

Abstract
Excess reactive oxygen species (ROS) play a crucial role under pathophysiological conditions, such as ischaemia/reperfusion and diabetes, potentially contributing to cardiac arrhythmia. hERG1 (KCNH2) potassium channels terminate the cardiac action potential and malfunction can lead to long-QT syndrome and fatal arrhythmia. To investigate the molecular mechanisms of hERG1 channel alteration by ROS, hERG1 and mutants thereof were expressed in HEK293 cells and studied with the whole-cell patch-clamp method. Even mild ROS stress induced by hyperglycaemia markedly decreased channel current. Intracellular H2O2 or cysteine-specific modifiers also strongly inhibited channel activity and accelerated deactivation kinetics. Mutagenesis revealed that cysteine 723 (C723), a conserved residue in a structural element linking the C-terminal domain to the channel's gate, is critical for oxidative functional modification. Moreover, kinetics of channel closure strongly influences ROS-induced modification, where rapid channel deactivation diminishes ROS sensitivity. Because of its fast deactivation kinetics, the N-terminally truncated splice variant hERG1b possesses greater resistance to oxidative modification.
Introduction
Oxidative stress is a recognized risk factor in cardiac dysfunction. The generation of reactive oxygen species (ROS) is elevated under many pathophysiological conditions such as ischaemia/reperfusion (Kuzuya et al. 1990), but also during hyper- and hypoglycaemia and inflammatory insults (Van Wagoner, 2008). One of the functional consequences of such oxidative stress is a prolongation of the action potential duration (APD), which leads to an increased risk of cardiac arrhythmias. In particular for diabetic patients, a higher incidence of sudden fatal arrhythmias is observed (Robillon et al. 1999; Casis & Echevarria, 2004). Although the molecular events underlying the pathophysiological outcome are diverse, recent results have shown that K+ channels contributing to the termination of the action potential play a pivotal role (Zhang et al. 2003).
The human ether à go-go-related gene 1 (hERG1, Kv11.1, KCNH2) encodes the pore-forming subunit of the K+ channel complex that carries the rapid component of the delayed rectifier current (IKr) in the heart (Sanguinetti et al. 1995; Trudeau et al. 1995). The principal role of IKr is to terminate the cardiac action potential, and therefore mutations in the hERG1 gene causing impaired expression or altered channel function clinically manifest as congenital type-2 long-QT syndrome (LQTS-2) (Sanguinetti & Tristani-Firouzi, 2006). Acquired forms of LQTS typically arise from off-target pharmacological block of IKr (Fermini & Fossa, 2003; Thomas et al. 2006) but may also be induced by posttranslational modifications of the channel protein (Chen et al. 2009).
Investigations on IKr in cardiac cells and recombinantly expressed hERG1a channels indicate that elevation of ROS levels by various routes diminishes the activity of this channel type. In particular, excess ROS induced by hypo- and hyperglycaemia (Zhang et al. 2003, 2006), by tumour necrosis factor alpha (TNF-α, Wang et al. 2004) and by ceramide (Bai et al. 2007) decreased the activity of hERG1 channels and antioxidants antagonized these effects. Direct application of the physiological oxidant H2O2 to hERG1a channels expressed in a mammalian cell line revealed multiple functional effects such as acceleration of channel activation and slowly developing acceleration of deactivation, which could account for an initial APD shortening and a subsequent APD prolongation when cardiac cells are exposed to H2O2 (Bérubéet al. 2001). Thus far, however, the molecular mechanism by which ROS modulate hERG1 channels remains elusive. In a study on hERG1a expressed in Xenopus oocytes, Taglialatela et al. (1997) showed that ROS produced by extracellular iron/ascorbic acid increases hERG1a outward current because of a shift in the voltage dependence of channel inactivation. This phenomenon could be abolished by mutagenesis of two extracellularly accessible histidine residues, but it remains unclear if these histidine residues are in fact modified by ROS (Pannaccione et al. 2002).
ROS may modify a variety of amino acids in proteins, but the sulfur-containing amino acids methionine and cysteine are most susceptible to oxidative modification. An earlier study showed that application of chloramine-T, an oxidizing agent capable of modifying methionine to methionine sulfoxide, potently inhibits hERG1a channels in mammalian cells (Su et al. 2007). The presence of the enzyme methionine sulfoxide reductase in the cytoplasm attenuated this effect and this finding was interpreted to indicate that methionine residues in the hERG1a protein, once oxidized, could lead to channel closure (Su et al. 2007). The contribution of cysteine modification within hERG1a remains unknown.
Even mild elevation of intracellular ROS levels, as occurring under hyperglycaemic situations, can adversely affect the heart rhythm and will most probably be associated with a modification of cysteine residues. In this study we show how hyperglycaemia-induced ROS stress and intracellular cysteine-modifying agents exquisitely regulate hERG1a channels. The exceptionally high sensitivity of hERG1a to intracellular ROS is mainly conferred by cysteine residues in the C-terminal domain, and their modification chiefly depends on the conformational state of the channel. As a consequence, the splice variant hERG1b, which shows faster deactivation than hERG1a, is much less sensitive to cysteine modification. Physiologically, the ratio of expressing hERG1a/hERG1b variants in particular cardiac cell types therefore not only determines the speed of deactivation of the heteromeric channels, but also defines the ROS sensitivity of the local IKr.
Methods
Channel constructs
In this study we used the human variant of KCNH2 (hERG1a). A splice variant (hERG1b) with an alternative N terminus of only 29 residues (Lees-Miller et al. 1997) was isolated from cDNA based on SH-SY5Y neuroblastoma cells. Site-specific mutagenesis of hERG1 was performed to replace methionine with leucine and cysteine with serine at positions indicated in the text. In addition, we used constructs in which part (hERG1aΔ2–15) or the entire N terminus (hERG1aΔ2–373, Schönherr & Heinemann, 1996) or part of the C terminus (hERG1aΔ866–1159) was deleted. Deletion constructs and single-site mutations were combined as indicated in the text. Single-site mutations were introduced by QuikChange mutagenesis (Agilent Technologies, San Diego, CA, USA) and all constructs were verified by DNA sequencing.
Cell culture
HEK293 (CAMR, Porton Down, Salisbury, UK) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) mixed 1:1 with Ham's F12 medium and supplemented with 10% fetal calf serum in a 5% CO2 incubator. Cells were trypsinized, diluted with culture medium and grown in 35 mm dishes. Electrophysiological experiments were performed 1–5 days after plating. HEK293 cells were transfected with the respective plasmids using the Rotifect (Roth, Karlsruhe, Germany) transfection reagent following the instructions of the supplier. A plasmid coding for CD8 was cotransfected for visualization of transfected cells by means of anti-CD8-coated microbeads (Dynabeads, Deutsche Dynal GmbH, Hamburg, Germany).
Electrophysiological measurements
Whole-cell voltage-clamp experiments were performed as described previously (Gessner & Heinemann, 2003). Briefly, patch pipettes with resistances of 0.7–1.2 MΩ were used. The series resistance was compensated by more than 70% in order to minimize voltage errors. A patch-clamp amplifier EPC9 or EPC10 was operated by PatchMaster software (both HEKA Elektronik, Lambrecht, Germany). Holding potential was −60 mV. Leak and capacitive currents were corrected with a p/n method with a leak holding voltage of −100 mV. Currents were low-pass filtered at 2 kHz and sampled at a rate of 10 kHz. All experiments were performed at constant temperature 19–21°C.
The patch pipettes contained (mm): 130 KCl, 2.56 MgCl2, 10 EGTA, 10 Hepes (pH 7.4 with KOH). The bath solution contained (mm): 110 NaCl, 40 KCl, 2 CaCl2, 2 MgCl2, 10 Hepes (pH 7.4 with NaOH). Deviations from this protocol are indicated in the text.
Oxidizing agents
Chloramine-T (ChT), hydrogen peroxide (H2O2) and tert-butyl hydrogen peroxide (tBHP) were diluted in the respective bath solution immediately before extracellular application. The cysteine-specific modifiers DTNB (5,5′-dithiobis-(2-nitrobenzoic acid)), MTSEA ((2-aminoethyl) methanethiosulfonate) and MTSES ((2-sulfonatoethyl) methanethiosulfonate) (MTS reagents from Toronto Research Chemicals, Toronto, Canada) were applied intracellularly by adding them to the pipette solution at 1 mm concentration.
Data analysis
Data were analysed with FitMaster (HEKA Elektronik) and IgorPro (WaveMetrics, Lake Oswego, OR, USA). Averaged data are presented as means ±s.e.m (n, number of independent measurements) unless specified otherwise. Averaged data were compared with a two-sided Student's t test assuming unequal variances. The resulting P values are specified.
Structural modelling
The hERG C-terminal structure was modelled with SWISS model (Guex & Peitsch, 1997) using the structure of mouse HCN2J (1Q3E) as the template.
Results
hERG channel inhibition by extracellularly applied H2O2
Oxidative stress, such as that induced by an elevated level of glucose, leads to cardiac abnormalities by exerting a negative impact on hERG channel function (Zhang et al. 2003; Wang et al. 2004). To shed light on the underlying molecular mechanisms, we measured the impact of extracellular H2O2 (1 mm), a physiological oxidant, on hERG1 channels expressed in HEK293 cells. Channels were activated by depolarization to 50 mV, followed by hyperpolarization to −40 mV and −100 mV for an assessment of the speed of channel deactivation during the repolarization phase of an action potential and at rest, respectively (Fig. 1A). Application of H2O2 produced a biphasic effect on the size of tail current at −40 mV: a transient and small increase (∼10%) occurring within 60 s, followed by a slower decline (Fig. 1C left, open circles). At −100 mV, the tail current did not show any increase but only decreased slowly by about 20% after 250 s (Fig. 1C right, open squares). In addition, consistent with an earlier report (Bérubéet al. 2001), treatment with H2O2 for 200 s accelerated the tail current kinetics at −100 mV; the time constant decreased from 39.3 ± 1.4 ms to 33.9 ± 1.9 ms (n = 8, P = 0.035).
Figure 1. Effect of extracellularly applied H2O2 and tBHP on hERG1a currents.
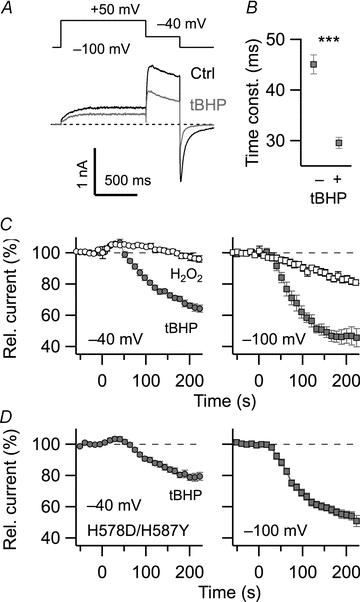
A, pulse protocol used to activate and deactivate hERG1 channels expressed in HEK293 cells. Superposition of two current sweeps, control (black) and 200 s after application of 1 mm tBHP (grey). B, time constant of channel deactivation at −100 mV before (−) and 200 s after application of tBHP: 45.1 ± 1.9 ms vs. 29.6 ± 1.1 ms (n = 8, P < 0.001). Corresponding values for H2O2: 39.3 ± 1.4 ms vs. 33.9 ± 1.9 ms (n = 8, P = 0.035). C, time course of normalized current at −40 mV (left) and −100 mV (right) with application of 1 mm H2O2 (open symbols) or tBHP (grey) at time zero; n = 8. D, similar experiment with 1 mm tBHP as shown in C for channel mutant H578D/H587Y. The bath solution contained (in mm): 145 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 Hepes (pH 7.4).
H2O2 only slowly crosses the cell membrane while the oxidant tBHP is more membrane permeable. Application of tBHP (1 mm) produced a transient increase in tail current at −40 mV similar to that observed with H2O2 but subsequently decreased the tail current much more rapidly (Fig. 1C left, filled circles). The current decrease by tBHP was more pronounced at −100 mV (about 50% after 2 min; Fig. 1C right, filled square). The kinetics of the current at −100 mV after treatment with tBHP was also significantly faster than that before treatment (45.1 ± 1.9 ms vs. 29.6 ± 1.1 ms; Fig. 1B, n = 8, P < 0.001).
The finding that extracellular application of H2O2 and tBHP, which differ in membrane permeability, produced a similar transient increase in current at −40 mV implicates an oxidative modification at an extracellularly accessible site. Pannaccione et al. (2002) in fact suggested that two histidine residues in the external mouth of the pore (H578 and H587) are crucial for the upregulation of hERG1 activity by extracellular iron/ascorbic acid, which is expected to induce ROS stress. When the two histidine residues were mutated (H578D/H587Y), the transient increase in current at −40 mV immediately after application of 1 mm tBHP greatly diminished while the slower current decrease persisted almost to the same extent as in the wild type (Fig. 1D). Thus, the slow but marked decrease in hERG1-mediated current prominently observed with the membrane-permeant tBHP is most probably caused by an oxidative modification, taking place at the cytosolic side of the channel protein. The two effects of oxidative treatment with H2O2 or tBHP, a decrease in peak tail current and acceleration of deactivation kinetics, should lead to a strong reduction of the total K+ current component (IKr), prominently contributing to the repolarization of cardiac cells.
hERG channel inhibition by intracellular cysteine-modifying agents
The results above suggest that the profound inhibition in the hERG1 activity by H2O2 or tBHP involves oxidative modification of intracellular targets. As cysteine is easily oxidized, we utilized the specific cysteine-modifying reagents DTNB, MTSEA and MTSES to identify the potential target residues. The cysteine modifiers were included in the whole-cell internal solution at 1 mm and the tail current magnitude at −120 mV following a 500 ms depolarization to 50 mV was monitored. This measurement protocol was necessary because inactivating hERG currents run down very rapidly in excised patch configurations. Inclusion of the modifiers in the internal solution caused a substantial and progressive decrease of peak tail current compared to the control situation (Fig. 2A and B). For example, the most effective modifier MTSES decreased the peak tail current size down to ∼25% (Fig. 2A and B). In addition, the kinetics of channel deactivation became noticeably faster, as illustrated for MTSES (Fig. 2C), resulting in a marked decrease in K+ current carried during the tail current period (Fig. 2A). The qualitative similarity of functional impacts of these Cys-specific modifiers and H2O2/tBHP suggests that the peroxides also work by cysteine modification. In addition, because different cysteine modifiers effected similar changes, it is suggested that the lack of free thiol groups rather than the specific adduct is important for the functional modification. Extracellular application of 1 mm MTSES was without effect on hERG1a (data not shown).
Figure 2. Effect of intracellular cysteine-modifying agents on hERG1a currents.
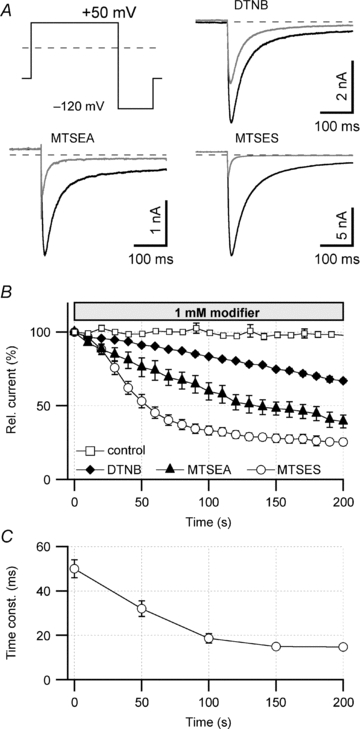
A, pulse protocol used to activate and deactivate hERG1a channels expressed in HEK293 cells and superposition of tail current traces at −120 mV before (black) and 200 s after (grey) cell dialysis with 1 mm of the indicated cysteine-modifying agents. B, time course of tail current reduction upon cell dialysis with control saline, 1 mm DTNB, MTSEA or MTSES. Relative remaining currents after 200 s were: control, I200 = 98 ± 2% (n = 6); DTNB, I200 = 67 ± 1% (n = 7); MTSEA, I200 = 39 ± 4% (n = 6); MTSES, I200 = 25 ± 2% (n = 6). Straight lines connect the data points. C, time constant of deactivation at −120 mV for hERG1 currents during MTSES (1 mm) dialysis (n = 6).
To rule out a general effect of cysteine-modifying agents to all cardiac K+ channels, we demonstrated that intracellular MTSES does not affect the voltage-gated K+ channel hKv1.5 (Supplementary Fig. S1). Since MTSES specifically modifies cysteines, the number of Cys residues in the respective channel proteins may reflect the differences of the channel types. hERG1a proteins harbour 24 cysteine residues (Fig. 3A) and hKV1.5 only 10, providing some first indication of the sensitivity of ERG channels.
Figure 3. Models of hERG1 subunits.
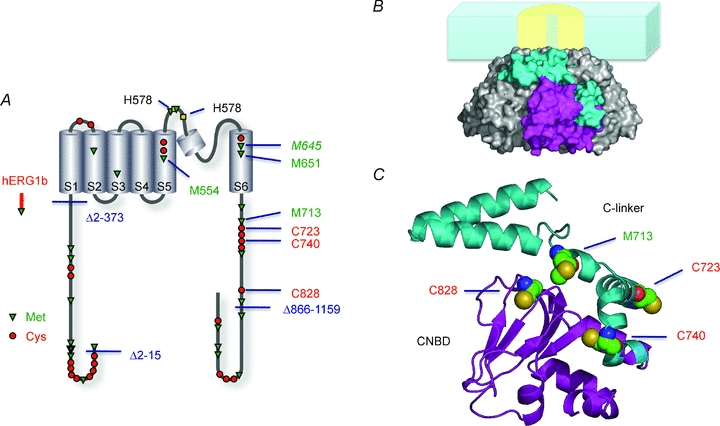
A, schematic presentation of hERG1a and hERG1b topological models indicating the deletions introduced to hERG1a as well as cysteine and methionine residues. Cys, Met and His residues of particular importance are highlighted. B, surface representation of four C-terminal domains forming a tetramer with one subunit coloured. The membrane and the channel's core domain is only indicated as cartoon. C, model of the C-terminal domain of a single subunit. The linker between the last transmembrane segment (S6 gate) and the area traditionally called the binding domain for cyclic nucleotides (CNBD) is referred to as ‘C-linker’. The hERG C-terminal structure was modelled using the structure of mouse HCN2J (1Q3E; Zagotta et al. 2003) as the template.
hERG1b and N-terminal deletions
To identify the critical cysteine residue(s) required for the action of peroxides and the cysteine modifiers, we individually mutated every N-terminal cysteine residue to serine and assayed the mutant's sensitivity towards MTSES. None of the mutants showed diminished MTSES sensitivity (not shown). Since a mutant without any cysteine in the N terminus was not electrophysiologically functional, we next utilized a naturally occurring splice variant that differs in the N terminus. The splice variant hERG1b is expressed in cardiac tissue (Jones et al. 2004; Sale et al. 2008) and, compared with the more common variant hERG1a, has a truncated N terminus with a unique sequence of only 29 residues (Fig. 3A). We found that homomeric hERG1b channels expressed in HEK293 cells, showing much faster deactivation kinetics, were dramatically less sensitive to MTSES; ≥85% of the tail current remained in the presence of MTSES and no change in the kinetics was observed (Fig. 4A and C). To determine if the diminished MTSES sensitivity of hERG1b is conferred by the short size of the N-terminus, we utilized a hERG1a mutant, in which almost the entire N terminus was deleted (hERG1aΔ2–373). This mutant showed noticeably faster deactivation compared with that of hERG1a and was markedly less sensitive to MTSES (Fig. 4A). Since 10 cysteine residues were deleted in fast deactivating hERG1aΔ2–373, the diminished MTSES sensitivity of the deletion mutant could be attributed to the lack of those residues. Alternatively, the rapid deactivation shown in hERG1aΔ2–373 as well as in hERG1b (Fig. 4C) could underlie the decreased MTSES sensitivity. We therefore generated mutant hERG1aΔ2–15; this mutant does not lack any cysteine residue but it exhibits rapid channel deactivation kinetics indistinguishable from that of hERG1b or hERG1aΔ2–373 (Fig. 4A and C). The sensitivity of hERG1aΔ2–15 to MTSES was similar to that of hERG1b and hERG1aΔ2–373; ≥65% of the tail current remained with MTSES without effects on deactivation kinetics (Fig. 4A). This diminished sensitivity of rapidly deactivating hERG1aΔ2–15 with a full complement of cysteine residues to MTSES therefore argues that (1) channel gating may be a key factor in the cysteine-directed modification of hERG channels and (2) that 10 N-terminal cysteine residues are of minor importance for the channel modulation by ROS.
Figure 4. hERG1b and N-terminal deletions.
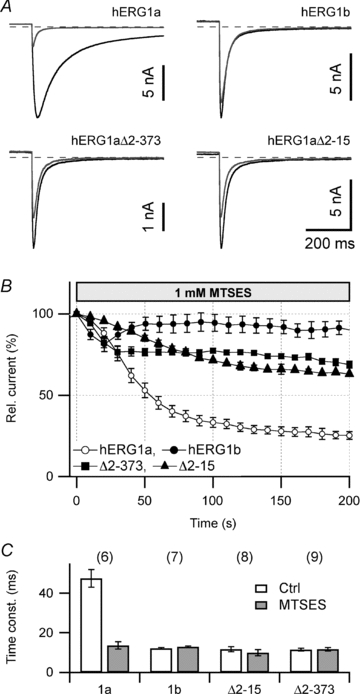
A, superposition of tail current traces at −120 mV before (black) and after 200 s cell dialysis with 1 mm MTSES (grey) for the indicated channel constructs expressed in HEK293 cells. B, time course of tail current reduction upon cell dialysis with 1 mm MTSES for the indicated channel types. Relative remaining current after 200 s: hERG1a, 25 ± 2% (n = 6); hERG1b, 89 ± 4% (n = 6); hERG1aΔ2–373, 68 ± 2% (n = 5); hERG1aΔ2–15, 63 ± 1% (n = 6). C, deactivation time constants at −120 mV of the indicated channel constructs before (open) and 200 s after cell dialysis with 1 mm MTSES (grey bars). n indicated in parentheses.
The apparent correlation of speed of channel deactivation with the amount of MTSES effect on the channels suggests that channel modification by MTSES may depend on the channel conformation. We thus tested if the effect of MTSES on hERG1a is affected by the holding potential by comparing the degree of current reduction after 100 s at −80 and −20 mV. A lack of significant difference (not shown) suggests that MTSES modification is not strongly use-dependent.
The short N-terminal variant hERG1b is abundantly expressed in SH-SY5Y neuroblastoma cells (Crociani et al. 2003), rendering these cells an excellent system to measure native hERG currents with a major contribution from hERG1b. MTSES (1 mm, intracellular) had only a small effect on the large outward K+ currents mediated by channels other than hERG in these cells while the tail currents – which mainly are mediated by hERG1b channels – are strongly depressed. Comparison of the time course of hERG current reduction obtained for endogenous channels in SH-SY5Y cells and for recombinant hERG1a in HEK293 cells, however, clearly showed that hERG in neuroblastoma cells is less sensitive to MTSES (Supplementary Fig. S2). Moreover, deactivation kinetics of hERG currents in SH-SY5Y cells was intermediate between recombinant hERG1a and hERG1b and MTSES accelerated deactivation at −120 mV to the same value as in hERG1a (Supplementary Fig. S2C), arguing that SH-SY5Y cells express a mixture of hERG1a and hERG1b isoforms.
Involvement of C-terminal cysteine residues
Of the 24 cysteine residues present in hERG1a, the 10 N-terminal cysteine residues are unlikely to play essential roles in the MTSES sensitivity as indicated by the results obtained from the N-terminal deletion mutants. The membrane-spanning S1–S6 domain possesses five cysteine residues but they are unlikely to be accessible from the cytosolic side. The C terminus contains the remaining nine cysteine residues (Fig. 3A). Complete deletion of the cytoplasmic C-terminal domain to remove all nine cysteine residues yielded an electrophysiologically non-functional protein. However, a smaller deletion encompassing the distal half of the C terminus (ΔC: hERG1aΔ866–1159) retaining five cysteine residues produced a functional channel, whose sensitivity towards MTSES was indistinguishable from that of the wild type (Fig. 5A). To assess the functional importance of the five cysteine residues remaining in the C terminus, each was mutated to serine and assayed for the fractional current remaining after MTSES treatment. Mutant C723S showed the least MTSES sensitivity with ∼60% of the current remaining, followed by C740S and C828S (Fig. 5A). Mutations C729S and C750S were without effect (Fig. 5A). The triple mutation C723/740/828S did not reduce the sensitivity any further suggesting that C723 plays a major role.
Figure 5. Involvement of C-terminal cysteine residues.
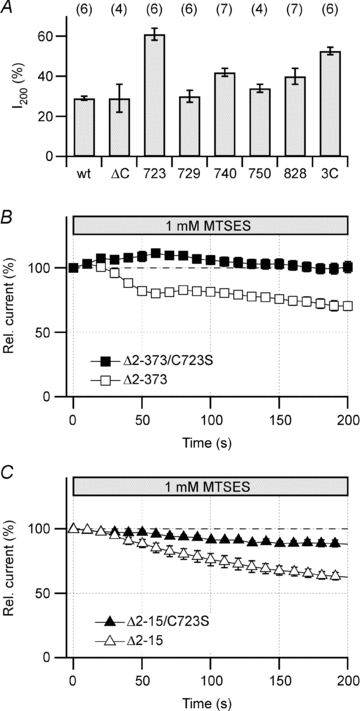
A, relative remaining hERG tail current after 200 s cell dialysis with 1 mm MTSES for HEK293 cells expressing hERG1a wild type and the indicated mutants. Numbers refer to positions of cysteine–serine replacements; ‘3C’ refers to C723/740/828S. B, time course of tail current reduction upon cell dialysis with 1 mm MTSES for hERG1aΔ2–373 (n = 5) and the combined mutant hERG1aΔ2–373/C723S (n = 6). C, as in B for hERG1aΔ2–15 (n = 5) and the combined mutant hERG1aΔ2–15/C723S (n = 6).
When mutation C723S was combined with the N-terminal deletion Δ2–373 as found in hERG1b, the MTSES sensitivity was completely absent (Fig. 5B). Even with a smaller deletion in the N terminus (hERG1aΔ2–15), C723S substantially diminished the sensitivity (Fig. 5C), arguing that C723 must have an important regulatory function for the channel complex.
Sensitivity of hERG channels to glucose-induced ROS stress
The results presented above show that C723 in the C terminus is the major determinant of the exquisite sensitivity of the channel to thiol-specific protein modification. To infer if this modification is relevant under mild physiological oxidative stress imposed by hyperglycaemia, we measured hERG1 current amplitudes in transiently transfected HEK293 cells under low and high glucose conditions. Cells were cultured in a medium containing 5 mm glucose and then prior to electrophysiological recording cells were kept for 3 h in a medium with 5 mm glucose (low glucose) or 25 mm glucose (high glucose). The median of the current density for the wild type decreased to 60% from −257 pA pF−1 (n = 29) to −155 pA pF−1 (n = 28) when exposed to hyperglycaemic stress. The mean current density (Fig. 6) also shows a significant reduction under high glucose conditions. A similar experiment performed with mutant C723S (Fig. 6) showed no significant change in current density (median of −70.8 pA pF−1 (n = 38) and −73.7 pA pF−1 (n = 35), respectively), affirming that C723 mediates the current decrease and that the MTSES sensitivity reported above is of physiological relevance.
Figure 6. Glucose stress and hERG1 channels.
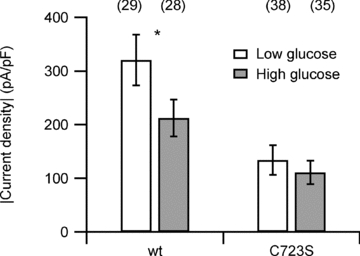
Current densities measured for hERG1a and mutant C723S under low and high glucose conditions. Values are means ±s.e.m. The asterisk denotes a statistically significant difference between low and high glucose conditions for the wild type with P = 0.035 according to a Student's t test. For mutant C723S the means are not statistically different (P = 0.26). The number of cells indicated in parentheses.
hERG channel inhibition by the oxidant chloramine-T
An earlier study (Su et al. 2007) showed that hERG channels at 37°C undergo a loss of function when exposed to the mild oxidant chloramine-T (ChT) and that the enzyme methionine sulfoxide reductase A, which repairs oxidized methionine to methionine, partially antagonized the inhibitory effect, implicating that methionine oxidation within hERG1 may impair its function. To assess relative contributions of methionine and cysteine oxidation, we thus tested the effect of 300 μm ChT, capable of oxidizing both cysteine and methionine, on hERG1 channels in HEK293 cells under the experimental conditions used thus far (20°C). Application of ChT led to a progressive loss of function described by a single exponential with a time constant of 80 ± 4.8 s and a remaining fraction of active channels of 24.8 ± 1.4% (Fig. 7).
Figure 7. Methionine oxidation affects hERG channel function.
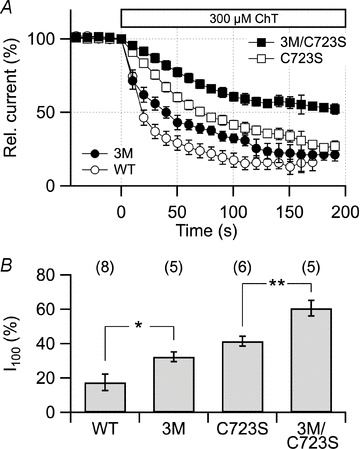
A, time course of tail current reduction induced by extracellular application of 300 μm ChT to HEK293 cells expressing hERG1a and the indicated mutants thereof. B, relative remaining currents 100 s after ChT application (I100) were: hERG1a, 17 ± 5% (n = 8); 3M, 32 ± 3% (n = 5); C723S, 42 ± 3% (n = 6); 3M/C723S, 61 ± 5% (n = 5). *P < 0.05; **P < 0.01. Significances: WT vs. 3M: P = 0.024; C723S vs. 3M/C723S: P = 0.0097. The bath solution contained (in mm): 140 NaCl, 10 KCl, 2 CaCl2, 2 MgCl2, 10 Hepes (pH 7.4 with NaOH); holding potential for the wild type and C723S was −100 mV, for 3M and 3M/C723S it was −120 mV.
This effect is in part mediated by C723 as mutant C723S was significantly (P < 0.01) less sensitive than the wild type (Fig. 7A, open symbols). However, a noticeable decrease in current by application of ChT persisted in C723S, indicating that ChT may modify other targets, such as methionine residues as previously suggested (Su et al. 2007). The 24 methionine residues of hERG1 are distributed among the N terminus (8), the core domain (7) and the C terminus (9) (Fig. 3A). A systematic mutagenesis approach involving deletion mutants and methionine-to-leucine exchanges (Supplementary Fig. S3) showed that three methionine residues located in the C terminus contribute to the inhibitory effect of ChT: M554, M651 and M713. The triple mutation M554/651/713L, named 3M hereafter, significantly slowed the kinetics of the inhibitory effect of ChT without affecting the steady-state level of inhibition. Since ChT is capable of oxidizing methionine as well as cysteine, we combined 3M with C723S and assessed the mutant's ChT sensitivity.
The triple mutant 3M reduces the effect of ChT with respect to the wild type, but the effect is stronger in the background of mutation C723S (Fig. 7). This becomes particularly apparent after application of ChT for an extended duration and clearly shows that methionine residues are also targets of oxidative modification of hERG1 channels, but the resulting effect is mostly superimposed by a stronger effect mediated by modification of C723. However, following long-term exposure to oxidative stress, methionine oxidation may indeed have an impact on IKr.
Discussion
ROS-mediated cardiac arrhythmias
Cardiac complications caused by severe oxidative insults such as ischaemia/reperfusion injury are well documented; however, relatively mild, but prolonged elevations from a normal ROS level may also have pathophysiological consequences. This may be particularly important for diabetic patients who experience prolonged and/or repeated episodes of hyper- or hypoglycaemia. Both abnormalities of glucose metabolism can lead to excess intracellular ROS (Zhang et al. 2003) and affect the physiological properties of the myocardium. One of such ROS-induced changes is APD prolongation, which may involve dysfunction of hERG1 channels responsible for the repolarizing IKr current. The hERG channel activity is inhibited by a wide spectrum of ROS-generating manipulations such as alterations of the glucose level (Zhang et al. 2003), application of TNF-α (Wang et al. 2004) or ceramide (Bai et al. 2007). The molecular mechanisms and the sites of action of ROS-induced hERG dysfunction, however, remained elusive. Our study reveals that a single cysteine residue, C723, in the cytoplasmic domain plays a prominent role in the oxidation sensitivity of the channel and that the N-terminus structure and three methionine residues in the C-terminus are also contributing factors.
In this study we have demonstrated that extracellular application of H2O2 induces two distinct effects on hERG currents: a transient increase in outward currents and, more importantly, a subsequent slower decrease accompanied by an acceleration of the deactivation kinetics. The former observation is consistent with the earlier report by Taglialatela et al. (1997) that ROS stress by iron/ascorbic acid increased outward currents in Xenopus oocytes heterologously expressing hERG channels by causing a small shift in the voltage dependence of inactivation. Pannaccione et al. (2002) suggest that two histidine residues in the extracellular loop between transmembrane segment S5 and the pore region take part in this process; when these residues are mutated to the amino acids found in the related hEAG1 channel (H578D, H587Y) the initial increase in current induced by H2O2 or tBHP is reduced in size. Thus, these histidine residues play a role in functional upregulation of hERG1 by extracellular oxidants but the molecular mechanism still needs to be explored. Our experiments (Fig. 1C and D) showed that the histidine mutants slightly diminished the current-inhibiting effect of tBHP indicating that the histidine residues may also contribute to the oxidation sensitivity of hERG channels.
The most prominent effect of peroxides, especially with the membrane-permeable tBHP, is to inhibit the overall hERG activity. The inhibitory action requires C723 in the C-terminus of the protein, and the same residue is subject to modification by physiological and mild ROS stress, such as that caused by an elevation of the glucose concentration in the cell culture medium. While wild type currents were significantly decreased by the high glucose treatment as previously reported by Zhang et al. (2003), channel mutant C723S remained less affected.
C723 clearly plays the most important role in the sensitivity of hERG to oxidative stress; however, other residues do contribute. For example, mutation of C740 and C828 also slightly diminish the sensitivity. Based on results using the methionine-preferring oxidant chloramine-T and intracellular supplementation of methionine sulfoxide reductase, Su et al. (2007) speculated that methionine oxidation could contribute to the sensitivity of hERG towards ROS. Our study also confirms the importance of methionine oxidation in hERG1 although modification of cysteines appears to be a more prominent event. However, while cysteine modification is readily reversible, long-term oxidative stress may cause methionine oxidation and, because of its poor reversibility when buried in a membrane protein, can give rise to long-term, cysteine-independent hERG1 channel dysfunction.
Thiol-modification of select cysteine residues in hERG1 channels thus is an effective mechanism by which excursions of the intracellular ROS level modulate channel activity. Greater ROS-mediated inhibition, however, may result in non-physiological responses, such as cardiac arrhythmia, because of inadequate APD prolongation.
hERG1 is primarily expressed in the heart while the variants hERG2 and hERG3 play roles in neuronal cells (Schwarz & Bauer, 2004). All of the residues critical for oxidation-dependent channel modulation, i.e. M713, C723, C740 and C828, are conserved in all three hERG variants and therefore the conclusions drawn here for cardiac hERG1 may also hold true for the neuronal hERG channels.
Relevance of hERG1b
One surprising result of the present study was that a small deletion in the N-terminal structure, preserving all of the cysteine or methionine residues, strongly diminished the channel's susceptibility towards ROS-induced functional inhibition. This feature correlates well with the increased speed of deactivation observed in that channel mutant. Noteworthy, this functional difference is also true for the naturally occurring splice variant hERG1b. It has a substantially truncated N-terminal structure, exhibits much faster deactivation kinetics than hERG1a and it is less sensitive towards thiol-directed modification. Thus, the overall ROS sensitivity of IKr, comprised of hERG1a and hERG1b in the human heart (Jones et al. 2004), will depend on the ratio of both hERG variants in cardiac myocytes. Using splice variant-specific quantitative RT-PCR, Larsen et al. (2008) detected mRNA of hERG1b, both in ventricle and atrium of the human heart. The ratio of hERG1b/hERG1a mRNA in ventricle and atrium ranged between 10 and 20%. However, the authors noted a large variation of this ratio among individuals, ranging from 3 to 30%. Therefore, a significant variability in ROS susceptibility of IKr in individual subjects must be expected.
Structural and mechanistic considerations
The C terminus of the hERG channel is similar in amino acid sequence to that of the cyclic nucleotide-modulated channel (HCN). A structural study shows that the C terminus of HCN may form two distinct structural units, the C-linker region and the downstream cyclic nucleotide binding domain (CNBD), and that the four subunits together may comprise a large concentric cytoplasmic domain (Zagotta et al. 2003). This structure is postulated to form a so-called gating ring (Jiang et al. 2002), whose conformational change on ligand binding promotes opening of the ion conduction gate within the transmembrane segment of the channel (Johnson & Zagotta, 2005). The sequence similarity of the HCN C terminus to that of hERG suggests that the cytoplasmic domain of hERG also forms a gating ring-like structure made of C-linker and CNBD-like segments (Fig. 3B and C). While recent experiments suggest that cyclic nucleotides fail to bind to the isolated CNBD domain of hERG (Brelidze et al. 2009), our results unequivocally show that the putative C-linker segment of hERG is of critical importance for channel function. In the structural homology model, all four oxidation-sensitive residues identified in the C terminus, namely C723, C740, C828 and M713, are located at the interface between the C-linker segment and the so-called CNBD domain (Fig. 3C). Thus, a change in the spatial orientation of the two structural units by modification of the side chains of these cysteine and methionine residues could underlie the electrophysiological changes documented in this study.
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft (HE2993/8-1, S.H.H.), IZKF (Interdisziplinäres Zentrum für Klinische Forschung) (S.H.H.), Landesprogramm ‘ProExzellenz’ Freistaat Thüringen, PE-114 (S.H.H., T.H.) and NIH (T.H.).
Glossary
Abbreviations
- APD
action potential duration
- ChT
chloramine-T
- CNBD
cyclic nucleotide binding domain
- DTNB
5,5′-dithiobis-(2-nitrobenzoic acid)
- HCN
cyclic nucleotide-modulated channel
- I200
current amplitude remaining 200 s after substance application
- IKr
rapidly activating outwardly rectifying potassium current in the heart
- LQTS
long-QT syndrome
- MTSEA
(2-aminoethyl) methanethiosulfonate
- MTSES
(2-sulfonatoethyl) methanethiosulfonate
- ROS
reactive oxygen species
- tBHP
tert-butyl hydrogen peroxide
- WT
wild type
Author contributions
Conception and design of the experiments (S.H.H., R.S., T.H), mutagenesis and data collection (K.K., N.S., G.G., R.S.), data analysis (K.K., S.H.H.), molecular modelling (T.H.); all authors contributed to drafting and revising the article and approved the final version. All experiments were performed at the Centre for Molecular Biomedicine, Friedrich Schiller University of Jena.
Supplemental material
Suppl. Figure 1
Suppl. Figure 2
Suppl. Figure 3
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Bai Y, Wang J, Shan H, Lu Y, Zhang Y, Luo X, Yang B, Wang Z. Sphingolipid metabolite ceramide causes metabolic perturbation contributing to HERG K+ channel dysfunction. Cell Physiol Biochem. 2007;20:429–440. doi: 10.1159/000107527. [DOI] [PubMed] [Google Scholar]
- Bérubé J, Caouette D, Daleau P. Hydrogen peroxide modifies the kinetics of HERG channel expressed in a mammalian cell line. J Pharm Exp Therap. 2001;297:96–102. [PubMed] [Google Scholar]
- Brelidze TI, Carlson AE, Zagotta WN. Absence of direct cyclic nucleotide modulation of mEAG1 and hERG1 channels revealed with fluorescence and electrophysiological methods. J Biol Chem. 2009;284:27989–27997. doi: 10.1074/jbc.M109.016337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casis O, Echevarria E. Diabetic cardiomyopathy: electromechanical cellular alterations. Curr Vasc Pharm. 2004;2:237–248. doi: 10.2174/1570161043385655. [DOI] [PubMed] [Google Scholar]
- Chen J, Sroubek J, Krishnan Y, Li Y, Bian J, McDonald TV. PKA phosphorylation of HERG protein regulates the rate of channel synthesis. Am J Physiol Heart Circ Physiol. 2009;296:H1244–H1254. doi: 10.1152/ajpheart.01252.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crociani O, Guasti L, Balzi M, Becchett A, Wanke E, Olivotto M, Wymore RS, Arcangeli A. Cell cycle-dependent expression of HERG1 and HERG1B isoforms in tumor cells. J Biol Chem. 2003;278:2947–2955. doi: 10.1074/jbc.M210789200. [DOI] [PubMed] [Google Scholar]
- Fermini B, Fossa AA. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat Rev Drug Discov. 2003;2:439–447. doi: 10.1038/nrd1108. [DOI] [PubMed] [Google Scholar]
- Gessner G, Heinemann SH. Inhibition of hEAG1 and hERG1 potassium channels by clofilium and its tertiary analogue LY97241. Br J Pharmacol. 2003;138:161–171. doi: 10.1038/sj.bjp.0705025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- Johnson JP, Jr, Zagotta WN. The carboxyl-terminal region of cyclic nucleotide-modulated channels is a gating ring, not a permeation path. Proc Natl Acad Sci U S A. 2005;102:2742–2747. doi: 10.1073/pnas.0408323102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EMC, Roti Roti EC, Wang J, Delfosse SA, Robertson GA. Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. J Biol Chem. 2004;279:44690–44694. doi: 10.1074/jbc.M408344200. [DOI] [PubMed] [Google Scholar]
- Kuzuya T, Hoshida S, Kim Y, Nishida M, Fuji H, Kitabatake A, Tada M, Kamada T. Detection of oxygen-derived free radical generation in the canine post-ischemic heart during late phase of reperfusion. Circ Res. 1990;66:1160–1165. doi: 10.1161/01.res.66.4.1160. [DOI] [PubMed] [Google Scholar]
- Larsen AP, Olesen S-P, Grunnet M, Jespersen T. Characterization of hERG1a and hERG1b potassium channels – a possible role for hERG1b in the IKr current. Pflüg Arch. 2008;456:1137–1148. doi: 10.1007/s00424-008-0476-7. [DOI] [PubMed] [Google Scholar]
- Lees-Miller JP, Kondo C, Wang L, Duff HJ. Electrophysiological characterization of an alternatively processed ERG K+ channel in mouse and human hearts. Circ Res. 1997;81:719–726. doi: 10.1161/01.res.81.5.719. [DOI] [PubMed] [Google Scholar]
- Pannaccione A, Castaldo P, Ficker E, Annunziato L, Taglialatela M. Histidines 578 and 587 in the S5-S6 linker of the human ether-a-gogo related gene-1 K+ channels confer sensitivity to reactive oxygen species. J Biol Chem. 2002;277:8912–8919. doi: 10.1074/jbc.M111353200. [DOI] [PubMed] [Google Scholar]
- Robillon JF, Sadoul JL, Benmerabet S, Joly-Lemoine L, Fredenrich A, Canivet B. Assessment of cardiac arrhythmic risk in diabetic patients using QT dispersion abnormalities. Diab Metab. 1999;25:419–423. [PubMed] [Google Scholar]
- Sale H, Wang J, O’Hara TJ, Tester DJ, Phartiyal P, He J-Q, Rudy Y, Ackermann MJ, Robertson GA. Physiological properties of hERG 1a/1b heteromeric currents and a hERG 1b-specific mutation associated with long-QT syndrome. Circ Res. 2008;103:e81–95. doi: 10.1161/CIRCRESAHA.108.185249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jian C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Schönherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol. 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JR, Bauer CK. Functions of erg K+ channels in excitable cells. J Cell Mol Med. 2004;8:22–30. doi: 10.1111/j.1582-4934.2004.tb00256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Limberis J, Martin RL, Xu R, Kolbe K, Heinemann SH, Hoshi T, Cox BF, Gintant G. Functional consequences of methionine oxidation of hERG potassium channels. Biochem Pharmacol. 2007;74:702–711. doi: 10.1016/j.bcp.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela M, Castaldo P, Iossa S, Pannaccione A, Fresi A, Ficker E, Annunziato L. Regulation of the human ether-a-gogo related gene (HERG) K+ channels by reactive oxygen species. Proc Natl Acad Sci U S A. 1997;94:11698–11703. doi: 10.1073/pnas.94.21.11698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Karle CA, Kiehn J. The cardiac hERG/IKr potassium channel as pharmacological target: structure, function, regulation, and clinical applications. Curr Pharm Des. 2006;12:2271–2283. doi: 10.2174/138161206777585102. [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- Van Wagoner DR. Oxidative stress and inflammation in atrial fibrillation: role in pathogenesis and potential as a therapeutic target. J Cardiovasc Pharm. 2008;52:306–313. doi: 10.1097/FJC.0b013e31817f9398. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang H, Zhang Y, Gao H, Nattel S, Wang Z. Impairment of HERG K+ channel function by tumor necrosis factor-α. J Biol Chem. 2004;279:13289–13292. doi: 10.1074/jbc.C400025200. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Han H, Wang J, Wang H, Yang B, Wang Z. Impairment of human ether-à-go-go-related gene (HERG) K+ channel function by hypoglycemia and hyperglycemia. J Biol Chem. 2003;278:10417–10426. doi: 10.1074/jbc.M211044200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiao J, Wang H, Luo X, Wang J, Villeneuve LR, Zhang H, Bai Y, Yang B, Wang Z. Restoring depressed HERG K+ channel function as a mechanism for insulin treatment of abnormal QT prolongation and associated arrhythmias in diabetic rabbits. Am J Physiol Heart Circ Physiol. 2006;291:H1446–H1455. doi: 10.1152/ajpheart.01356.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.