

Abstract
The role of atrial natriuretic peptide (ANP) in regulating fetal cardiac growth is poorly understood. Angiotensin II (Ang II) stimulates proliferation in fetal sheep cardiomyocytes when growth is dependent on the activity of the mitogen-activated protein kinase (MAPK) and phosphoinositol-3-kinase (PI3K) pathways. We hypothesized that ANP would suppress near-term fetal cardiomyocyte proliferation in vitro and inhibit both the MAPK and PI3K pathways. Forty-eight hour 5-bromodeoxyuridine (BrdU) uptake (used as an index of proliferation) was measured in cardiomyocytes isolated from fetal sheep (135 day gestational age) in response to 100 nm Ang II with or without ANP (0.003–100 nm) or 1 μm 8-bromo-cGMP. The effects of these compounds on the MAPK and PI3K pathways were assessed by measuring extracellular signal-regulated kinase (ERK) and AKT phosphorylation following 10 min of treatment with Ang II, ANP or 8-bromo-cGMP. In right ventricular myocytes (RV), the lowest dose of ANP (0.003 nm) inhibited Ang II-stimulated BrdU uptake by 68%. Similarly, 8-bromo-cGMP suppressed Ang II-stimulated proliferation by 62%. The same effects were observed in left ventricular (LV) cardiomyoytes but the RV was more sensitive to the inhibitory effects of ANP than the LV (P < 0.0001). Intracellular cGMP was increased by 4-fold in the presence of 100 nm ANP. Ang II-stimulated ERK and Akt phosphorylation was inhibited by 100 nm ANP. The activity of ANP may in part be cGMP dependent, as 8-bromo-cGMP had similar effects on the cardiomyocytes.
Introduction
In adult mammals, atrial natriuretic peptide (ANP) is released from the atria in response to stretch. ANP is a 28 amino acid peptide that binds to a guanylyl cyclase-coupled receptor (natriuretic peptide receptor; NPRA or Npr1). It is found on the plasmalemma of many cell types. When bound by ligand, NPRA stimulates natriuresis, diuresis and vasodilatation (reviewed by McGrath et al. 2005). ANP also binds to NPRC, a non-guanylyl cyclase-coupled receptor that has been thought to act as a clearance receptor for natriuretic peptides, but may also play a role in cell growth (Rose & Giles, 2008).
During fetal life, ANP is produced by both atrial and ventricular myocardial cells and its actions are similar to those in the adult (Brace & Cheung, 1987; Brace et al. 1988; Jaekle et al. 1995). However, the circulating levels of ANP in the fetus are 5-fold higher than in the pregnant ewe (Cheung et al. 1987; Rosenfeld et al. 1992). Although ANP is not ordinarily detected in the adult ventricular myocardium, its production can be stimulated in response to cardiac disease states; its elevated gene expression in the ventricle has become a reliable marker of hypertrophic growth (Gardner, 2003). ANP is not a pro-hypertrophic factor despite its increased expression in hypertrophic myocardium. Rather, ANP suppresses the enlargement of both neonatal and adult cardiomyocytes in response to growth factors such as angiotensin II (Ang II) (Rosenkranz et al. 2003; Hayashi et al. 2004).
Evidence from gene-disrupted mice suggests that ANP regulates normal heart growth in the womb. In separate reports, Knowles et al. (2001) and Holtwick et al. (2002) observed that two different strains of NPRA knockout mice have larger hearts at birth than wild-type controls. These findings imply that ANP prevents myocardial overgrowth in normal cardiac development. Nevertheless, these experiments offer no insight into mechanisms by which ANP might moderate growth promoting processes. Holtwick et al. (2002) found that the 30% increase in cardiomyocyte size associated with the absence of ANP receptor function did not fully account for the 130% increase in cardiac mass at birth. The logical conclusion was that myocyte proliferation played a greater role in this cardiac growth than cardiomyocyte enlargement.
Cardiomyocyte proliferation accounts for most of fetal heart growth during the first half of gestation in sheep but as gestation proceeds, cells undergo ‘terminal differentiation’, the process by which mononucleated cardiomyocytes become binucleated by karyokinesis (Barbera et al. 2000; Jonker et al. 2007b). Mitotic activity is permanently suppressed in the resulting binucleated cardiomyocytes. In precocious species, such as sheep, some 80–90% of cardiomyocytes are binucleated at birth (Jonker et al. 2007b). Intrauterine events that suppress myocyte proliferation rates may therefore diminish the cardiomyocyte endowment at birth and compromise the future cardiac health of the offspring. Experiments in rats suggest that the number of heart cells is set at birth in offspring exposed to intrauterine hypoxia even though plastic growth remains theoretically possible during the first 2 weeks of postnatal life before terminal differentiation is complete (Bae et al. 2003; Li et al. 2004). Thus, the set point for cardiomyocyte endowment may be determined around the time of birth.
Several factors are known to stimulate the proliferation of immature ovine cardiomyocytes in vivo and/or in vitro. These include cardiac load (Giraud et al. 2005), Ang II (Sundgren et al. 2003b), cortisol (Giraud et al. 2006) and insulin-like growth factor-1 (IGF-1) (Sundgren et al. 2003a). It is also known that thyroid hormone inhibits proliferation of near-term fetal cardiomyocytes (Chattergoon et al. 2007).
The pro-proliferative actions of Ang II and IGF-1 operate through two signalling pathways that regulate the cell cycle in the fetal cardiomyocyte: the extracellular signal-regulated kinase (ERK) branch of the mitogen-activated protein kinase (MAPK) cascade and the phosphoinositol-3 kinase (PI3K) pathway (Sundgren et al. 2003a,b;). Blockade of either pathway prevents cardiomyocyte proliferation in fetal sheep (Sundgren et al. 2003a,b;). Hayashi et al. (2004) found that ANP strongly inhibited the MAPK–ERK pathway in immature rat cardiomyocytes. Thus, ANP may restrain heart growth via inhibition of the ERK pathway in the cardiomyocyte. Conversely, Silberbach et al. (1999) found that ANP stimulated the ERK system in neonatal rat cardiomyocytes. ANP inhibited immature cardiomyocyte hypertrophy in both of these rodent studies. Considering these contradictory data, the role of ANP in regulating the MAPK–ERK pathway in immature myocytes remains uncertain.
Both in vivo and in vitro studies in the rodent model suggest an important role for ANP in the regulation of immature cardiac muscle cell growth. However, early cardiomyocyte growth differs between small rodents and larger species such as sheep and humans, making it difficult to determine the degree to which rodent findings may apply to large mammals. Two important questions remain regarding the role of ANP in regulating the proliferation of the fetal cardiomyocyte in the large mammal: (1) Does ANP suppress proliferation in the immature cardiomyocyte? (2) Does ANP inhibit the MAPK–ERK and/or the PI3K pathways in the immature cardiomyocyte? The answers to these questions are highly important because they are key to understanding myocyte endowment at birth.
In this study, we hypothesized that ANP would suppress fetal cardiomyocyte proliferation and inhibit both the MAPK–ERK pathway and the PI3K pathway in immature cardiac myocytes from fetal sheep. In order to test these hypotheses, we determined the effect of ANP on Ang II-stimulated proliferative activity of fetal cardiomyocytes in vitro and studied the degrees to which activation of members of the MAPK–ERK and PI3K pathways were affected by physiological levels of ANP.
Methods
Ethical approval
All procedures were approved by the Oregon Health and Science University Institutional Animal Care and Use Committee. At 135 ± 1 days of gestation (mean ±s.d.; term, 145 days), hearts from fetal sheep were collected from control animals which included instrumented fetuses used as vehicle controls for other studies (slowly infused with lactated Ringer's solution into the right atrium), and non-instrumented fetuses. No differences were detected in experiments between cells isolated from instrumented (N= 5) and non-instrumented (N= 12) fetuses.
Cardiomyocyte isolation
Following killing of the ewe with an overdose of sodium pentobarbital which crosses the placenta (Euthasol ∼85 mg kg−1, Virbac, TX, USA), the heavily anaesthetized fetus received 10,000 U of heparin sulfate, followed by 10 ml of saturated KCl into the umbilical vein to arrest the heart in diastole. The fetal hearts were harvested and enzymatically dissociated as described in detail previously (Jonker et al. 2007a). Briefly, the hearts were retrogradely perfused via the coronary arteries with a series of gassed solutions (95% O2 and 5% CO2 at 39°C). First, they were perfused with Tyrodes buffer for 5–10 min (no calcium added; 140 mm NaCl, 5 mm KCl, 1 mm MgCl2 6H2O, 10 mm glucose, 10 mm Hepes; pH 7.35) until the vessels were cleared of blood; 5–10 min with 160 U ml−1 Type II collagenase (Worthington Biochemicals, Lakewood, NJ, USA) and 0.78 U ml−1 Type XIV protease (Sigma) in Tyrode buffer to digest the tissue; 5–10 min with a high potassium (KB) solution (74 mm glutamic acid, 30 mm KCl, 30 mm KH2PO4, 20 mm taurine, 3 mm MgSO4, 0.5 mm EGTA, 10 mm Hepes, 10 mm glucose; pH 7.37). The right ventricle (RV) and left ventricle (LV) free walls were separately removed with sterile scissors and gently agitated in KB solution to release the cells. Cardiac myocytes formed the bulk of the cell slurry; however, endothelial cells, fibroblasts and blood cells were also present.
Cardiomyocyte culture
The cells were cultured as previously described by our laboratory (Sundgren et al. 2003b; Chattergoon et al. 2007) with some modifications. LV and RV cells were studied separately. The freshly isolated cell slurry rested for 30 min at room temperature before centrifugation (at 2000 g for 5 min) and resuspension in sterile serum media (Gibco DMEM low glucose: 5.56 mm d-glucose, 4 mm l-glutamine, 1 mm sodium pyruvate, 5.33 mm KCl, 0.4 mm glycine, pH 7.4 with 10 mg l−1 insulin–transferrin–sodium selenite supplement (Sigma), 1 ml l−1 antibiotic–antimycotic solution (Sigma), and final concentration 10% fetal bovine serum (FBS; Invitrogen)). Serum-free media were the same composition as serum media without FBS. Cells were pre-plated twice to remove non-myocyte cells (2 h each time, 39°C; 95% air and 5% CO2) and plated on 22 mm × 22 mm glass coverslips (at a density of 500,000 cells per coverslip for later measurements of myocyte proliferation) or in 6-well plates (at a density of 1,000,000 cells per well for later protein isolation). Coverslips and plates were coated with laminin (3–5 mg ml−1) at least 4 h prior and then aspirated immediately before cell seeding. Cells were incubated in serum media at 39°C for 24 h. Cells were then incubated for 48 h in serum-free media, and the media was changed again to fresh serum-free for another 24 h. Twenty-four hours after the last media change, cells were treated with experimental drugs (day 5). For intracellular cGMP production experiments, passage 1 fetal sheep cardiomyocytes (N= 6 fetuses) were thawed quickly at 37°C and reconstituted to 1 million cells ml−1 in serum media. Frozen cells behaved identically to freshly isolated cells. They were plated in 6-well plates at a density of 500,000 cells per well for cGMP studies. Cells were allowed to attach for 24 h before the experimental protocol was started. The intracellular cGMP production experiments did not require a large number of fetal cardiomyocytes. In order to reduce the number of animals used for these experiments, we utilized cardiomyocytes previously isolated from late gestation (135 day) fetal sheep and frozen. Each ‘N’ value (or data point) is represented by a different fetus.
Treatment groups
To determine the effect of ANP on fetal cardiomyocyte proliferation we analysed three courses of cell treatment using 5-bromodeoxyuridine (BrdU) incorporation as an index of cell proliferation: (1) 48 h in serum-free media with ANP at a range of doses; (2) 48 h in serum-free media, 100 nm Ang II and ANP at a range of doses; (3) 48 h in serum-free media, 100 nm Ang II and 1 μm of the cGMP analogue 8-bromo-cyclic-GMP. The doses of ANP used were designed to include both the physiological range found within the circulation of the fetal sheep (0.03–0.33 nm) (Cheung et al. 1987; Brace & Cheung, 1987) and concentrations of ANP typically used within the literature (100 nm) (Silberbach et al. 1999).We have previously shown that 100 nm Ang II optimally stimulates fetal sheep cardiomyocyte BrdU uptake in vitro (Sundgren et al. 2003b) and this same dose was chosen for similar studies (Pandey et al. 2000). 8-Bromo-cGMP was used to identify possible downstream pathways of ANP activity.
BrdU uptake experiments
Fetal cardiomyocytes were cultured with 10 μm BrdU (Sigma) to determine proliferation rates in response to increasing doses of human/porcine 1–28 ANP (AnaSpec, Fremont, CA, USA) (0.003, 0.03, 0.33, 3.25, 32.5 and 100 nm) or 1 μm 8-bromo-cGMP (Bachem, Torrance, CA, USA) with and without 100 nm human Ang II (Bachem). Following 4 h of treatment, the media was aspirated and cells were fixed in acidified ethanol and stored in PBS with 0.1% sodium azide at 4°C until staining. Cardiomyocytes were stained with anti-myosin (mouse anti-myosin, 1:5000, Abcam, Cambridge, MA, USA) and anti-BrdU (rat anti-BrdU, 1:500, Abcam) overnight at 4°C as previously described (Chattergoon et al. 2007). Cells were washed with 1× PBS and incubated in anti-mouse rhodamine red (1:200, Jackson ImmunoResearch, West Grove, PA, USA) and anti-rat fluorescein isothiocyanate (FITC) (1:200, Jackson ImmunoResearch) secondary antibodies for 2 h at room temperature. Slides were mounted using Vectashield Hardset mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA) and stored overnight in the dark at 4°C to allow for the mount to dry. A minimum sample of 300 myocytes per treatment group was assessed for BrdU incorporation using fluorescence microscopy. Cardiomyocytes staining for myosin (rhodamine red) with green (FITC) punctate nuclei indicating BrdU incorporation, were counted as BrdU positive (Fig. 1).
Figure 1. BrdU uptake in proliferating cultured fetal sheep cardiomyocytes.
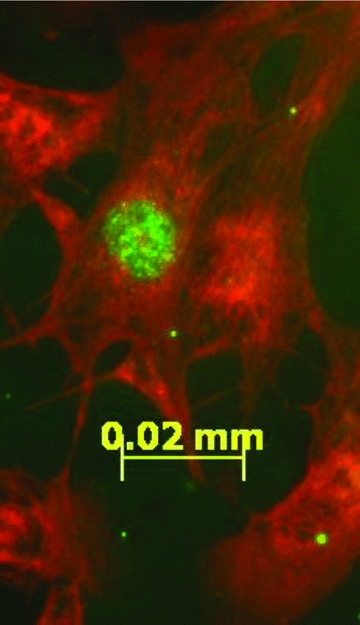
Cardiomyocytes isolated from the left and right ventricles of late gestation fetal sheep were cultured with the thymidine analogue BrdU for 48 h. Myosin was visualized with rhodamine red (red); BrdU-positive nuclei were visualized with FITC (green).
Intracellular cGMP production
Cells were incubated for 10 min in serum-free media with 0, 0.03, 0.33, 3.3 and 100 nm ANP. Cells were lysed (0.1 m HCl) and intracellular cGMP levels were determined following manufacturer's instructions (Cyclic GMP EIA, Catalogue no. 581021; Cayman Chemicals, Ann Arbor, MI, USA). Protein concentration was quantified by BCA assay following the manufacturer's instructions (Pierce, Rockford, IL, USA). Results were normalized to the amount of cellular protein per well and expressed as pmol cGMP (mg protein)−1.
Cell stimulation and Western blot analysis
To identify signalling pathways which may be altered by ANP, we pre-incubated cells with 100 nm ANP or 1 μm 8-bromo-cGMP for 10 min prior to addition of 100 nm Ang II for 10 min (39°C, 95% air and 5% CO2). Cells were also treated for 10 min with 100 nm ANP, 1 μm cGMP and 100 nm Ang II separately. Cells incubated for 10 min in serum-free or 10% serum media were used as negative and positive controls, respectively. Following treatment, cardiomyocytes were lysed (5 mm Tris-HCl, 5 mm EDTA, 0.06% SDS, Protease inhibitor Mini-complete tablet (Roche), and Phosphatase Inhibitor Cocktail 1 and 2 (Sigma)) and collected into pre-chilled tubes. Protein concentration was quantified as described above. Equal amounts of total protein/sample (10 μg) were separated by SDS-PAGE on a 10% gel and transferred to Optitran BA-S 83 nitrocellulose membrane. Membranes were blocked with 5% milk PBS-T (PBS + 0.01% Tween) buffer for 1 h. Membranes were incubated with primary antibodies rabbit anti-pERK (1:1000, Cell Signaling), mouse anti-ERK2 (1:1000, Cell Signaling), rabbit anti-pAKT (1:1000 Cell Signaling) and rabbit anti-total AKT (1:1000) overnight at 4°C. Membranes were washed in PBS-T before exposure to the anti-mouse or anti-rabbit secondary antibody (1:4000, Cell Signaling) for 1 h at room temperature. Antibody binding was detected using a chemiluminescence system (SuperSignal, Pierce); protein expression was quantified from a digitized image of the blot and expressed as a ratio of phosphorylated to total protein.
Statistical analysis
BrdU-positive cardiomyocytes were reported as a percentage of total number of cardiomyocytes counted (minimum of 300 per slide). Densitometry of Western blots was quantified using Image J 1.42q software (National Institutes of Health, USA). One-way repeated measures ANOVA was used to analyse differences between treatment groups with post hoc differences determined by Tukey's multiple comparison test. Differences between ventricles were analysed by two-way ANOVA. Significance was defined as P < 0.05. Data are expressed as mean ±s.e.m.
Results
BrdU uptake
When cultured in serum-free media, fetal sheep cardiomyocytes had low rates of BrdU uptake over a 48 h period (2.9 ± 0.2% in LV and 2.1 ± 0.1% in RV), as previously reported (Sundgren et al. 2003b). Neither physiological (0.03–3.3 nm) nor pharmacological (30–100 nm) concentrations of ANP altered rates of BrdU uptake in cardiomyocytes isolated from LV (5.0 ± 0.5%) or RV (3.0 ± 0.7%). However, there were important differences in behaviour of cardiomyocytes between the two ventricles. Overall, BrdU uptake rates were consistently higher in cells from the LV than from RV (P < 0.05). Exposure to either serum or Ang II (100 nm) treatment caused a significant increase in BrdU uptake in both LV (Fig. 2B) and RV (Fig. 2A) cardiomyocytes.
Figure 2. Inhibition of Ang II-stimulated BrdU uptake in cardiomyocytes.
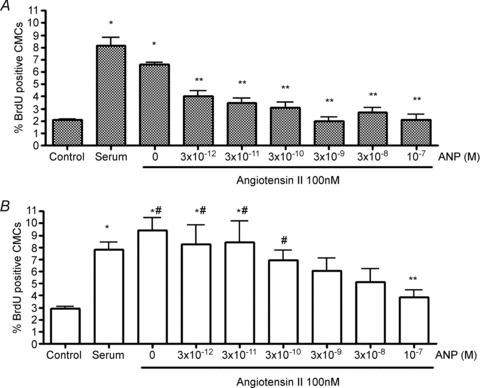
The Ang II-stimulated BrdU uptake in cardiomyocytes isolated from the right (A) and left (B) fetal ventricles were inhibited in a dose-dependent manner (48 h treatment). Left ventricular cardiomyocytes were less sensitive to ANP than were right ventricular cardiomyocytes. Data are means ±s.e.m. N= 6 fetuses per group. #P < 0.05 vs. matching treatment in RV; *P < 0.05 vs. serum-free control in same ventricle; **P < 0.05 vs. Ang II alone in same ventricle.
The key question of this study was to determine whether ANP affects proliferation rates of cardiomyocytes. Figure 2 shows that ANP inhibited the stimulatory action of Ang II on BrdU uptake in both LV and RV cardiomyocytes in a dose-dependent manner. Ang II-stimulated cardiomyocyte proliferation was inhibited at lower doses of ANP in the RV (0.003–0.3 nm) than in the LV (P < 0.01). Thus, cardiomyocytes from the RV appear to be more sensitive to the inhibitory effects of ANP compared to those of the LV (two-way ANOVA, P < 0.0001).
The role of cGMP
In order to determine whether ANP suppression was acting through the second messenger cGMP, we incubated cardiomyocytes with 1 μm of the cGMP analogue, 8-bromo-cGMP, with and without 100 nm Ang II for 48 h and determined BrdU uptake. Figure 3 shows that cGMP suppressed the Ang II-stimulated cardiomyocyte BrdU uptake in a manner indistinguishable from 100 nm ANP, whereas cGMP had no effect on basal rates of proliferation (RV: 2.4 ± 1.5%; LV: 2.0 ± 0.8%).
Figure 3. Inhibition of Ang II-stimulated BrdU uptake in cardiomyocytes with 8-bromo-cGMP.
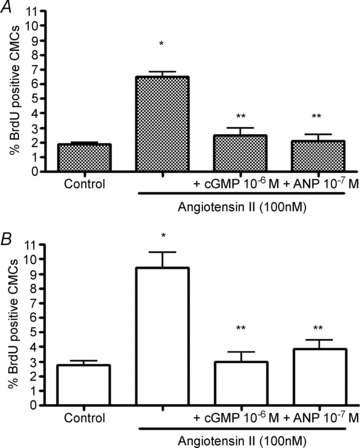
Ang II-stimulated BrdU uptake in cardiomyocytes isolated from right (A) and left (B) fetal ventricles was inhibited by 48 h of treatment with the cGMP analogue, 8-bromo-cGMP. Control cells were cultured in serum-free conditions. Data are means ±s.e.m. N= 5 or 6 fetuses per group. *P < 0.05 vs. control; **P < 0.05 vs. Ang II alone.
To further understand the differences in sensitivity to ANP between the ventricles, cGMP production by myocytes isolated from LV and RV was determined in response to varying doses of ANP (0.03, 0.3, 3.3, 100 nm). Intracellular cGMP levels were normalized to protein levels of the cells within the same well. In the absence of ANP for a 10 min period, intracellular levels of cGMP were very low and not different between ventricles (RV: 2.2 ± 1.3 pmol mg−1; LV: 1.4 ± 0.6 pmol mg−1). However, cGMP levels increased in both ventricles over the same period of time with the addition of 3.3 nm ANP (RV: 9.3 ± 7.2 pmol mg−1; LV: 4.5 ± 2.0 pmol mg−1) and 100 nm ANP (RV: 12.6 ± 9.5 pmol mg−1; LV: 6.8 ± 2.7 pmol mg−1) (P < 0.05 vs. no ANP). Intracellular cGMP levels varied highly between individual hearts, as seen in other species and cell types (Tsuruda et al. 2002). The results were expressed as changes compared to untreated controls to normalize the varied baseline expression between animals (Fig. 4). No differences were observed in cGMP production rates between ventricles in response to ANP.
Figure 4. Intracellular cGMP levels increased in response to 10−7m ANP in cardiomyocytes isolated from right and left fetal ventricles.
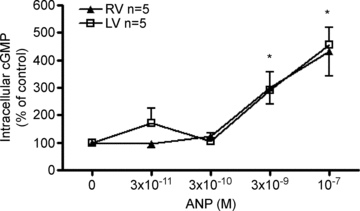
The percentage change from baseline intracellular cGMP levels in cardiomyocytes isolated from both left and right fetal ventricles as a function of ANP dose are shown. No effect of ventricle was detected. All treatments were 10 min. Data are means ±s.e.m. *P < 0.05 vs. no ANP control.
Effect of ANP on ERK and PI3K pathway signalling
The levels of phospho-ERK relative to total protein were increased in cardiomyocytes treated for 10 min with serum media (data not shown). Right ventricular cardiomyocytes showed a similar increase in phospho-ERK levels following treatment with 100 nm Ang II for 10 min (Fig. 5A). Left ventricular myocytes showed the same response profile but did not reach statistical significance (Fig. 5B). Treatment with 100 nm of ANP alone after 5, 10, 30 or 60 min had no effect on ERK phosphorylation in either ventricle (data not shown). Neither physiological nor pharmacological doses of ANP alone had any effect on ERK phosphorylation after 10 min (data not shown). Similar to the pattern observed in the BrdU experiments, however, a 10 min pre-incubation with either 100 nm ANP or 1 μm 8-bromo-cGMP inhibited the Ang II-induced ERK phosphorylation in cardiomyocytes isolated from the fetal RV (Fig. 5A). The trends in LV cardiomyocytes were similar to those in the RV. 8-bromo-cGMP significantly inhibited Ang II-induced ERK phosphorylation in both ventricles.
Figure 5. ANP inhibits Ang II-induced ERK phosphorylation in fetal cardiomyocytes isolated from the right ventricle (A) with a similar trend for the left ventricle (B).
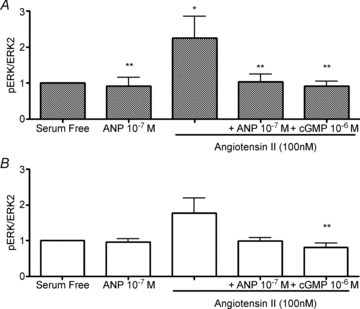
cGMP suppresses the generation of phospho-ERK in right and left ventricular cardiomyocytes. Data are means ±s.e.m. N= 5 or 6 fetuses per group. All treatments were 10 min. *P < 0.05 vs. serum-free control; **P < 0.05 vs. Ang II alone.
To investigate other mitogenic pathways in the fetal cardiomyocyte that may be influenced by ANP, we assessed the phosphorylation of a member of the PI3K pathway, AKT, in response to Ang II, ANP and cGMP. A 10 min treatment of 100 nm ANP had no effect on AKT phosphorylation in either ventricle (Fig. 6). Treatment with 100 nm of Ang II for the same period of time increased AKT phosphorylation in LV cardiomyocytes (P < 0.05) but pre-incubation with either 100 nm ANP or 1 μm 8-bromo-cGMP completely inhibited this effect. AKT phosphorylation in cardiomyocytes isolated from the RV was not measurably affected by isolated treatment with Ang II, ANP or cGMP.
Figure 6. The effects of ANP on Ang II-induced AKT phosphorylation in fetal cardiomyocytes isolated from the right (A) and left (B) ventricles.
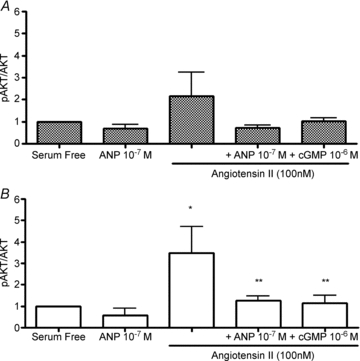
AKT phosphorylation was significantly increased in the presence of Ang II, an effect which was suppressed by ANP in the left ventricle (B) with a similar trend in the right ventricle (A). All treatments were 10 min. Data are means ±s.e.m. N= 5 or 6 fetuses per group. *P < 0.05 vs. serum-free controls; **P < 0.05 vs. Ang II alone.
Discussion
The primary finding of this study was that ANP is a powerful inhibitor of Ang II-induced proliferation in near-term fetal sheep cardiomyocytes in a dose-dependent manner. We also showed that intracellular cGMP increased by 4-fold in the presence of ANP and that the suppressive effect of ANP on proliferation was mimicked by the cGMP analogue, 8-bromo-cGMP. ANP also had effects on known mitogenic signalling pathways. The normal increases in ERK and PI3K activation under the influence of Ang II were suppressed by ANP. These findings supported our hypothesis that ANP acts as an anti-proliferative factor in the fetal heart by dampening growth factor signalling pathways that ordinarily stimulate cardiomyocyte proliferation.
The anti-proliferative effects of ANP in fetal cardiomyocytes have not to our knowledge previously been demonstrated. However, our findings are consistent with many reports demonstrating that ANP inhibits stimulated growth in other cell types including vascular smooth muscle cells (Neuser et al. 1993; Hashim et al. 2006), fibroblasts (Fujisaki et al. 1995; Glenn et al. 2009), mesangial cells (Sugimoto et al. 1993, 1996; Pandey et al. 2000) adipose cells (Sarzani et al. 2008) and astrocytes (Prins et al. 1996). The long-term consequence of having too many cardiomyocytes in the heart has not been thoroughly investigated. One could speculate that it would lead to wall thickening because of the apparently obligatory minimal cardiomyocyte size that is maintained in all mammals. Hence, ANP might play a critical role in regulating cell number by suppression of proliferation.
ANP may exert its anti-proliferative effects via cGMP signalling in fetal myocytes
ANP binding to a membrane-bound, guanylyl cyclase-linked receptor (NPRA) leads to a conformational change of the receptor and initiates the production of cyclic guanosine monophosphate (cGMP) (Silberbach & Roberts, 2001). cGMP in turn stimulates cGMP-activated protein kinase which is thought to mediate most of the cellular effects of ANP. We confirmed that ANP treatment stimulates cGMP production and that the cGMP analogue 8-bromo-cGMP was able to imitate ANP's effects on BrdU uptake and associated signalling cascades in cultured fetal sheep cardiomyocytes. Thus, consistent with previous studies in other cell types (Wolf et al. 1992; Fujisaki et al. 1995; Hamad et al. 1999; Pandey et al. 2000), we provide evidence that suppression of mitotic activity by ANP occurs through signalling of the second messenger cGMP in the fetal cardiomyocyte.
While the type A receptor (NPRA) has been credited with mediating the majority of ANP's cardioprotective actions via cGMP release, ANP also binds to another receptor, the type C natriuretic peptide receptor (NPRC) found ubiquitously throughout the body. NPRC is now known to be coupled to the Gi protein which has inhibitory effects on adenylate cyclase and leads to downstream inhibition of ERK and PI3K pathways (as reviewed by Rose & Giles, 2008). NPRC mediates ANP's growth inhibitory effects in several cell types, either in association with, or independent of, NPRA-mediated cGMP release (Cahill & Hassid, 1991; Prins et al. 1996; Hamad et al. 1999; Hashim et al. 2006; Li et al. 2006). Conceivably, there is a cGMP-independent component to the anti-mitotic actions of ANP in the fetal cardiomyocyte. Though ANP effectively inhibited Ang II-induced proliferation at low doses in RV myocytes over 48 h, these same doses, given acutely, did not measurably increase cGMP levels. Importantly, the NPRA/cGMP signalling pathway accounted for ANP's anti-proliferative effects in studies of other cell types (Haneda et al. 1993; Pandey et al. 2000; Hayashi et al. 2004) suggesting that the ANP anti-growth effects may be entirely cGMP dependent. NPRC gene-disrupted mice develop skeletal abnormalities and hypotension due to chronically elevated ANP levels, but maintain normal renal and cardiovascular function (Matsukawa et al. 1999). In contrast, NPRA gene-disrupted mice have larger hearts at birth and are more susceptible to heart damage in response to cardiovascular challenges (Oliver et al. 1997; Knowles et al. 2001; Holtwick et al. 2002; Kuhn et al. 2002). While NPRC could mediate some of ANP's anti-mitotic effects in fetal myocytes, the mimicry of ANP's effects on proliferation and signalling by the cGMP analogue leads us to speculate that the guanylyl cyclase-linked NPRA is a primary pathway by which ANP suppresses fetal sheep cardiomyocyte growth. Future studies should address the role of NPRC-mediated signalling in fetal myocytes.
ANP inhibits ERK and AKT phosphorylation in fetal cardiomyocytes
As in this study, ANP has also been found to inhibit growth factor-stimulated ERK phosphorylation in other studies (Sugimoto et al. 1993; Pandey et al. 2000; Hashim et al. 2006). We have previously reported that the inhibition of either MEK in the ERK pathway or PI3K completely suppresses cardiomyocyte proliferation in immature ovine cardiomyocytes (Sundgren et al. 2003a,b;). It now appears that ANP also acts to suppress ERKs and PI3K which prevents the normal stimulatory growth actions of Ang II.
The cardiomyocyte-specific deletion of NPRA in adult mice was associated with higher basal levels of cardiac ERK phosphorylation in vivo (Bubikat et al. 2005), suggesting that ANP chronically inhibits ERK activation. However, Silberbach et al. (1999) found that ANP increased ERK phosphorylation in a dose- and time-dependent fashion in neonatal rat myocytes. This finding contradicts that of Hayashi et al. (2004), who showed that ANP inhibited basal ERK phosphorylation in neonatal rat myocytes in a dose- and time-dependent manner. We did not detect a measurable effect of ANP (0.003–100 nm) on basal ERK phosphorylation at 5, 10, 30 or 60 min in fetal sheep myocytes. However, unlike immature ovine cardiomyocytes, those from neonatal rats are isolated post-birth and lose their ability to take up BrdU after 48 h in culture (Sadoshima et al. 1997). Cardiomyocytes isolated from fetal sheep maintain their proliferative potential even after weeks in culture and they are thus appropriate for studying cardiomyocyte proliferation.
ANP may exert anti-proliferative effects via inhibition of the PI3K pathway in the fetal heart. In addition to the ERK pathway, PI3K-derived signals are also necessary for increased cardiomyocyte proliferation in the fetal heart in vitro (Sundgren et al. 2003a). Stimulation of PI3K at the cell membrane leads to phosphorylation and activation of its downstream effector, AKT. Phosphorylated AKT activates several signalling pathways within the cardiomyocyte, affecting cell survival, metabolism, inflammation and cell growth (Matsui et al. 2003). AKT is activated by pro-growth factors such as IGF-I in adult (Kulik et al. 1997) as well as fetal cardiomyocytes (Sundgren et al. 2003a). In this study, AKT was activated under the influence of Ang II. Others have shown that ANP and cGMP stimulate the PI3K/AKT pathway in cardiomyocytes from the neonatal rat (Kato et al. 2005), as well as human and bovine endothelial cells (Kawasaki et al. 2003). In fetal sheep cardiomyocytes, ANP did not affect basal levels of phospho-AKT, though it did suppress the activation of AKT when stimulated by Ang II. The latter data are consistent with that of Hashim et al. (2006), who reported that ANP inhibited Ang II-stimulated proliferation by disrupting ERK and AKT phosphorylation in rat vascular smooth muscle cells.
It is not clear how the ANP/cGMP and Ang II signalling pathways interact in the immature cardiomyocyte. Both ANP and 8-bromo-cGMP stimulate the expression of MAPK phosphatase 1 (MKP-1) in neonatal rodent cardiomyocytes (Sugimoto et al. 1996; Hayashi et al. 2004). MKP-1 is a dual serine/threonine and tyrosine phosphatase that specifically inactivates members of the MAPK family (reviewed by Boutros et al. 2008). It is possible that the growth-inhibitory actions of ANP may be due to the induction of MKP-1 expression (Sugimoto et al. 1996; Pandey et al. 2000; Hayashi et al. 2004). The role of MKP-1 in the actively proliferating cardiomyocyte has not yet been investigated.
ANP sensitivity differs between fetal ventricles
We found a significant difference in sensitivity to ANP between the left and right fetal ventricles. This is not completely surprising because we and others have shown that the fetal ventricles are different from one another, in many ways. The functions of the two ventricles differ considerably (Thornburg & Morton, 1986; Pinson et al. 1987; Thornburg & Reller, 1999). The right fetal ventricle is more sensitive than the left to plasma protein infusion, anaemia and aortic banding in vivo (Olson et al. 2006a,b; Jonker et al. 2007a). Conversely, Segar et al. (2001) reported that LV, but not RV, weights increased in response to chronic Ang II infusion of fetal sheep. We did not detect a difference in BrdU uptake between cardiomyocytes isolated from LV or RV in response to Ang II treatment in the present study or in previous work (Sundgren et al. 2003b). Interestingly, we also noted differences in activity of the ERK and PI3K signalling pathways between ventricles. Where the ERK pathway is stimulated by Ang II and inhibited by ANP treatment in the RV, it is the P13K pathway in the LV that is most significantly altered. However, only the highest dose of ANP (10−7m) suppressed the proliferative effect of Ang II on LV myocytes while the lowest dose of ANP (0.003 nm) was effective in the RV cells; this may explain why Segar et al. found that the LV, but not RV responded to Ang II in vivo.
ANP does not affect basal cardiomyocyte proliferation
ANP had no measurable effect on basal BrdU uptake, ERK or PI3K signalling in isolated fetal sheep cardiomyocytes. In the near-term fetal sheep, ANP concentration is ∼2 ng mg−1 ventricular protein or 3 mg per ventricle and ∼200 ng mg−1 protein in the atria (Cheung & Roberts, 1993). Circulating levels of ANP are 4- to 5-times higher in the fetus compared to the adult – ranging from 150 to 265 pg ml−1 (50–90 pm) in the healthy, late-gestation sheep fetus (Cheung et al. 1987). These high levels are probably due to a robust constitutive ANP secretion from the fetal ventricles. Thus, it appears that high ANP levels in the fetus do not interfere with normal growth of the heart but offer some protection against runaway growth of the myocardium. Since chronic administration of growth factors like IGF-1 is known to cause cardiomyocyte proliferation in utero (Sundgren et al. 2003a) even in the presence of high circulating levels of ANP, it remains unclear how growth factor regulatory signals are integrated in vivo. We suggest that cardiac growth in this pre-partum period relies on an intricate balance between pro- and anti-proliferative stimuli, and perturbations to this balance can greatly affect cardiomyocyte endowment.
Conclusions
Our data support the hypothesis that ANP suppresses stimulated proliferation in immature cardiomyocytes. The component of cardiomyocyte proliferation stimulated by Ang II was completely inhibited by ANP and was accompanied by a suppression of ERK and AKT phosphorylation. The mitotic suppression by ANP may be mediated by NPRA via cGMP. The differences in ANP and Ang II signalling between the heart ventricles are noteworthy and warrant further investigation.
Acknowledgments
This work at Oregon Health and Science University was supported by the National Institute of Child Health and Human Development Program Project Grant (P01HD34430), an American Heart Association Fellowship (O’Tierney), the Tartar Trust Foundation and the Department of Veterans Affairs. The authors have no conflicts of interest to declare.
Glossary
Abbreviations
- Ang II
angiotensin II
- ANP
atrial natriuretic peptide
- BrdU
5-bromodeoxyuridine
- cGMP
cyclic guanosine monophosphate
- CMC
cardiomyocyte
- ERK
extracellular signal-regulated kinase
- IGF-1
insulin-like growth factor-1
- LV
left ventricle
- MAPK
mitogen-activated protein kinase
- MKP-1
MAPK phosphatase-1
- NPRA
natriuretic peptide receptor type A
- PI3K
phosphoinositol-3 kinase
- RV
right ventricle
Author contributions
P.F.O'T. conducted all experiments. P.F.O’T. and K.L.T. were involved in study conception and design, interpretation of data, writing and revising the manuscript. N.N.C., S.L. and G.D.G. contributed to study design, data interpretation and manuscript revision. All authors have approved the final version to be published. Experiments were performed at Oregon Health & Science University, Portland, OR, USA.
References
- Bae S, Xiao Y, Li G, Casiano CA, Zhang L. Effect of maternal chronic hypoxic exposure during gestation on apoptosis in fetal rat heart. Am J Physiol Heart Circ Physiol. 2003;285:H983–H990. doi: 10.1152/ajpheart.00005.2003. [DOI] [PubMed] [Google Scholar]
- Barbera A, Giraud GD, Reller MD, Maylie J, Morton MJ, Thornburg KL. Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1157–R1164. doi: 10.1152/ajpregu.2000.279.4.R1157. [DOI] [PubMed] [Google Scholar]
- Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev. 2008;60:261–310. doi: 10.1124/pr.107.00106. [DOI] [PubMed] [Google Scholar]
- Brace RA, Cheung CY. Cardiovascular and fluid responses to atrial natriuretic factor in sheep fetus. Am J Physiol Regul Integr Comp Physiol. 1987;253:R561–R567. doi: 10.1152/ajpregu.1987.253.4.R561. [DOI] [PubMed] [Google Scholar]
- Brace RA, Miner LK, Siderowf AD, Cheung CY. Fetal and adult urine flow and ANF responses to vascular volume expansion. Am J Physiol Regul Integr Comp Physiol. 1988;255:R846–R850. doi: 10.1152/ajpregu.1988.255.5.R846. [DOI] [PubMed] [Google Scholar]
- Bubikat A, De Windt LJ, Zetsche B, Fabritz L, Sickler H, Eckardt D, Godecke A, Baba HA, Kuhn M. Local atrial natriuretic peptide signalling prevents hypertensive cardiac hypertrophy in endothelial nitric-oxide synthase-deficient mice. J Biol Chem. 2005;280:21594–21599. doi: 10.1074/jbc.M501103200. [DOI] [PubMed] [Google Scholar]
- Cahill PA, Hassid A. Clearance receptor-binding atrial natriuretic peptides inhibit mitogenesis and proliferation of rat aortic smooth muscle cells. Biochem Biophys Res Commun. 1991;179:1606–1613. doi: 10.1016/0006-291x(91)91758-5. [DOI] [PubMed] [Google Scholar]
- Chattergoon NN, Giraud GD, Thornburg KL. Thyroid hormone inhibits proliferation of fetal cardiac myocytes in vitro. J Endocrinol. 2007;192:R1–R8. doi: 10.1677/JOE-06-0114. [DOI] [PubMed] [Google Scholar]
- Cheung CY, Gibbs DM, Brace RA. Atrial natriuretic factor in maternal and fetal sheep. Am J Physiol Endocrinol Metab. 1987;252:E279–E282. doi: 10.1152/ajpendo.1987.252.2.E279. [DOI] [PubMed] [Google Scholar]
- Cheung CY, Roberts VJ. Developmental changes in atrial natriuretic factor content and localization of its messenger ribonucleic acid in ovine fetal heart. Am J Obstet Gynecol. 1993;169:1345–1351. doi: 10.1016/0002-9378(93)90303-z. [DOI] [PubMed] [Google Scholar]
- Fujisaki H, Ito H, Hirata Y, Tanaka M, Hata M, Lin M, Adachi S, Akimoto H, Marumo F, Hiroe M. Natriuretic peptides inhibit angiotensin II-induced proliferation of rat cardiac fibroblasts by blocking endothelin-1 gene expression. J Clin Invest. 1995;96:1059–1065. doi: 10.1172/JCI118092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner DG. Natriuretic peptides: markers or modulators of cardiac hypertrophy? Trends Endocrinol Metab. 2003;14:411–416. doi: 10.1016/s1043-2760(03)00113-9. [DOI] [PubMed] [Google Scholar]
- Giraud GD, Faber JJ, Jonker S, Davis L, Anderson DF. Intravascular infusions of plasma into fetal sheep cause arterial and venous hypertension. J Appl Physiol. 2005;99:884–889. doi: 10.1152/japplphysiol.01429.2004. [DOI] [PubMed] [Google Scholar]
- Giraud GD, Louey S, Jonker S, Schultz J, Thornburg KL. Cortisol stimulates cell cycle activity in the cardiomyocyte of the sheep fetus. Endocrinology. 2006;147:3643–3649. doi: 10.1210/en.2006-0061. [DOI] [PubMed] [Google Scholar]
- Glenn DJ, Rahmutula D, Nishimoto M, Liang F, Gardner DG. Atrial natriuretic peptide suppresses endothelin gene expression and proliferation in cardiac fibroblasts through a GATA4-dependent mechanism. Cardiovasc Res. 2009;84:209–217. doi: 10.1093/cvr/cvp208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamad AM, Johnson SR, Knox AJ. Antiproliferative effects of NO and ANP in cultured human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 1999;277:L910–L918. doi: 10.1152/ajplung.1999.277.5.L910. [DOI] [PubMed] [Google Scholar]
- Haneda M, Kikkawa R, Koya D, Sakamoto K, Nakanishi S, Matsuda Y, Shigeta Y. Biological receptors mediate anti-proliferative action of atrial natriuretic peptide in cultured mesangial cells. Biochem Biophys Res Commun. 1993;192:642–648. doi: 10.1006/bbrc.1993.1463. [DOI] [PubMed] [Google Scholar]
- Hashim S, Li Y, Anand-Srivastava MB. Small cytoplasmic domain peptides of natriuretic peptide receptor-C attenuate cell proliferation through Gialpha protein/MAP kinase/PI3-kinase/AKT pathways. Am J Physiol Heart Circ Physiol. 2006;291:H3144–H3153. doi: 10.1152/ajpheart.00327.2006. [DOI] [PubMed] [Google Scholar]
- Hayashi D, Kudoh S, Shiojima I, Zou Y, Harada K, Shimoyama M, Imai Y, Monzen K, Yamazaki T, Yazaki Y, Nagai R, Komuro I. Atrial natriuretic peptide inhibits cardiomyocyte hypertrophy through mitogen-activated protein kinase phosphatase-1. Biochem Biophys Res Commun. 2004;322:310–319. doi: 10.1016/j.bbrc.2004.07.119. [DOI] [PubMed] [Google Scholar]
- Holtwick R, Baba HA, Ehler E, Risse D, Vobeta M, Gehrmann J, Pierkes M, Kuhn M. Left but not right cardiac hypertrophy in atrial natriuretic peptide receptor-deficient mice is prevented by angiotensin type 1 receptor antagonist losartan. J Cardiovasc Pharmacol. 2002;40:725–734. doi: 10.1097/00005344-200211000-00010. [DOI] [PubMed] [Google Scholar]
- Jaekle RK, Sheikh AU, Berry DD, Washburn L, Rose JC. Hemodynamic and hormonal responses to atrial distension in the ovine fetus. Am J Obstet Gynecol. 1995;173:694–701. doi: 10.1016/0002-9378(95)90325-9. [DOI] [PubMed] [Google Scholar]
- Jonker SS, Faber JJ, Anderson DF, Thornburg KL, Louey S, Giraud GD. Sequential growth of fetal sheep cardiac myocytes in response to simultaneous arterial and venous hypertension. Am J Physiol Regul Integr Comp Physiol. 2007a;292:R913–R919. doi: 10.1152/ajpregu.00484.2006. [DOI] [PubMed] [Google Scholar]
- Jonker SS, Zhang L, Louey S, Giraud GD, Thornburg KL, Faber JJ. Myocyte enlargement, differentiation, and proliferation kinetics in the fetal sheep heart. J Appl Physiol. 2007b;102:1130–1142. doi: 10.1152/japplphysiol.00937.2006. [DOI] [PubMed] [Google Scholar]
- Kato T, Muraski J, Chen Y, Tsujita Y, Wall J, Glembotski CC, Schaefer E, Beckerle M, Sussman MA. Atrial natriuretic peptide promotes cardiomyocyte survival by cGMP-dependent nuclear accumulation of zyxin and Akt. J Clin Invest. 2005;115:2716–2730. doi: 10.1172/JCI24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki K, Smith RS, Jr, Hsieh CM, Sun J, Chao J, Liao JK. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Mol Cell Biol. 2003;23:5726–5737. doi: 10.1128/MCB.23.16.5726-5737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles JW, Esposito G, Mao L, Hagaman JR, Fox JE, Smithies O, Rockman HA, Maeda N. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A-deficient mice. J Clin Invest. 2001;107:975–984. doi: 10.1172/JCI11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M, Holtwick R, Baba HA, Perriard JC, Schmitz W, Ehler E. Progressive cardiac hypertrophy and dysfunction in atrial natriuretic peptide receptor (GC-A) deficient mice. Heart. 2002;87:368–374. doi: 10.1136/heart.87.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik G, Klippel A, Weber MJ. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Bae S, Zhang L. Effect of prenatal hypoxia on heat stress-mediated cardioprotection in adult rat heart. Am J Physiol Heart Circ Physiol. 2004;286:H1712–H1719. doi: 10.1152/ajpheart.00898.2003. [DOI] [PubMed] [Google Scholar]
- Li Y, Hashim S, Anand-Srivastava MB. Intracellular peptides of natriuretic peptide receptor-C inhibit vascular hypertrophy via Gqα/MAP kinase signalling pathways. Cardiovasc Res. 2006;72:464–472. doi: 10.1016/j.cardiores.2006.08.012. [DOI] [PubMed] [Google Scholar]
- McGrath MF, de Bold ML, de Bold AJ. The endocrine function of the heart. Trends Endocrinol Metab. 2005;16:469–477. doi: 10.1016/j.tem.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Matsui T, Nagoshi T, Rosenzweig A. Akt and PI 3-kinase signalling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2003;2:220–223. [PubMed] [Google Scholar]
- Matsukawa N, Grzesik WJ, Takahashi N, Pandey KN, Pang S, Yamauchi M, Smithies O. The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc Natl Acad Sci U S A. 1999;96:7403–7408. doi: 10.1073/pnas.96.13.7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuser D, Stasch JP, Knorr A, Kazda S. Inhibition by atrial natriuretic peptide of endothelin-1-stimulated proliferation of vascular smooth-muscle cells. J Cardiovasc Pharmacol. 1993;22:S257–S261. doi: 10.1097/00005344-199322008-00068. [DOI] [PubMed] [Google Scholar]
- Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL, Pandey KN, Milgram SL, Smithies O, Maeda N. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci U S A. 1997;94:14730–14735. doi: 10.1073/pnas.94.26.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson AK, Protheroe KN, Scholz TD, Segar JL. The mitogen-activated protein kinases and Akt are developmentally regulated in the chronically anemic fetal sheep heart. J Soc Gynecol Investig. 2006a;13:157–165. doi: 10.1016/j.jsgi.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Olson AK, Protheroe KN, Segar JL, Scholz TD. Mitogen-activated protein kinase activation and regulation in the pressure-loaded fetal ovine heart. Am J Physiol Heart Circ Physiol. 2006b;290:H1587–H1595. doi: 10.1152/ajpheart.00984.2005. [DOI] [PubMed] [Google Scholar]
- Pandey KN, Nguyen HT, Li M, Boyle JW. Natriuretic peptide receptor-A negatively regulates mitogen-activated protein kinase and proliferation of mesangial cells: role of cGMP-dependent protein kinase. Biochem Biophys Res Commun. 2000;271:374–379. doi: 10.1006/bbrc.2000.2627. [DOI] [PubMed] [Google Scholar]
- Pinson CW, Morton MJ, Thornburg KL. An anatomic basis for fetal right ventricular dominance and arterial pressure sensitivity. J Dev Physiol. 1987;9:253–269. [PubMed] [Google Scholar]
- Prins BA, Weber MJ, Hu RM, Pedram A, Daniels M, Levin ER. Atrial natriuretic peptide inhibits mitogen-activated protein kinase through the clearance receptor. Potential role in the inhibition of astrocyte proliferation. J Biol Chem. 1996;271:14156–14162. doi: 10.1074/jbc.271.24.14156. [DOI] [PubMed] [Google Scholar]
- Rose RA, Giles WR. Natriuretic peptide C receptor signalling in the heart and vasculature. J Physiol. 2008;586:353–366. doi: 10.1113/jphysiol.2007.144253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld CR, Samson WK, Roy TA, Faucher DJ, Magness RR. Vasoconstrictor-induced secretion of ANP in fetal sheep. Am J Physiol Endocrinol Metab. 1992;263:E526–E533. doi: 10.1152/ajpendo.1992.263.3.E526. [DOI] [PubMed] [Google Scholar]
- Rosenkranz AC, Woods RL, Dusting GJ, Ritchie RH. Antihypertrophic actions of the natriuretic peptides in adult rat cardiomyocytes: importance of cyclic GMP. Cardiovasc Res. 2003;57:515–522. doi: 10.1016/s0008-6363(02)00667-3. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Aoki H, Izumo S. Angiotensin II and serum differentially regulate expression of cyclins, activity of cyclin-dependent kinases, and phosphorylation of retinoblastoma gene product in neonatal cardiac myocytes. Circ Res. 1997;80:228–241. doi: 10.1161/01.res.80.2.228. [DOI] [PubMed] [Google Scholar]
- Sarzani R, Marcucci P, Salvi F, Bordicchia M, Espinosa E, Mucci L, Lorenzetti B, Minardi D, Muzzonigro G, Dessi-Fulgheri P, Rappelli A. Angiotensin II stimulates and atrial natriuretic peptide inhibits human visceral adipocyte growth. Int J Obes (Lond) 2008;32:259–267. doi: 10.1038/sj.ijo.0803724. [DOI] [PubMed] [Google Scholar]
- Segar JL, Dalshaug GB, Bedell KA, Smith OM, Scholz TD. Angiotensin II in cardiac pressure-overload hypertrophy in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2001;281:R2037–R2047. doi: 10.1152/ajpregu.2001.281.6.R2037. [DOI] [PubMed] [Google Scholar]
- Silberbach M, Gorenc T, Hershberger RE, Stork PJ, Steyger PS, Roberts CT., Jr Extracellular signal-regulated protein kinase activation is required for the anti-hypertrophic effect of atrial natriuretic factor in neonatal rat ventricular myocytes. J Biol Chem. 1999;274:24858–24864. doi: 10.1074/jbc.274.35.24858. [DOI] [PubMed] [Google Scholar]
- Silberbach M, Roberts CT., Jr Natriuretic peptide signalling: molecular and cellular pathways to growth regulation. Cell Signal. 2001;13:221–231. doi: 10.1016/s0898-6568(01)00139-5. [DOI] [PubMed] [Google Scholar]
- Sugimoto T, Haneda M, Togawa M, Isono M, Shikano T, Araki S, Nakagawa T, Kashiwagi A, Guan KL, Kikkawa R. Atrial natriuretic peptide induces the expression of MKP-1, a mitogen-activated protein kinase phosphatase, in glomerular mesangial cells. J Biol Chem. 1996;271:544–547. doi: 10.1074/jbc.271.1.544. [DOI] [PubMed] [Google Scholar]
- Sugimoto T, Kikkawa R, Haneda M, Shigeta Y. Atrial natriuretic peptide inhibits endothelin-1-induced activation of mitogen-activated protein kinase in cultured rat mesangial cells. Biochem Biophys Res Commun. 1993;195:72–78. doi: 10.1006/bbrc.1993.2011. [DOI] [PubMed] [Google Scholar]
- Sundgren NC, Giraud GD, Schultz JM, Lasarev MR, Stork PJ, Thornburg KL. Extracellular signal-regulated kinase and phosphoinositol-3 kinase mediate IGF-1 induced proliferation of fetal sheep cardiomyocytes. Am J Physiol Regul Integr Comp Physiol. 2003a;285:R1481–R1489. doi: 10.1152/ajpregu.00232.2003. [DOI] [PubMed] [Google Scholar]
- Sundgren NC, Giraud GD, Stork PJ, Maylie JG, Thornburg KL. Angiotensin II stimulates hyperplasia but not hypertrophy in immature ovine cardiomyocytes. J Physiol. 2003b;548:881–891. doi: 10.1113/jphysiol.2003.038778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg KL, Morton MJ. Filling and arterial pressures as determinants of left ventricular stroke volume in fetal lambs. Am J Physiol. 1986;251:H961–H968. doi: 10.1152/ajpheart.1986.251.5.H961. [DOI] [PubMed] [Google Scholar]
- Thornburg KL, Reller MD. Coronary flow regulation in the fetal sheep. Am J Physiol. 1999;277:R1249–R1260. doi: 10.1152/ajpregu.1999.277.5.R1249. [DOI] [PubMed] [Google Scholar]
- Tsuruda T, Boerrigter G, Huntley BK, Noser JA, Cataliotti A, Costello-Boerrigter LC, Chen HH, Burnett JC., Jr Brain natriuretic Peptide is produced in cardiac fibroblasts and induces matrix metalloproteinases. Circ Res. 2002;91:1127–1134. doi: 10.1161/01.res.0000046234.73401.70. [DOI] [PubMed] [Google Scholar]
- Wolf G, Thaiss F, Schoeppe W, Stahl RA. Angiotensin II-induced proliferation of cultured murine mesangial cells: inhibitory role of atrial natriuretic peptide. J Am Soc Nephrol. 1992;3:1270–1278. doi: 10.1681/ASN.V361270. [DOI] [PubMed] [Google Scholar]