs, both probably disynaptic. There were, however, even faster excitatory inputs, although not strong, possibly direct from thalamus, which is also illustrated by a thinner line from thalamus to L2/3 in the schematic diagram on the left. Black and green arrowheads indicate the onset of EPSPs (black) and IPSPs (green). B, hyperpolarizing responses in L4 as well as L2/3 disappeared (open green arrowheads) in the presence of bicuculline (1 μm). C, in the presence of bicuculline, L2/3 spikes preceded L4 spikes consistently, and thus removal of feedforward inhibition reversed the spike sequences. D and E, another example of bicuculline effect on spike sequences from a different pair. Both cells were slightly depolarized (−53 mV and −44 mV for L4 and L2/3 cells, respectively) to induce spike activities in the absence of bicuculline (D). All L4 spikes appeared before those of L2/3 in 30 trials of thalamic stimulation. When bicuculline was applied (2 μm), later spikes in L4 as well as earlier spikes in L2/3 appeared, and thus spike sequences of “L4 leading L2/3” were destroyed (E).
To test whether thalamic stimulation indeed elicited inhibitory responses in L2/3 cells, we recorded from 15 L2/3 pyramidal cells in response to thalamic stimulation in mice from P11 to 21. Of the 15 cells tested, we could reliably record IPSPs in 12 L2/3 cells. Figure 9 shows an example of such recordings, where L4 and L2/3 cells were simultaneously recorded. In this case, thalamic stimulation elicited EPSPs followed by IPSPs in the L4 cell, which was consistent with what we have described so far. In the L2/3 cell, thalamic stimulation generated IPSPs first, followed by EPSPs, which was in agreement with previous in vivo experiments with whisker stimulation in mice (Foeller et al. 2005) and electrical stimulation of the thalamus in cats (Ferster & Lindstrom, 1983). Subsequent application of bicuculline (1 μm), a GABAA receptor antagonist, completely blocked the hyperpolarizing response, confirming that it was mediated by GABAA receptors (Fig. 9B). Previous to bicuculline application, thalamic stimulation produced action potentials only occasionally, while in the presence of bicuculline, thalamic stimulation consistently produced action potentials followed by prolonged depolarization in both L4 and L2/3 cells (Fig. 9C), indicating strong suppression had been elicited by thalamic stimulation. Surprisingly, bicuculline reversed the spike sequences in these cells, and action potentials in the L2/3 cell preceded those of the L4 cell consistently (Fig. 9C). The observed mean onset latency of IPSPs in L2/3 cells was 4.8 ± 0.5 ms (n= 9, in animals aged P15–P21) suggesting that the IPSPs were disynaptic. As described earlier, thalamic latency for GABA cells was 2.1 ± 0.1 ms, and spike generation and monosynaptic inhibitory connection takes 1.43 ± 0.41 and 0.87 ± 0.08 ms, respectively (Figs 2 and 3), all of which sum to about 4.4 ms, which matches well with our observation (4.8 ± 0.5 ms). We next focused on the effect of removal of inhibitory inputs on spike sequences in L4 and L2/3 neurons. We found, however, that both L4 and L2/3 RS cells rarely fire in control conditions, as consistent with previous studies (Porter et al. 2001; Gabernet et al. 2005; Sun et al. 2006; Cruikshank et al. 2007). Since one obvious reason is the feedforward suppression, we tried to enhance the spike probability either by depolarization or low dose of bicuculline (0.5–4 μm), or combination of both manipulations. In a total of 17 pairs of simultaneous recording from L4 and L2/3 cells tested, both cells fired in only seven pairs in the ‘quasi’ control conditions, in all of which L4 firings led that of L2/3 cells. Of the seven pairs, reversals of spike sequences were tested in the higher dose of bicuculline, as shown in Fig. 9D and E. In this case, both cells, which turned out to be RS cells, did not fire at all at the resting membrane potentials (−63 mV and −65 mV for L4 and L2/3, respectively). When slightly depolarized (−53 mV and −44 mV for L4 and L2/3, respectively), however, these cells fired 12 and 6 times in 30 trials, for L4 and L2/3, respectively (Fig. 9D, no bicuculline). The mean peak spike latencies were 20.0 ± 2.7 ms (mean ±s.d., range: 14.3–23.6 ms for L4 cell) and 34.6 ± 13.4 ms (mean ±s.d., range: 23.8–57.1 for L2/3 cell), and thus all L4 spikes preceded those of L2/3 cell. In the presence of bicuculline (2 μm), both cells increased their firing probabilities and mean spike latencies became 22.2 ± 7.0 ms (mean ±s.d., range: 14.1–54.2 ms for L4 cell) and 31.3 ± 17.6 (mean ±s.d., range: 13.6–85.1 ms for L2/3 cell). Importantly, late spikings (later than 23.6 ms) in L4, and early spikings (earlier than 23.8 ms) in L2/3 that had conceivably been suppressed by feedforward inhibition appeared in bicuculline application and actual reversal of spike sequences were observed 12 times out of 22 stimulations with spikes in both cells in the total of 62 stimulations in this pair. Similar reversal of spike sequences were observed in every single pair of the seven recordings. The reversal of spike sequences due to the appearance of late spikings in L4 was observed in all seven recordings, and that due to the appearance of preceding spikes in L2/3 was observed in 2 of the 7 paired recordings. These results strongly indicate that feedforward inhibition is involved with the regulation of spike sequences into occurring ‘L4 leading L2/3’.
One weakness of this bicuculline experiment is that not only the fast feedforward but also all the inhibitory effects are removed by bicuculline, and so we cannot necessarily ascribe the effect to the reduction of fast feedforward suppression. Another way to test the relationship between fast feedforward suppression and its regulation on spike sequences would be to look at the development of spike sequences elicited by whisker stimulation, because fast feedforward suppression was accomplished at P12–14 (Fig. 6), so one can ask if the spike sequences between L4 and L2/3 changes in parallel. To examine this, we performed simultaneous extracellular recording of whisker responses using 16 vertically aligned probes from cells in L2/3 to L5, and found that the predicted sequences of L4 cell leading L2/3 cell were generally preserved at P23 animal (Supplemental Fig. 2). We also found that such layer specific temporal pattern of whisker responses developed in parallel with accomplishment of preceding activation of inhibitory cells (Supplemental Figs 3, 4 and 5). All of these results support the idea that fast feedforward inhibition based on the preceding activation of thalamocortical GABAergic cells plays at least some role in the formation of spike sequences of ‘L4 leading L2/3’.
Discussion
In the present study, we first described our observation that thalamic stimulation often produces IPSPs within a very short delay relative to monosynaptic EPSPs. From our own results of monosynaptic conduction time, we reasoned that thalamocortical activation of inhibitory cells would have to precede that of excitatory cells. We confirmed that this was actually the case by directly testing this. We also found that differential conduction velocities of thalamic axons, which depended on target cell types, underlies this latency difference, possibly based on differences in axon diameters, but only after the end of the second postnatal week during development. We then argued that such fast feedforward suppression through the preceding activation of GABAergic neurons could theoretically regulate the spike timing of L4 and L2/3 cells so that their firings occur in the order of ‘L4 leading L2/3’ in response to thalamic input. We demonstrated that thalamic stimulation evoked disynaptic IPSPs not only in L4 cells but also in L2/3 cells, and in the latter case, even prior to disynaptic EPSPs. When IPSPs were suppressed by a GABAA receptor antagonist, L4–L2/3 spike sequences became disordered or even reversed.
Recent studies revealed that hippocampal FS cells have specific dendritic membrane properties that optimize fast activation (Hu et al. 2010; Norenberg et al. 2010). Such postsynaptic mechanisms certainly play an important role in producing fast activation and termination of action potentials and thus contribute to generating a short window of excitation. However, such postsynaptic mechanisms cannot account for the shorter latency of thalamic activation in FS cells.
In another recent study (Cruikshank et al. 2010), channelrhodopsin-2 (ChR2) was selectively expressed in thalamocortical cells, and then focal laser stimulations to ChR-2-expressing thalamocortical terminal areas in cortical L4 were shown to produce excitation followed by inhibition in nearly all recordings from the cells in the same layer. Interestingly, the mean latency from the onset of the excitation to the onset of the inhibitory current was 2.38 ± 0.18 ms, which is substantially longer than previous reports using electrical stimulation of thalamus (Agmon & Connors, 1992; Porter et al. 2001; Gabernet et al. 2005; Hull & Scanziani, 2007), including their own previous report (Cruikshank et al. 2007) and our present results: 1.5 ± 0.2 and 1.9 ± 0.1 for excitatory and inhibitory cells (Fig. 8A and C). Since in the direct activation of thalamocortical terminals by laser stimulation, all the thalamocortical terminals on both excitatory and inhibitory cell are simultaneously activated, shorter delay of fast feedforward inhibition by electrical stimulation of thalamus should be attributed to the latency difference that appeared between inhibitory and excitatory cells from thalamus, as the current study demonstrated. In addition, from the quantitative standpoint, our direct dual patch experiments predict that if there are no latency differences in excitatory and inhibitory cells, feedforward IPSP would appear with a delay of 2.30 ± 0.49 ms from the onset of the thalamic EPSP (1.43 ± 0.41 for spike initiation and 0.87 ± 0.08 for inhibitory conduction, Fig. 2B and C), which fits very well with what Cruikshank et al. (2010) observed in direct activation of thalamic terminals (2.38 ± 0.18 ms). Thus, their study strongly supports our results.
One previous in vivo study indicated that the thalamic neurons most responsive to whisker stimulation also have the fastest conduction times (Simons et al. 2007), and these fast conducting, strongly responsive thalamic neurons are thought to synapse on inhibitory but not excitatory neurons in the cortex. It seems very likely that they may correspond to the large diameter thalamic fibres driving fast spiking GABAergic neurons that we suspect to exist.
Involvement of axo-axonic inhibitions?
Among excitatory pyramidal cells in L2/3, one recent paper (Ren et al. 2007) described that local terminals of pyramidal cell may synapse onto inhibitory cell terminals, and thus excitation of presynaptic pyramidal cells could produce fast IPSPs in the postsynaptic pyramidal cells through axo-axonic synapses. As we have shown ample evidence that fits quite well (timing, development, conduction velocity, threshold), there is no doubt that preceding activation of fast spiking GABAergic cells from thalamus would be the main reason for producing fast feedforward suppression. Nevertheless, is there any possibility that these axo-axonic inhibitions contribute to fast feedforward inhibition driven by thalamic inputs? Although we cannot completely rule out the involvement of such synapses, we believe that its involvement is unlikely for the following reasons. First, as described in the Supplemental Material, for an identification of GABAergic neurons, we performed an extensive number of simultaneous multiple recordings (262 pairs). Of these, all 16 inhibitory responses came from presynaptic GFP-positive GABAergic cells and all 32 excitatory responses came from presynaptic GFP-negative cells, and thus such axo-axonic connections may contribute little, if at all, to suppression within barrels in L4. Second, axo-axonic inhibition alone cannot account for some of our observations. For example, delays of the onset of disynaptic IPSPs from thalamic EPSPs were dependent on cell types and those of excitatory cells were shorter than those of inhibitory cells (Fig. 8), which would not occur if axo-axonic inhibition dominates. Finally, as we as well as a previous study (Ferster & Lindstrom, 1983) have shown, in L2/3 disynaptic IPSPs often precede disynaptic EPSPs (Fig. 9), which is probably important for spike regulation, and could only be possible by preceding activation of GABA cells, as we discussed before. Axo-axonic inhibition through L4 excitatory cells activating inhibitory terminals onto L2/3 cells cannot be faster than disynaptic EPSPs in L2/3 cells because such IPSPs need to activate an additional synapse, even if it might be axo-axonic, and thus it should be delayed from disynaptic EPSPs.
Cell type specificity
Of the 16 inhibitory thalamo-recipient neurons that were simultaneously recorded with excitatory thalamo-recipient neurons (Fig. 3E), 14 cells were classified as FS and only two cells were RSNP. This is consistent with the reports that the main thalamo-recipient GABA neurons are FS in layer 4, and thus fast feedforward inhibition is mediated by this subtype in the cortical network (Beierlein et al. 2000, 2003; Gabernet et al. 2005; Inoue & Imoto, 2006; Cruikshank et al. 2007). On the contrary, other reports claim that both FS and RSNP cells receive thalamic inputs to a similar extent (Porter et al. 2001). One possible explanation for this discrepancy might depend on the definition of FS and other cell types. Recent reports using transgenic mice demonstrated that classification of GABAergic cell types with a combination of three properties of the cells (spike adaptation ratio, spike width, input resistance) makes clearer separation, but the range of each property has substantial overlaps (Tan et al. 2008). Considering this, two RSNP cells showed a modest SR, within a range of FS variation (Tan et al. 2008), and thus could possibly be atypical FS cells.
Removal of GABA inhibition in L4, L2/3 cells
As predicted, thalamic stimulation evoked disynaptic IPSPs prior to possible disynaptic EPSPs in some L2/3 cells (Fig. 9). This finding raises an interesting issue. Such prior appearance of disynaptic IPSPs to possible disynaptic EPSPs supports our primary finding that thalamic activation of GABA cells precedes that of excitatory cells (Figs 3–5), provided that monosynaptic excitatory and inhibitory conduction times are comparable (Fig. 2A and B). Why do L2/3 cells need an inhibition before excitation? Is there any chance of possible ‘unwanted’ inputs entering L2/3 cells? In most cases, L4 activity is likely to precede L2/3 activity simply because of the well-established anatomical fact that thalamic axons innervate L4 more densely than L2/3. Ferster & Lindstrom (1983), however, demonstrated that a considerable number of thalamic inputs directly terminated in layer 3; they further reported that all the cells (n= 18) recorded in L3 received monosynaptic inputs from the thalamus. Under such condition, L3 cells may well have a chance to fire before L4 cells by direct thalamic inputs, resulting in the reversed sequence of activation, ‘L3 followed by L4.’ Thus, preceding inhibition would be necessary in L2/3; otherwise, the spiking order of ‘L4 leading L2/3’ could not be guaranteed, or sometimes even reversed, as if it were in the presence of a GABAA receptor antagonist (Fig. 9C).
Thalamocortical innervation to layer 4 cells
Our results indicated that GABAergic neurons are likely to be innervated by thalamic fibres with larger diameters than those innervating excitatory cells. Although this may appear inconsistent with previous studies reporting that a single thalamic fibre synapses on both excitatory and inhibitory cells (Gabernet et al. 2005; Inoue & Imoto, 2006; Cruikshank et al. 2007), it does not at all contradict the previous observations. Instead, both observations would be consistent if an additional group of thalamic axons of relatively small number but with larger diameters connected exclusively to inhibitory cells. Then, if fine stimulating electrodes were used to activate single fibres (Gabernet et al. 2005; Inoue & Imoto, 2006; Cruikshank et al. 2007) with minimum current, smaller axons innervating both excitatory and inhibitory cells could be activated individually, which would result in recording GABA and excitatory cells with the same latency (Gabernet et al. 2005; Inoue & Imoto, 2006; Cruikshank et al. 2007). Since we did not intend to activate single fibres, we used larger stimulating electrodes (tip diameter, 25–150 μm), which resulted in the activation of multiple fibres. Consequently, axons with thicker diameters that supposedly innervate inhibitory cells should have already been activated at a stimulus strength above the threshold of smaller diameter fibres that innervate excitatory (and perhaps inhibitory, too) neurons, as is consistent with our experiments (Fig. 7). A differential threshold for excitatory and fast spiking inhibitory neurons was observed previously in a similar experiment that obtained results comparable to ours (see Fig. 3 g of Cruikshank et al. 2007). Nevertheless, Cruikshank et al. ascribed a lower threshold of inhibitory cells to the more convergent inputs from the thalamus to inhibitory neurons as compared with excitatory neurons (Swadlow, 2002). Interestingly, they also described that thalamocortical latency to fast spiking cells is shorter than to regular spiking cells (2.2 ± 0.1 vs. 2.6 ± 0.1 ms, respectively), although the difference was slightly smaller than we observed. In fact, although not explicitly stated, almost all of the previous studies reported that thalamic latencies to GABA cells are, more or less, shorter than to excitatory cells across the species. Mean latencies (ms) to GABA vs. excitatory cells for mice are 3.2 vs. 3.6 (Inoue & Imoto, 2006), 2.06 vs. 2.3 (Hull et al. 2009) and 3.5 vs. 4.0 (Beierlein et al. 2002); for rats are 2.2 vs. 2.6 (Cruikshank et al. 2007) and 1.9 vs. 2.4 (Beierlein & Connors, 2002); and for cats, 1.3–1.5 vs. 1.6–2.8 (Yamamoto et al. 1988). A single exception is the report that latencies for FS, low threshold spiking (LTS) and excitatory cells are 2.9, 3.1 and 3.0, respectively in rats (Beierlein et al. 2003). Thus, our value of 0.7 ms difference on average is slightly larger than most of other studies except one case in cats. This difference might be due to the fact that most of the previous studies used a minimum stimulation protocol, in which those cases where finer fibres innervating both excitatory and inhibitory cells are activated with the same latency are included. This would tend to minimize the mean latency differences.
Relevance to plasticity in the somatosensory cortex
We have shown that preceding activation of GABAergic neurons by thalamic inputs could play a role in regulating the spike sequence of ‘L4 leading L2/3’. Since regulation of spike sequences between L4 and L2/3 cells should have great influence on the direction and magnitude of plastic changes at these synapses (Feldman, 2000; Celikel et al. 2004; Nevian & Sakmann, 2006), which are characterized as spike timing dependent (STDP, or spike timing dependent plasticity), the establishment of preceding activation of GABAergic cells from thalamus could also be relevant to the expression of plasticity at these synapses. This also fits with our finding that preceding activation of GABAergic cells was established around the end of the second postnatal week, because receptive field map plasticity starts also at this age during development. In the barrel cortex, an activation of GABAergic neurons produces depolarization instead of hyperpolarization until P5. It is only after P6–7 that GABA receptor activation starts to generate hyperpolarizing responses (Daw et al. 2007). However, another week is necessary for the initiation of cortical map plasticity, suggesting that the presence of feedforward suppression is not in itself enough for the initiation of plasticity. Several more days of further maturation are required, during which the mechanism of preceding activation of feedforward GABAergic neurons is accomplished, as our results indicated (Fig. 6A and B). The importance of GABAergic inhibition in the initiation of plasticity was first suggested in the visual cortex (Hensch et al. 1998). GABA positive interneurons consist of highly diverse populations (Kawaguchi & Kubota, 1997; Markram et al. 2004). Among these, large basket cells that are parvalbumin-positive, fast spiking neurons are suggested to play a crucial role in driving visual cortical plasticity (Fagiolini et al. 2004). It has been suggested that a balance of excitation and inhibition determines the initiation of the critical period during development (Hensch et al. 1998; Hensch, 2005; Katagiri et al. 2007). However, the exact mechanism by which fast spiking inhibitory circuits drive plasticity is still unknown. Our results may imply that the essential substance of the ‘balance’ of excitation and inhibition may be the precisely timed excitation in L4 followed by L2/3 cells separated by feedforward inhibition based on the preceding activation of GABA cells from thalamic input.
In conclusion, we have described precise neural mechanisms for the creation of fast feedforward inhibition, its developmental course, and its relevance to sequential activation of ‘L4 leading L2/3’ and the initiation of plasticity.
Acknowledgments
Authors are grateful for helpful discussions with Drs. H. Sawai, T. Miyoshi, and Y. Okamura and members of Okamura lab. This work was supported by grants-in-aid for scientific research on priority areas (F.K., C.I. and I.F.), and Core Research for Evolutional Science and Technology by Japan Science and Technology Agency (I.F.).
Glossary
Abbreviations
- AHP
afterhyperpolarization
- ChR2
channelrhodopsin-2
- EPSP(C)
excitatory postsynaptic potential (current)
- (E)GFP
(enhanced) green fluorescent protein
- FS
fast-spiking
- GAD
glutamate decarboxylase
- IPSP
inhibitory postsynaptic potential
- RS
regular spiking
- RSNP
regular spiking non-pyramidal
- SR
spike ratio
- STDP
spike timing-dependent plasticity
Author contributions
F.K. designed and performed all the experiments and wrote the paper, C.I. and M.O. helped with some of the slice experiments and histological work, K.I., H.T. and I.F. helped with in vivo recording experiments described in the Supplemental Material, and Y.Y. and K.O. produced transgenic mice. Physiological experiments were conducted in the Osaka University Graduate School of Medicine, Laboratory for Cognitive Neuroscience, Graduate School of Engineering Science, and immunohistochemistry was done in the Department of Physiology, Faculty of Medicine, Saitama Medical University.
Supplemental material
Supplemental document
Figure 1
Figure 2
Figure 3
Figure 4
Figure 5
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- Agmon A, Connors BW. Correlation between intrinsic firing patterns and thalamocortical synaptic responses of neurons in mouse barrel cortex. J Neurosci. 1992;12:319–329. doi: 10.1523/JNEUROSCI.12-01-00319.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong-James M, Fox K, Das-Gupta A. Flow of excitation within rat barrel cortex on striking a single vibrissa. J Neurophysiol. 1992;68:1345–1358. doi: 10.1152/jn.1992.68.4.1345. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Connors BW. Short-term dynamics of thalamocortical and intracortical synapses onto layer 6 neurons in neocortex. J Neurophysiol. 2002;88:1924–1932. doi: 10.1152/jn.2002.88.4.1924. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Fall CP, Rinzel J, Yuste R. Thalamocortical bursts trigger recurrent activity in neocortical networks: layer 4 as a frequency-dependent gate. J Neurosci. 2002;22:9885–9894. doi: 10.1523/JNEUROSCI.22-22-09885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, Gibson JR, Connors BW. A network of electrically coupled interneurons drives synchronized inhibition in neocortex. Nat Neurosci. 2000;3:904–910. doi: 10.1038/78809. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Gibson JR, Connors BW. Two dynamically distinct inhibitory networks in layer 4 of the neocortex. J Neurophysiol. 2003;90:2987–3000. doi: 10.1152/jn.00283.2003. [DOI] [PubMed] [Google Scholar]
- BeMent SL, Ranck JB., Jr A quantitative study of electrical stimulation of central myelinated fibers. Exp Neurol. 1969;24:147–170. doi: 10.1016/0014-4886(69)90012-0. [DOI] [PubMed] [Google Scholar]
- Blakemore WF. Observations on remyelination in the rabbit spinal cord following demyelination induced by lysolecithin. Neuropathol Appl Neurobiol. 1978;4:47–59. doi: 10.1111/j.1365-2990.1978.tb00528.x. [DOI] [PubMed] [Google Scholar]
- Brumberg JC, Pinto DJ, Simons DJ. Cortical columnar processing in the rat whisker-to-barrel system. J Neurophysiol. 1999;82:1808–1817. doi: 10.1152/jn.1999.82.4.1808. [DOI] [PubMed] [Google Scholar]
- Celikel T, Szostak VA, Feldman DE. Modulation of spike timing by sensory deprivation during induction of cortical map plasticity. Nat Neurosci. 2004;7:534–541. doi: 10.1038/nn1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ, Connors BW. Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat Neurosci. 2007;10:462–468. doi: 10.1038/nn1861. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Urabe H, Nurmikko AV, Connors BW. Pathway-specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron. 2010;65:230–245. doi: 10.1016/j.neuron.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Ashby MC, Isaac JT. Coordinated developmental recruitment of latent fast spiking interneurons in layer IV barrel cortex. Nat Neurosci. 2007;10:453–461. doi: 10.1038/nn1866. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol Lond. 2009;587.4:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Fritschy JM, Low K, Mohler H, Rudolph U, Hensch TK. Specific GABAA circuits for visual cortical plasticity. Science. 2004;303:1681–1683. doi: 10.1126/science.1091032. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Ferster D, Lindstrom S. An intracellular analysis of geniculo-cortical connectivity in area 17 of the cat. J Physiol Lond. 1983;342:181–215. doi: 10.1113/jphysiol.1983.sp014846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foeller E, Celikel T, Feldman DE. Inhibitory sharpening of receptive fields contributes to whisker map plasticity in rat somatosensory cortex. J Neurophysiol. 2005;94:4387–4400. doi: 10.1152/jn.00553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M, Scanziani M. Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron. 2005;48:315–327. doi: 10.1016/j.neuron.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Gil Z, Amitai Y. Properties of convergent thalamocortical and intracortical synaptic potentials in single neurons of neocortex. J Neurosci. 1996;16:6567–6578. doi: 10.1523/JNEUROSCI.16-20-06567.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Efficacy of thalamocortical and intracortical synaptic connections: quanta, innervation, and reliability. Neuron. 1999;23:385–397. doi: 10.1016/s0896-6273(00)80788-6. [DOI] [PubMed] [Google Scholar]
- Hall SM. The effect of injections of lysophosphatidyl choline into white matter of the adult mouse spinal cord. J Cell Sci. 1972;10:535–546. doi: 10.1242/jcs.10.2.535. [DOI] [PubMed] [Google Scholar]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science. 1998;282:1504–1508. doi: 10.1126/science.282.5393.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Martina M, Jonas P. Dendritic mechanisms underlying rapid synaptic activation of fast-spiking hippocampal interneurons. Science. 2010;327:52–58. doi: 10.1126/science.1177876. [DOI] [PubMed] [Google Scholar]
- Hull C, Isaacson JS, Scanziani M. Postsynaptic mechanisms govern the differential excitation of cortical neurons by thalamic inputs. J Neurosci. 2009;29:9127–9136. doi: 10.1523/JNEUROSCI.5971-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Scanziani M. It's about time for thalamocortical circuits. Nat Neurosci. 2007;10:400–402. doi: 10.1038/nn0407-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Imoto K. Feedforward inhibitory connections from multiple thalamic cells to multiple regular-spiking cells in layer 4 of the somatosensory cortex. J Neurophysiol. 2006;96:1746–1754. doi: 10.1152/jn.00301.2006. [DOI] [PubMed] [Google Scholar]
- Itami C, Kimura F, Nakamura S. Brain-derived neurotrophic factor regulates the maturation of layer 4 fast-spiking cells after the second postnatal week in the developing barrel cortex. J Neurosci. 2007;27:2241–2252. doi: 10.1523/JNEUROSCI.3345-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itami C, Samejima K, Nakamura S. Improved data processing for optical imaging of developing neuronal connectivity in the neonatal mouse barrel cortex. Brain Res Protoc. 2001;7:103–114. doi: 10.1016/s1385-299x(01)00048-4. [DOI] [PubMed] [Google Scholar]
- Jacobson S. Sequence of myelination in the brain of the albino rat A. cerebral cortex, thalamus and related structures. J Comp Neurol. 1963;121:5–29. doi: 10.1002/cne.901210103. [DOI] [PubMed] [Google Scholar]
- Katagiri H, Fagiolini M, Hensch TK. Optimization of somatic inhibition at critical period onset in mouse visual cortex. Neuron. 2007;53:805–812. doi: 10.1016/j.neuron.2007.02.026. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex. 1997;7:476–486. doi: 10.1093/cercor/7.6.476. [DOI] [PubMed] [Google Scholar]
- Koester J, Siegelbaum SA. Local signaling: Passive electrical properties of the neuron. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principles of Neural Science. 4th edn. McGraw-Hill; 2000. pp. 140–149. [Google Scholar]
- Kyriazi H, Carvell GE, Brumberg JC, Simons DJ. Laminar differences in bicuculline methiodide's effects on cortical neurons in the rat whisker/barrel system. Somatosens Mot Res. 1998;15:146–156. doi: 10.1080/08990229870871. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norenberg A, Hu H, Vida I, Bartos M, Jonas P. Distinct nonuniform cable properties optimize rapid and efficient activation of fast-spiking GABAergic interneurons. Proc Natl Acad Sci U S A. 2010;107:894–899. doi: 10.1073/pnas.0910716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, Johnson CK, Agmon A. Diverse types of interneurons generate thalamus-evoked feedforward inhibition in the mouse barrel cortex. J Neurosci. 2001;21:2699–2710. doi: 10.1523/JNEUROSCI.21-08-02699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren M, Yoshimura Y, Takada N, Horibe S, Komatsu Y. Specialized inhibitory synaptic actions between nearby neocortical pyramidal neurons. Science. 2007;316:758–761. doi: 10.1126/science.1135468. [DOI] [PubMed] [Google Scholar]
- Salami M, Itami C, Tsumoto T, Kimura F. Change of conduction velocity by regional myelination yields constant latency irrespective of distance between thalamus and cortex. Proc Natl Acad Sci U S A. 2003;100:6174–6179. doi: 10.1073/pnas.0937380100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons DJ, Carvell GE, Kyriazi HT, Bruno RM. Thalamocortical conduction times and stimulus-evoked responses in the rat whisker-to-barrel system. J Neurophysiol. 2007;98:2842–2847. doi: 10.1152/jn.00800.2007. [DOI] [PubMed] [Google Scholar]
- Staiger JF, Zilles K, Freund TF. Distribution of GABAergic elements postsynaptic to ventroposteromedial thalamic projections in layer IV of rat barrel cortex. Eur J Neurosci. 1996;8:2273–2285. doi: 10.1111/j.1460-9568.1996.tb01191.x. [DOI] [PubMed] [Google Scholar]
- Sun QQ, Huguenard JR, Prince DA. Barrel cortex microcircuits: thalamocortical feedforward inhibition in spiny stellate cells is mediated by a small number of fast-spiking interneurons. J Neurosci. 2006;26:1219–1230. doi: 10.1523/JNEUROSCI.4727-04.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swadlow HA. Thalamocortical control of feed-forward inhibition in awake somatosensory ‘barrel’ cortex. Philos Trans R Soc Lond B Biol Sci. 2002;357:1717–1727. doi: 10.1098/rstb.2002.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Tan Z, Hu H, Huang ZJ, Agmon A. Robust but delayed thalamocortical activation of dendritic-targeting inhibitory interneurons. Proc Natl Acad Sci U S A. 2008;105:2187–2192. doi: 10.1073/pnas.0710628105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff RH, Franklin RJ. Demyelination and remyelination of the caudal cerebellar peduncle of adult rats following stereotaxic injections of lysolecithin, ethidium bromide, and complement/anti-galactocerebroside: a comparative study. Glia. 1999a;25:216–228. doi: 10.1002/(sici)1098-1136(19990201)25:3<216::aid-glia2>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Woodruff RH, Franklin RJ. The expression of myelin protein mRNAs during remyelination of lysolecithin-induced demyelination. Neuropathol Appl Neurobiol. 1999b;25:226–235. doi: 10.1046/j.1365-2990.1999.00172.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Samejima A, Oka H. Short latency activation of local circuit neurons in the cat somatosensory cortex. Brain Res. 1988;461:199–203. doi: 10.1016/0006-8993(88)90742-1. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T, Tsumoto T, Kimura F. Transiently higher release probability during critical period at thalamocortical synapses in the mouse barrel cortex: relevance to differential short-term plasticity of AMPA and NMDA EPSCs and possible involvement of silent synapses. Eur J Neurosci. 2004;20:3006–3018. doi: 10.1111/j.1460-9568.2004.03756.x. [DOI] [PubMed] [Google Scholar]
- Yazaki-Sugiyama Y, Kang S, Cateau H, Fukai T, Hensch TK. Bidirectional plasticity in fast-spiking GABA circuits by visual experience. Nature. 2009;462:218–221. doi: 10.1038/nature08485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.