

Abstract
Corticostriatal projections constitute the main input to the basal ganglia, an ensemble of interconnected subcortical nuclei involved in procedural learning. Thus, long-term plasticity at corticostriatal synapses would provide a basic mechanism for the function of basal ganglia in learning and memory. We had previously reported the existence of a corticostriatal anti-Hebbian spike timing-dependent plasticity (STDP) at synapses onto striatal output neurons, the medium-sized spiny neurons. Here, we show that the blockade of GABAergic transmission reversed the time dependence of corticostriatal STDP. We explored the receptors and signalling mechanisms involved in the corticostriatal STDP. Although classical models for STDP propose NMDA receptors as the unique coincidence detector, the involvement of multiple coincidence detectors has also been demonstrated. Here, we show that corticostriatal STDP depends on distinct coincidence detectors. Specifically, long-term potentiation is dependent on NMDA receptor activation, while long-term depression requires distinct coincidence detectors: the phospholipase Cβ (PLCβ) and the inositol-trisphosphate receptor (IP3R)-gated calcium stores. Furthermore, we found that PLCβ activation is controlled by group-I metabotropic glutamate receptors, type-1 muscarinic receptors and voltage-sensitive calcium channel activities. Activation of PLCβ and IP3Rs leads to robust retrograde endocannabinoid signalling mediated by 2-arachidonoyl-glycerol and cannabinoid CB1 receptors. Interestingly, the same coincidence detectors govern the corticostriatal anti-Hebbian STDP and the Hebbian STDP reported at cortical synapses. Therefore, LTP and LTD induced by STDP at corticostriatal synapses are mediated by independent signalling mechanisms, each one being controlled by distinct coincidence detectors.
Introduction
The basal ganglia are involved in learning of contextual cognitive and motor sequences allowing adaptive control of behaviour (Graybiel, 2005; Yin & Knowlton, 2006). The striatum, the major input nucleus of the basal ganglia, processes information from the entire cerebral cortex. The striatal output neurons, the medium-sized spiny neurons (MSNs), act as detectors of distributed patterns of cortical activity (Calabresi et al. 1987; Nisenbaum et al. 1994). In turn, MSNs relay the integrated cortical information towards the basal ganglia output nuclei. As long-term synaptic efficacy changes are believed to underlie learning and memory (Martin et al. 2000; Martin & Morris, 2002; Lynch, 2004), the corticostriatal long-term plasticity provides a fundamental mechanism for the function of the basal ganglia in procedural learning (Di Filippo et al. 2009; Wickens, 2009; Yin et al. 2009).
We have investigated the activity-dependent long-term plasticity at the corticostriatal synapses (Fino et al. 2005, 2008, 2009a). The temporal relationship of activity in pre- and postsynaptic neurons is determinant for the induction of activity-dependent long-term synaptic plasticity, a process named spike timing-dependent plasticity (STDP) (for reviews see: Sjöström et al. 2008; Caporale & Dan, 2008). We investigated the STDP at the level of MSNs on rat brain corticostriatal slices and reported a bidirectional STDP whose spike-timing dependence displayed an anti-Hebbian learning rule (Fino et al. 2005) compared to the classical STDP described in different mammalian brain structures (Markram et al. 1997; Magee & Johnston, 1997; Bi & Poo, 1998; Debanne et al. 1998; but see Bell et al. (1997) and Tzounopoulos et al. (2004, 2007) for anti-Hebbian STDP, respectively, in the cerebellum-like structure of the mormyrid electric fish and in the dorsal cochlear nucleus in rodents). Indeed, when a MSN action potential (AP) was evoked slightly before cortical activation (post–pre pairing), a robust LTP was observed. Conversely, when cortical activity preceded a MSN back-propagating AP (bAP), a LTD was induced. We explored here the effects of the blockade of the GABAergic transmission on the spike-timing dependence at corticostriatal synapses. In order to fully characterize the corticostriatal STDP, we addressed the question of the molecular components engaged in these long-term synaptic efficacy changes. We determined which intracellular pathways and cellular coincidence detectors were underlying MSN STDP. Classical models for STDP propose NMDA receptors (NMDARs) as the unique coincidence detector (Nishiyama et al. 2000; Shouval et al. 2002). However, the involvement of multiple coincidence detectors to induce STDP has been reported (Sjöström et al. 2003; Bender et al. 2006; Nevian & Sakmann, 2006). Here we sought to determine the molecular components involved in corticostriatal STDP and whether they rely on one or multiple coincidence detectors.
Methods
Ethical approval
Animals, OFA rats (Charles River, L’Arbresle, France) (postnatal days 15–21), were killed by decapitation and brains were immediately removed. All experiments were performed in accordance with local animal welfare committee (Institute of Biology, Center for Interdisciplinary Research in Biology and College de France) and EU guidelines (directive 86/609/EEC). Every precaution was taken to minimize stress and the number of animals used in each series of experiments.
Electrophysiological recordings
Connections between the somatosensory cortex (layer 5) and the striatum are preserved in a horizontal plane (Fino et al. 2005). Accordingly, the present patch-clamp recordings of MSNs were performed on horizontal brain slices (330 μm) from OFA rats (postnatal days 15–21). These horizontal slices included the somatosensory cortical area and the corresponding corticostriatal projection field (Fino et al. 2005, 2008, 2009a,b;) and were prepared with a vibrating blade microtome (VT1000S and VT1200S, Leica Micosystems, Nussloch, Germany). Patch-clamp recordings were made as previously described (Venance et al. 2004; Fino et al. 2005, 2008). Briefly, borosilicate glass pipettes of 4–7 MΩ resistance contained for perforated patch-clamp recordings (mm): 105 potassium gluconate, 30 KCl, 10 Hepes and 0.3 EGTA (adjusted to pH 7.35 with KOH); and for whole-cell recordings (mm): 105 potassium gluconate, 30 KCl, 10 Hepes, 10 phosphocreatine, 4 ATP-Mg, 0.3 GTP-Na, 0.3 EGTA (adjusted to pH 7.35 with KOH). The composition of the extracellular solution was (mm): 125 NaCl, 2.5 KCl, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 10 μm pyruvic acid bubbled with 95% O2–5% CO2. In a subset of experiments, the Mg2+ was removed and the composition of the external solution was the following (mm): 127 NaCl, 2.5 KCl, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, and 10 μm pyruvic acid. All recordings were performed at 34°C using a temperature control system (Bioptechs ΔTC3, Butler, PA, USA) and slices were continuously superfused at 2–3 ml min−1 with the extracellular solution. Individual neurons were identified using infrared-differential interference contrast microscopy with CCD camera (Hamamatsu C2400-07; Hamamatsu, Japan). Signals were amplified using EPC9-2 and EPC10-2 amplifiers (HEKA Elektronik, Lambrecht, Germany). Current-clamp recordings were filtered at 2.5 kHz and sampled at 5 kHz and voltage-clamp recordings were filtered at 5 kHz and sampled at 10 kHz using the programs Pulse-8.53 and Patchmaster v2x32 (HEKA Elektronik). The series resistance was compensated at 75–80%.
Under control condition, recordings were performed without any pharmacological treatments or ionic modifications to preserve the local striatal microcircuits involved in corticostriatal transmission. Most of electrophysiological recordings were realized in whole-cell patch-clamp configuration, except when specified (see below the ‘perforated patch-clamp recordings’ paragraph). dl-2-Amino-5-phosphono-pentanoic acid (d-AP5, 40 μm) (Tocris Bioscience, Bristol, UK), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μm) (Tocris), bicuculline methiodide (20 μm) (Sigma), N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251, 3 μm) (Tocris), LY367385 (100 μm) (Tocris), pirenzepine (1 μm) (Sigma) (1Z,2E)-1-(2-fluorophenyl)-3-(4-hydroxyphenyl)-prop-2-en-one-O-(2-dimethylamino-ethyl)-oxime-hemifumarate (SR46349B, 1 μm) (Sanofi-Synthelabo, Montpellier, France) and mibefradil (20 μm) (Sigma) were bath applied. AM251 was dissolved in ethanol and then added in the external solution at a final concentration of ethanol of 0.03%. BAPTA (10 mm) (Sigma) and MK801 (1 mm) (Tocris) were dissolved directly into the intracellular solution and applied via the patch-clamp pipette. U73122 (5 μm) (Tocris) and ryanodine (100 μm) (Tocris) were dissolved in ethanol, and then added into the intracellular solution; the final concentration of ethanol was 0.033% or 0.1%, respectively. Thapsigargin (0.5 μm) (Tocris) and tetrahydrolipstatin (10 μm) (THL, referenced also as Orlistat, Sigma) were dissolved in DMSO and applied internally via the patch-clamp pipette at a final concentration of DMSO of 0.0025% or 0.1%, respectively.
Perforated patch-clamp recordings
Amphotericin B (Sigma) was used to perform perforated patch-clamp experiments, as previously described (Fino et al. 2008, 2009a,b;). The concentration of amphotericin B in the patch-clamp pipette solution was 200 μg ml−1. Perforated patch-clamp recordings were obtained in a subset of cells to confirm, without affecting the cytoplasm content of the cells, the spike-timing dependence feature of MSN STDP.
Spike timing-dependent plasticity induction protocols
Electrical stimulations of the cerebral cortex were performed with a bipolar electrode (Phymep, Paris, France) placed in layer 5 of the somatosensory cortex (Fino et al. 2005). Electrical stimulations were monophasic at constant current (stimulator from WPI, Stevenage, UK, or ISO-Flex stimulator controlled by a Master-8, AMPI, Jerusalem, Israel). There was no significant difference in the current intensities of cortical stimulations between each stimulation protocol group: post–pre and pre–post pairings. This indicates that the spike-timing dependence of induced synaptic plasticities (LTP versus LTD) was not related to the stimulation intensity. Currents were adjusted in order to evoke striatal excitatory postsynaptic currents (EPSCs) ranging from 50 to 200 pA amplitudes. Repetitive control stimuli were applied at 0.1 Hz, a frequency for which neither short- nor long-term synaptic efficacy changes in EPSC amplitudes were induced (Fino et al. 2005).
STDP protocols consisted in pairings of pre- and postsynaptic stimulations (100 times at 1 Hz) with a time shifting (Δt) of several milliseconds. Presynaptic stimulations correspond to cortical stimulations and the postsynaptic stimulation to an AP evoked by a direct application of a depolarizing current step (30 ms duration) in the MSN. Δt was measured as the time interval between the peak of the postsynaptic AP and the stimulation artifact (due to presynaptic electrical stimulation) recorded in the MSN. Neurons were recorded for 10 min during baseline and for at least 60 min after the cellular conditioning protocol; long-term synaptic efficacy changes were measured at approximately 60 min. Series resistance was monitored and calculated from the response to a hyperpolarizing potential (−5 mV) step during each sweep throughout the experiments and a variation above 20% led to the rejection of the experiment. Repetitive control stimuli were applied at a frequency of 0.1 Hz for 60 min. Stability of the EPSC amplitudes was investigated during 70 min and no significant variations were detected (Fino et al. 2005). Indeed, there was no significant variation between normalized EPSC amplitudes during baseline (100.0 ± 3.0%) and at 70 min (95.9 ± 2.7%) (n = 5). Drugs were applied in the bath, after recording 10 min of baseline in control condition and 10 min before cellular conditioning protocol, and were present continuously until the end of the recording. Throughout this paper, the letter ‘i’ before the name of a drug indicates that the drug was applied intracellularly through the patch-clamp pipette. Under these conditions, the pipettes were systematically tip-filled with regular internal solution and back-filled with drug internal. Once the cell was patched, drugs were allowed to diffuse into the cell during at least 15 min before starting the recording of the 10 min baseline before the STDP protocol.
Data analysis
Off-line analysis was performed using Igor-Pro (Wavemetrics, Lake Oswego, OR, USA). All results were expressed as the mean ±s.e.m. To assess the significance of the induced STDP, we performed a column statistic analysis for each group of values. We compared each group of experiments to a theoretical value of 100 using the non-parametric Wilcoxon's signed-rank test. In two cases (AM251 effect on t-LTP and the i-MK801 NMDA:AMPA charge ratio analysis) we performed a Mann–Whitney non-parametric test. Statistical analysis was performed using Prism 5.0 software (GraphPad Software Inc., La Jolla, CA, USA).
To assess the stability of the baseline, we averaged the EPSC amplitudes every 2 min during the baseline (10 min), a variation of the EPSC amplitude (comparison of the 5 values) greater than 30% led to the rejection of the experiment. EPSC mean amplitudes, measured 60 min after the induction protocol, were the average of 30 evoked EPSCs. Each 30 EPSCs was normalized to the mean of the baseline and the 30 normalized amplitudes were averaged. Synaptic efficacy changes were classified as either LTP or LTD when the mean of normalized EPSC amplitudes was significantly different from the control baseline.
NMDA:AMPA current ratios were calculated with the use of 0-Mg2+ external solution. NMDA and AMPA currents were both measured at MSN resting potential in the absence of Mg2+ in the external solution before and after d-AP5 (40 μm). The NMDA:AMPA current charge ratios were calculated with the currents integrals of the evoked currents in control and d-AP5 conditions (in regular and i-MK801 intracellular solutions), using MiniAnalysis 6.0.7 software (Synaptosoft, Fort Lee, NJ, USA). i-MK801 experiments were conducted as follow: MSNs were first recorded during 15–20 min in a Mg2+ free external solution (to increase the efficiency of the i-MK801 blockage of the NMDA channels), then the regular external solution was applied and the STDP experiments were performed (10 min of baseline followed by at least 60 min of recording).
Results
Characterization of the corticostriatal spike-timing dependence
MSNs were identified and distinguished from striatal interneurons based on electrophysiological properties (Kawaguchi et al. 1995; Fino et al. 2008, 2009a). Briefly, MSNs displayed a hyperpolarized resting membrane potential (RMP; −73.4 ± 0.6 mV, n = 171), a depolarizing ramp to spike threshold and an inward rectification of the I–V relationship (Fig. 1A). Electrical stimulations of cortical afferents evoked glutamatergic monosynaptic EPSCs (inhibited by CNQX 10 μm and d-AP5 50 μm, n = 5) in MSNs (Fig. 1B) with a success rate of 97% (n = 176).
Figure 1. Corticostriatal t-LTP and t-LTD.
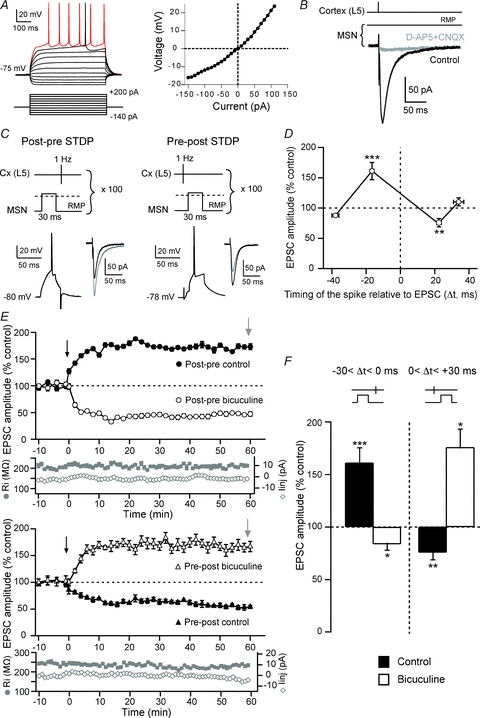
A, characteristic membrane properties and spiking pattern of a MSN: a hyperpolarized RMP, an inward rectification and a long depolarizing ramp to the AP threshold leading to a delayed spike discharge. Raw traces show individual voltage responses to series of 500 ms current pulses from −140 pA to +200 pA with 40 pA steps (black traces) and to +50 pA above AP threshold (red trace). The MSN steady-state I–V relationship illustrates the inward rectification. B, cortically evoked MSN EPSCs (averages of 10 traces) in control and with CNQX (10 μm) and d-AP5 (40 μm). C, schematic representation of STDP protocol and the corresponding raw traces of the postsynaptic MSN. An AP was evoked in the MSN a few milliseconds before (post–pre sequence) (left side) or after (pre–post sequence) (right side) a cortical stimulation (100 paired stimulations at 1 Hz). Post–pre pairings induced t-LTP and pre–post pairings induced t-LTD, as illustrated by EPSCs (averages of 10 raw traces) evoked before (black traces) or 60 min after (grey traces) the cellular conditioning protocol. D, spike timing-dependent changes in synaptic efficacy estimated 60 min after STDP induction. After post–pre pairings (−30 < Δt < 0 ms), a significant (P < 0.001) t-LTP (161.2 ± 14.1%, n = 21) occurred. Conversely, pre–post pairings (0 < Δt < +40 ms) induced a significant (P < 0.01) t-LTD (72.3 ± 6.9%, n = 14). No more LTP and LTD was observed for Δt > −30 ms (87.7±1%, n = 2) and Δt > +40 ms (110.2 ± 6.6%, n = 2), respectively. E, representative experiments illustrating the time courses of synaptic efficacy changes induced by post–pre (upper panel) and pre–post pairings (lower panel), respectively, in the absence (black circles and triangles) and presence of bicuculline (20 um) (open circles and triangles). The black arrow indicates the STDP protocol induction and the grey arrow the measurement of the induced plasticity. Evolution of input resistance and holding current illustrates the stability of the recordings with time. F, the inhibition of GABAergic transmission by bicuculline affects the spike-timing dependence. Post–pre pairings induced t-LTP in control condition but a t-LTD was observed in the presence of bicuculline, in the presence of bicuculline. Conversely, pre-post pairings induced t-LTD in control conditions but a t-LTP with bicuculline. *P < 0.05, **P < 0.01, ***P < 0.001.
Using STDP protocols (Fig. 1C), we examined the influence of the temporal relationship between the discharges of MSNs and the activation of their cortical afferents on the induction of long-term synaptic plasticity. Consistent with previous results (Fino et al. 2005), we found that STDP protocols induce efficient bidirectional long-term synaptic efficacy changes in MSNs with a success rate of 77% (n = 35) for time intervals between pre- and postsynaptic activation (Δt) between −30 and +30 ms (Fig. 1D). Post–pre pairings (Fig. 1C) induced significant (P < 0.001) t-LTP with a success rate of 81% (17 t-LTP out of 21 post–pre pairing experiments) for −30 < Δt < 0 ms (Fig. 1D–F). After potentiation, the mean value of the EPSC amplitude was 161.2 ± 14.1% (Δt = −16.6 ± 1.1 ms, n = 21) (Fig. 1F). Pre–post pairings (Fig. 1C) induced exclusively t-LTD, since significant EPSC amplitude depression occurred with a success rate of 71% (10 t-LTD out of 14 pre–post pairing experiments) for 0 < Δt < +30 ms (Fig. 1D–F). The mean value of EPSC amplitude was 72.3 ± 6.9% (Δt = +23.1 ± 1.4 ms, n = 14) (Fig. 1F). Therefore, t-LTP or t-LTD can be induced in most of the MSNs depending strictly on the STDP protocol. This high occurrence is in agreement with corticostriatal STDP reported so far (Pawlak & Kerr, 2008; but see Shen et al. 2008 in which LTD cannot be induced in MSNs expressing the D1 receptor in mice). There was no significant difference between STDP obtained in whole-cell configuration (Fino et al. 2005) or in perforated patch-clamp recordings (t-LTP: 161.2 ± 14.1%, n = 21 and 214.7 ± 32.5%, n = 3; t-LTD: 75.8 ± 7%, n = 14 and 58.5 ± 11.9%, n = 3).
Since our aim was to elucidate the mechanisms that underpin corticostriatal STDP, we first address the spike-timing dependence discrepancy among studies. Namely, we have reported an anti-Hebbian STDP (post–pre and pre–post pairings induced, respectively, LTP and LTD) (Fino et al. 2005) while a Hebbian spike-timing dependence was also reported (Pawlak & Kerr, 2008; Shen et al. 2008). One of the most striking experimental difference was that GABAergic networks were either inhibited (Pawlak & Kerr, 2008; Shen et al. 2008) or unaffected (Fino et al. 2005, 2008; the present study). We observed that the inhibition of GABAA mediated transmission is sufficient to affect the physiological spike-timing dependence of corticostriatal STDP. When bicuculline (20 μm) was bath applied, post–pre pairings induced significant t-LTD with a success rate of 71% (n = 7) for −30 < Δt < 0 ms (Fig. 1E). After depression, the mean value of the EPSC amplitude was 83.7 ± 5.8% (Δt = −17.7 ± 0.4 ms, n = 7) (Fig. 1F). Following bicuculline treatment, pre–post pairings induced t-LTP, since significant EPSC amplitude potentiation occurred with a success rate of 100% (n = 4) for 0 < Δt < +30 ms (Fig. 1E). The mean value of EPSC amplitude was 176.0 ± 17.2% (Δt = +22.3 ± 4.3 ms, n = 4) (Fig. 1F). Here, we show that application of bicuculline reversed the physiological spike-timing dependence of corticostriatal STDP.
Spike timing-dependent LTD and LTP require postsynaptic calcium
We then sought to determine the signalling system required for STDP occurrence. STDP pharmacology was investigated for −30 < Δt < +30 ms since this Δt corresponded to a reliable induction window of t-LTP and t-LTD, under control conditions. We first explored the calcium dependence of the STDP. We observed that t-LTD and t-LTP induction were blocked with a fast calcium buffer, BAPTA (10 mm), applied intracellularly (i-BAPTA) through the patch-clamp pipette (Fig. 2). Indeed, as illustrated by a representative experiment (Fig. 2A), a lack of long-term plasticity was observed with i-BAPTA for 0 < Δt < +30 ms (Δt = +20.2 ± 2.8 ms, n = 5) and −30 < Δt < 0 ms (Δt = −13.8 ± 1.6 ms, n = 6). The synaptic efficacy changes measured 60 min after STDP protocols were not significantly different from the initial baseline for pre–post (94.4 ± 6.0%, n = 5) and for post–pre (104.3 ± 6.4%, n = 6) pairings (Fig. 2B). Consequently, the occurrence of both t-LTD and t-LTP required postsynaptic calcium.
Figure 2. t-LTD and t-LTP require postsynaptic calcium.
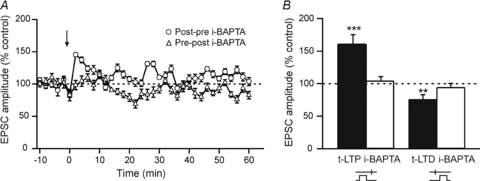
A, representative experiments illustrate the absence of induced plasticity with i-BAPTA (10 mm) after either post–pre (open circles) or pre–post (open triangles) sequences. The black arrow indicates the STDP protocol induction. B, t-LTP and t-LTD are blocked by postsynaptic i-BAPTA. **P < 0.01, ***P < 0.001.
t-LTD and t-LTP are differentially sensitive to NMDA receptor activation
The involvement of NMDARs, which act as coincidence detectors, has been reported in various forms of STDP (Dan & Poo, 2006; Caporale & Dan, 2008; Sjöström et al. 2008). Thus, we tested if NMDARs were a determinant calcium source for corticostriatal t-LTD or t-LTP. We investigated the involvement of NMDARs in the induction and maintenance of t-LTD by blocking NMDARs with d-AP5 treatment (40 μm). d-AP5 did not have a significant effect on t-LTD, suggesting that NMDARs are not involved. Specifically, after the conditioning protocol the mean EPSC amplitude value was 70.2 ± 10.7% (Δt = +25.0 ± 1.8 ms, n = 5) and significantly (P < 0.05) different from baseline (Fig. 3A). When d-AP5 was applied (n = 5), post–pre pairings did not induce t-LTP (Fig. 3B). The mean EPSC amplitude recorded 60 min after protocol induction (103.9 ± 4.7%, Δt = −15.0 ± 1.2 ms, n = 5) did not significantly differ from control baseline, indicating the involvement of NMDARs in the induction of t-LTP. Together, these data indicate that t-LTP, but not t-LTD, required NMDAR activation.
Figure 3. t-LTP is sensitive to postsynaptic NMDA receptors.
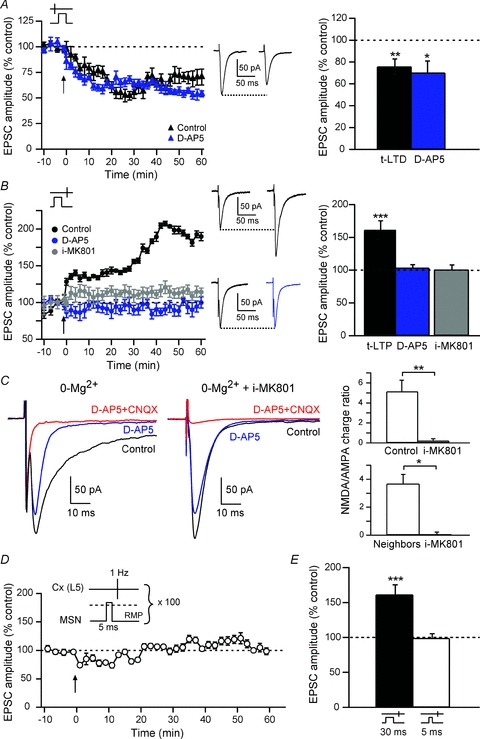
A, t-LTD, induced by pre–post pairings (black triangles), was not blocked by d-AP5 treatment (blue triangles), as illustrated by representative experiments. Bar graph of long-term synaptic efficacy changes shows the induction of significant LTD in control (72.3 ± 6.9%, n = 14) and in d-AP5 (70.2 ± 10.7%, n = 5) conditions. Insets: averaged EPSCs before (left) or after (right) STDP protocols. B, t-LTP induced by post–pre pairings (black circles) was totally blocked with d-AP5 (103.9 ± 4.7%, n = 5) (blue circles) or i-MK801 (98.6 ± 10.1%, n = 5) (grey circles). Note that d-AP5 and i-MK801 did not induce significant synaptic efficacy changes. Simultaneous recordings of MSNs with the regular intracellular solution while neighbours (<30 μm) were recorded with i-MK801 showed unaffected NMDA:AMPA charge ratios in control MSNs. This indicates that corticostriatal t-LTP was postsynaptic NMDAR-activation dependent. Insets: average EPSCs before (left) or after (right) STDP protocols, in control condition (top traces) or with a d-AP5 treatment. C, i-MK801 blocked the NMDA currents measured at −80 mV with a 0-Mg2+ intracellular solution. Representative EPSCs recorded at −80 mV in 0-Mg2+ (left side) or in 0-Mg2++ i-MK801 (right side); in both cases, averages of 10 EPSCs are shown in control external solution, d-AP5 or CNQX/d-AP5. Bar graphs show the quantification of the NMDA:AMPA current integral ratio for all MSNs recorded in 0-Mg2+ (top bar graph) and for neighbouring MSNs recorded with regular intracellular solution or with i-MK801 (bottom bar graph). D and E, 5 ms suprathreshold postsynaptic depolarization paired with presynaptic stimulation did not induce significant t-LTP. ns: not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
To determine the locus (pre- versus postsynaptic) of NMDARs necessary for t-LTP, we performed STDP experiments with MK801 (1 mm) added into the intracellular solution of the postsynaptic recording pipette (i-MK801). Although MK801 is classically applied in the external medium to block NMDARs, this molecule has a potent effect on NMDARs when applied into the cytoplasm of the cell (Bender et al. 2006; Nevian & Sakmann, 2006). The efficiency of the i-MK801 was assessed by estimating the NMDA:AMPA current ratios. Because i-MK801 is an open channel blocker, a 0-Mg2+ extracellular solution was applied to increase the efficiency of the NMDA channel block by i-MK801. Under these conditions, i-MK801 completely blocked NMDA currents (Fig. 3C). The ratio of NMDA:AMPA current integrals for MSNs recorded in control was 5.13 ± 1.14 (n = 5) and 0.21 ± 0.21 for MSNs with i-MK801 (n = 5). It represents a 95.9% decrease in NMDA:AMPA charge ratio. Moreover, in i-MK801 conditions, no further effect of d-AP5 was observed (n = 6) on the cortically-evoked EPSCs. i-MK801 (n = 6) prevented the induction of t-LTP (Fig. 3B), similarly to bath application of d-AP5. The mean EPSC amplitude value (100.5 ± 8.4%, Δt = −16.2 ± 0.5 ms, n = 6) was not significantly different from baseline. Thus, postsynaptic NMDAR activation is necessary for induction of t-LTP. We verified if i-MK801 could act extracellularly by spilling out in the extracellular medium, using double patch-clamp recording of neighbouring MSNs, one recorded with i-MK801 and the other one with the regular intracellular solution. Neighbouring MSNs were recorded simultaneously <30 μm away. A normal NMDA:AMPA charge ratio could still be induced in the control MSN (3.67 ± 0.67, n = 4) while no detectable effect of d-AP5 was observed in the neighbouring i-MK801 MSNs (NMDA:AMPA charge ratio: 0.01 ± 0.12, n = 4) (Fig. 3C).
In our STDP protocols, we used 30 ms suprathreshold postsynaptic depolarization, supposedly sufficient to remove the Mg2+ block of NMDARs. We tested if a shorter suprathreshold depolarization (5 ms versus 30 ms) was also able to remove the Mg2+ block. Here, we show that brief postsynaptic suprathreshold depolarizations paired with presynaptic stimulations (−30 < Δt < 0 ms) were not able to induce a significant plasticity (Fig. 3D and E). The mean EPSC amplitude recorded 60 min after protocol induction (98.9 ± 7.0%, Δt = −14.7 ± 0.5 ms, n = 5) did not significantly differ from control baseline, indicating the lack of induced synaptic plasticity (Fig. 3E). Therefore, a longer postsynaptic depolarization (30 ms), supra- (Fino et al. 2005) or subthreshold (Fino et al. 2009b), is needed to efficiently unblock the Mg2+ from NMDARs and induce a LTP. This is in accordance with the kinetics of physiological summation of EPSPs induced by cortical or thalamic activity leading to a spiking activity in MSNs, as observed in in vivo studies (Stern et al. 1998).
Together these results indicate that t-LTP required activation of postsynaptic NMDARs, and that t-LTD was not mediated by NMDARs. Accordingly, besides NMDARs, distinct coincidence detectors should sense the pre–post paired stimulations responsible for t-LTD.
PLCβ acts as coincidence detector for t-LTD
At hippocampal synapses, PLCβ acts as an efficient coincidence detector triggering retrograde endocannabinoid signalling to mediate short-term plasticity (Hashimotodani et al. 2005, 2008). Accordingly, we sought to test if PLCβ acts as a coincidence detector in corticostriatal STDP. In the presence of i-U73122 (5 μm), a PLC inhibitor applied intracellularly, pre–post pairings did not induce any significant t-LTD (Fig. 4A). Indeed, the mean EPSC amplitude value was 113.0 ± 7.3% (Δt = +16.2 ± 1.5 ms, n = 5), a value not significantly different from baseline (Fig. 4A). Therefore, t-LTD requires PLCβ activation.
Figure 4. t-LTD requires PLCβ activity.
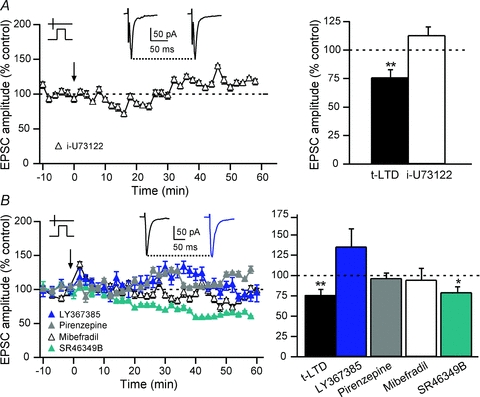
A, t-LTD induced by pre–post pairings was blocked by a specific PLC antagonist, i-U73122 (5 μm). i-U73122 blocked the induction of t-LTD (113.0 ± 7.3%, n = 5), indicating that the activation of PLC is required to induce t-LTD. Insets: averaged EPSCs before (left) or after (right) STDP protocols. B, summary of the effects of the specific group-I mGluR antagonist (LY367385, 100 μm), the M1R antagonist (pirenzepine, 1 μm), the 5-HT2AR antagonist (SR46349B, 1 μm) and the L- and T-type VSCC antagonist (mibefradil, 20 μm). t-LTD was blocked by treatment with LY367385 (135.2 ± 22.2%, n = 5), pirenzepine (96.6 ± 6.5%, n = 8) or mibefradil (94.6 ± 14.1%, n = 5). On the contrary, blockade of 5-HT2Rs did not preclude t-LTD occurrence (79.3 ± 6.9%, n = 6). Insets: averaged EPSCs before (black) or after (blue) STDP protocols in LY367385 treatment. ns: not significant, *P < 0.05, **P < 0.01.
The molecular details of how PLCβ acts as a coincidence detector involve elevation of calcium concentration, which is needed for its catalytic function, and its concomitant activation by Gq/11-coupled receptors, including group-I mGluRs, M1/M3 muscarinic and 5-HT2 serotoninergic receptors (5-HT2Rs) (Rebecchi & Pentyala, 2000; Rhee, 2001). Accordingly, we tested if VSCCs, group-I mGluRs, muscarinic receptors or 5-HT2Rs were involved in the induction and maintenance of the t-LTD. MSNs express various types of calcium channels (T-, N- and L-types) (Carter & Sabatini, 2004). L-type VSCCs have been reported to control the induction of changes in synaptic efficacy (Magee & Johnston, 2005). We found that in the presence of mibefradil (20 μm, n = 5), a blocker of T- and L-type VSCCs, pre–post pairings did not induce t-LTD (Fig. 4B). The mean EPSC amplitude value was 94.6 ± 14.1% (Δt = +20.6 ± 1.2 ms, n = 5), which was not significantly different from baseline (Fig. 4B). These results suggest that the increase in PLCβ activity requires calcium elevation through calcium entry via T- and/or L-type VSCCs.
We then tested if mGluRs were involved in the t-LTD. Indeed, mGluRs have been implicated in the induction of t-LTD (Nevian & Sakmann, 2006), of tetanic stimulation-induced LTD (Calabresi et al. 1992) and pharmacological LTD induced by mGluR agonists (Maejima et al. 2005). MSNs express mainly the group-I mGluRs (mGluR1,5) (Testa et al. 1994). They do not express detectable levels of mGluR1 but express widely mGluR5 in the somatodendritic elements (Uchigashima et al. 2007). In the presence of an antagonist of group-I mGluRs (LY367385, 100 μm), no more significant plasticity was observed (Fig. 4B). The mean EPSC amplitude value (135.2 ± 22.2%, Δt = +20.8 ± 2.8 ms, n = 5) was not significantly different from baseline (Fig. 4B). Accordingly, corticostriatal t-LTD depends on mGluR5 activation.
MSNs also express Gq/11-coupled M1Rs (Hersch et al. 1994). Therefore, we tested if M1R activity was needed for the activation of the PLCβ and the subsequent induction of a t-LTD. In the presence of pirenzepine (1 μm), an antagonist of M1Rs, no significant synaptic efficacy changes were observed after pre–post pairings (Fig. 4B). The mean EPSC amplitude value was 96.6 ± 6.5% (Δt = +19.1 ± 1.5 ms, n = 8), a value not significantly different from baseline (Fig. 4B).
We then tested if 5-HT2ARs were involved in the t-LTD. Indeed, 5-HT2ARs, Gq/11-coupled receptors that activate PLCβ, are expressed in striatum (Di Matteo et al. 2008). However, in the presence of SR46349B (1 μm), an antagonist of 5-HT2ARs, a pre–post pairing still induced significant t-LTD (Fig. 4B), indicating that 5-HT2Rs are not involved. The mean EPSC amplitude value was 79.3 ± 6.9% (Δt = +26.5 ± 0.4 ms, n = 6), a value significantly different (P < 0.05) from baseline (Fig. 4B).
Altogether, these experiments show that t-LTD induction required PLCβ activation, and this activation could be mediated by VSCCs, mGluR5 or M1Rs (but not by 5-HT2ARs).
The catalytic activity of the PLCβ allows the formation of diacylglycerol (DAG) and inositol trisphosphate (IP3) from phosphatidylinositol bisphosphate (PIP2). IP3 is a co-agonist, together with calcium, of IP3R. Consequently, IP3R acts as a coincidence detector since it needs PLCβ activation, with IP3 production, and a postsynaptic activity mediating a calcium entry (Bezprozvanny et al. 1991; Finch et al. 1991). Thus, we investigated the consequence for t-LTD of IP3 release using thapsigargin, an irreversible inhibitor of the Ca2+-ATPase responsible for the refilling of the IP3R-gated calcium stores. In the presence of i-thapsigargin (0.5 μm), pre–post pairings failed to induce t-LTD (Fig. 5). Indeed, the mean EPSC amplitude recorded after a pre–post pairing in i-thapsigargin was not significantly different from baseline (113.8 ± 13.3%, Δt = +16.8 ± 1.7 ms, n = 6). In contrast, i-ryanodine (100 μm, n = 7), a blocker of ryanodine receptors and consequently of calcium-induced calcium-release (CICR) from internal stores, did not prevent t-LTD after pre–post pairings (Fig. 5). The mean EPSC amplitude was significantly decreased from baseline (81.3 ± 9.7%, Δt = +20.6 ± 1.7 ms, n = 7, P < 0.01). Therefore, corticostriatal t-LTD required calcium from IP3R-dependent stores but not from the ryanodine receptor-dependent stores.
Figure 5. t-LTD requires the integrity of IP3R-gated calcium stores.
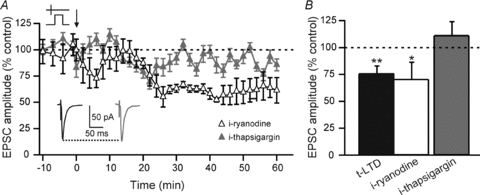
A, as illustrated by representative experiments, t-LTD induced by pre–post pairings was blocked when IP3R-gated calcium stores were emptying by the use of i-thapsigargin (0.5 μm), an antagonist of the Ca2+-ATPase responsible for the calcium refilling of IP3-R-gated calcium stores. When ryanodine-sensitive internal calcium stores (CICR stores) were calcium depleted with i-ryanodine (100 μm), t-LTD could still be induced after pre–post pairings. Insets: averaged EPSCs before (black) or after (grey) STDP protocols with i-thapsigargin treatment. B, summary of the effects of i-ryanodine and i-thapsigargin treatments. t-LTD was blocked by i-thapsigargin treatment (113.8 ± 13.3%, n = 6) but not by i-ryanodine treatment (81.3 ± 9.7%, n = 7). ns: not significant, *P < 0.05, **P < 0.01.
PLCβ and IP3 receptors trigger retrograde endocannabinoid signalling
Endocannabinoids have been identified as retrograde messengers mediating short- and long-term synaptic efficacy changes (Freund et al. 2003; Chevaleyre et al. 2006; Kano et al. 2009). Synaptically driven endo-cannabinoid signalling requires activation of group-I mGluRs and PLCβ (Jung et al. 2005; Maejima et al. 2005). Furthermore, endocannabinoid release depends on the activity of the postsynaptic element and modifies synaptic transmission via presynaptic CB1Rs. CB1Rs have been found presynaptically in the corticostriatal pathway (Herkenham et al. 1991; Katona et al. 2006). Considering these data, we investigated if the induction of t-LTD was dependent on the activation of presynaptic CB1Rs using the CB1R selective antagonist AM251 (3 μm). AM251 prevented the occurrence of t-LTD as illustrated by a representative experiment showing an absence of synaptic efficacy changes (Fig. 6A). Specifically, in the presence of AM251 the mean EPSC amplitude value (105.5 ± 6.1%, Δt = +23.4 ± 1.5 ms, n = 5) was not significantly different from baseline (Fig. 6B).
Figure 6. t-LTD requires endocannabinoid signalling.
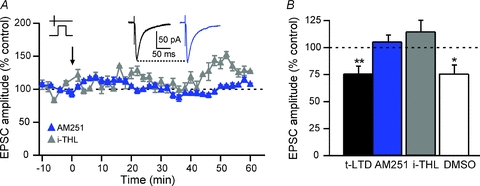
A, as illustrated by representative experiments, t-LTD induced by pre–post pairings was blocked after bath applied CB1R specific antagonist AM251 (3 μm) or i-THL (10 μm), an inhibitor the DGLα responsible for the synthesis of the 2-AG. The i-THL was dissolved in DMSO (0.1–0.025%); the DMSO by itself did not block the induction of the t-LTD. Insets: averaged EPSCs before (black) or after (blue) STDP protocols with AM251 treatment. B, the average of synaptic efficacy changes was 105.5 ± 6.1% after AM251 treatment (n = 5), 114.8 ± 10.4% with i-THL (n = 9) and 76.2 ± 8.2% with vehicle controls (DMSO 0.1–0.025%) (n = 8). ns: not significant, *P < 0.05, **P < 0.01.
We then tested which endocannabinoid, anandamide and/or 2-AG, activated CB1Rs. Activation of Gq/11-coupled receptors, such as mGluR5 or M1Rs, triggers the biosynthesis of 2-AG, but not anandamide, through PLCβ activation (Jung et al. 2005; Maejima et al. 2005). 2-AG is a product of conversion of 1,2-DAG by diacylglycerol lipase α (DGLα) (Stella et al. 1997; Bisogno et al. 2003; Hashimotodani et al. 2005, 2008). We found that t-LTD was precluded when tetrahydrolipstatin (THL, 10 μm) was added into the postsynaptic recording pipette to block DGLα (Fig. 6). An example of the absence of plasticity after a pre–post sequence in the presence of i-THL is illustrated by a representative experiment (Fig. 6A). The mean EPSC amplitude value (114.8 ± 10.4%, Δt = +22.6 ± 2.1 ms, n = 9) was not significantly different from baseline, indicating the absence of t-LTD. Note that DMSO (0.1–0.025%), the vehicle used to dilute i-THL, did not preclude t-LTD (76.2 ± 8.2%, Δt = +18.5 ± 1.5 ms, n = 8) (Fig. 6B). Specifically, significant t-LTD was observed with i-DMSO at 0.1% (n = 4, P < 0.05) or 0.025% (n = 4, P < 0.05).
Therefore, 2-AG, rather than anandamide, mediated long-term synaptic efficacy changes for t-LTD at corticostriatal synapses via activation of CB1Rs.
t-LTP is not modulated by endocannabinoid retrograde signalling
The synthesis and release of endocannabinoids require a large and sustained increase in intracellular calcium (Piomelli, 2003). Since such a calcium entry occurs when NMDARs are activated in t-LTP, we tested the putative effects of endocannabinoids on t-LTP, especially in light of the fact that endocannabinoids can modulate a CB1R-independent LTP (Chevaleyre et al. 2006; Kano et al. 2009). Indeed, CB1Rs are expressed at a high level in the striatum (Herkenham et al. 1991) and their activation inhibits glutamate release (Gerdeman & Lovinger, 2001; Huang et al. 2001). To test the hypothesis that endocannabinoids could be involved in corticostriatal t-LTP, we inhibited CB1Rs with AM251 (3 μm) during post–pre STDP experiments. The amplitude of the evoked t-LTP was not significantly different in the presence of AM251 (178.0 ± 27.7%, Δt = −16.9 ± 2.1, n = 7) compared to control conditions (161.2 ± 14.1%, Δt = −16.6 ± 1.1 ms, n = 21) (Fig. 7). Consequently, endocannabinoid retrograde signalling does not influence the corticostriatal t-LTP induction or maintenance.
Figure 7. t-LTP and t-LTD require distinct coincidence detectors.
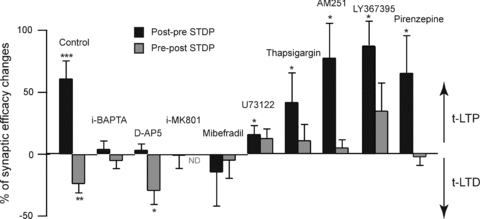
Inhibition of PLCβ, IP3Rs or CB1Rs does not preclude induction of t-LTP. Contrarily to t-LTD, a significant t-LTP could be observed after inhibition of PLCβ (i-U73122, 5 μm), Ca2+-ATPase responsible of the refilling of the IP3R-gated calcium stores (thapsigargin, 0.5 μm), CB1Rs (AM251, 3 μm), group-I mGluRs (LY367395, 100 μm) or M1Rs (pirenzepine, 1 μm). ND: not determined. *P < 0.05, **P < 0.01, ***P < 0.001.
Distinct coincidence detectors for t-LTP and t-LTD
Two distinct pathways, which include different coincidence detectors, govern the bidirectional corticostriatal STDP. Accordingly, we evaluated the relative contribution of each signalling pathway in either t-LTP or t-LTD. Namely, the involvement of the various coincidence detectors (NMDARs, PLCβ and IP3Rs) and some key elements of the cascade signalling (VSCCs, mGluRs, M1Rs and CB1Rs) were analysed.
As shown in Figs 3 and 7, NMDARs are involved exclusively in the t-LTP and not in the t-LTD. Indeed, d-AP5 and i-MK801 treatments blocked the induction of t-LTP after post–pre pairings, whereas d-AP5 did not prevent the induction of t-LTD after pre–post pairings (Figs 3 and 7). Then, we tested the influence of PLCβ and IP3R activation on the induction of t-LTP following post–pre pairings. When PLCβ was inhibited with i-U73122 (5 μm, n = 5), pre–post stimulations failed to induce t-LTD but significant LTP (116.2 ± 6.8%, n = 11, Δt = −13.2 ± 0.8 ms, P < 0.05) could still be elicited after post–pre pairings (Fig. 7). Therefore, PLCβ activity is necessary for neither the induction nor the maintenance of t-LTP, but instead affects the magnitude of the evoked potentiation. The PLCβ activity requires a concomitant activation by Gq/11-coupled receptors and an elevation of calcium. The blockade of group-I mGluRs and M1Rs precluded the induction of t-LTD but did not have any significant effect on t-LTP. Indeed, post–pre pairings after LY367385 (187.7 ± 20.0%, n = 3, Δt = −15.0 ± 0.9 ms) or pirenzepine (165.8 ± 29.9%, n = 5, Δt = −14.4 ± 1.4 ms) treatments induced significant t-LTP (P < 0.05 in both cases) (Fig. 7). We then investigated the role of IP3Rs, as a different source of calcium, in the induction of t-LTP by post–pre pairings. When IP3R-gated calcium stores were emptied by i-thapsigargin (0.5 μm), post–pre STDP pairings still induced significant t-LTP (142.2 ± 23.4%, Δt = −13.4 ± 1.4 ms, n = 5, P < 0.05), while no t-LTD was obtained after pre–post pairings (Fig. 7). Lastly, CB1R activation is involved in t-LTD, but did not significantly interfere with induction and maintenance of t-LTP (Fig. 7). Besides NMDARs and IP3Rs, VSCCs could also constitute a source of calcium. Therefore, we investigated if VSCCs were determinant in the induction of t-LTP. In the presence of bath applied mibefradil (20 μm, n = 9), post–pre pairings did not induce t-LTP any more (Fig. 7). The mean EPSC amplitude value was 84.9 ± 9.0% (Δt = −15.8 ± 1.6 ms, n = 9) and not significantly different from baseline. VSCC activation is therefore necessary for the induction of both t-LTP and t-LTD.
Altogether, these results show that different coincidence detectors are engaged in segregated pathways leading either to t-LTP (NMDARs) or to t-LTD (PLCβ and IP3Rs) (Figs 7 and 8).
Figure 8. Corticostriatal t-LTP and t-LTD rely on distinct coincidence detectors.
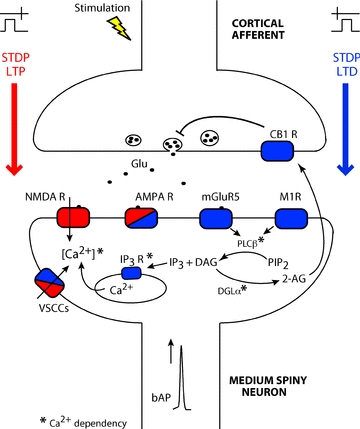
Schematic representation of the different pathways involved in the induction of the corticostriatal t-LTP (red) and t-LTD (blue). Separate coincidence detectors for t-LTP and t-LTP are represented. t-LTP relies on postsynaptic NMDARs and VSCCs activation, while t-LTD involves group-I mGluRs, M1Rs, VSCCs, PLCβ, IP3Rs and retrograde endocannabinoid signalling. Therefore, corticostriatal t-LTP and t-LTD are underlain by two independent cascade signalling pathways composed by distinct coincidence detectors. * indicates the calcium dependence of IP3Rs, PLCβ and DGLα. 2-AG: 2-arachidonoylglycerol; AMPA R: AMPA receptor; CB1Rs: CB1 receptor; DAG: diacylglycerol; DGLα: diacylglycerol lipase; Glu: glutamate; IP3: inositol trisphosphate; IP3R: inositol trisphosphate receptor; mGluR5: metabotropic glutamate type 5 receptor; NMDARs: NMDA receptors; PIP2: phosphatidylinositol bisphosphate; PLCβ: phospholipase Cβ; VSCCs: L- and T-type voltage sensitive calcium channels.
Discussion
The corticostriatal pathway provides the first step of cortical information processing in the basal ganglia. We previously reported that the corticostriatal pathway displays specific STDP properties: post–pre pairings induce t-LTP while pre–post pairings induce t-LTD using paired STDP stimulations repeated 100 times at 1 Hz (Fino et al. 2005). Discrepancies concerning the spike-timing dependence of plasticities among corticostriatal STDP studies (Fino et al. 2005; Pawlak & Kerr, 2008; Shen et al. 2008) could arise from the different experimental conditions. Indeed, GABAergic networks were either inhibited (Pawlak & Kerr, 2008; Shen et al. 2008) or unaffected (Fino et al. 2005, 2008; the present study). Here, we show here that inhibiting the GABAergic transmission affects the spike-timing dependence. Namely, post-pre pairings induced t-LTP in control condition but t-LTD was observed in the presence of bicuculine. Conversely, pre-post pairings induced t-LTD in control conditions and t-LTP with bicuculline treatment. A GABAergic induced shunt could affect the backpropagation of APs, which has been demonstrated to be a key parameter to orientate the STDP toward a potentiation or depression (Letzkus et al. 2006; Sjöström & Häusser, 2006). Although we did not observe any significant effect of bicuculline on the AP afterhyperpolarization, an unspecific effect of bicuculline on SK channels cannot be excluded (Debarbieux et al. 1998). This highlights the role of GABAergic circuits within the striatum controlling the integration of cortical inputs and the time dependence of the orientation of STDP.
Classical models for STDP propose NMDARs as the unique coincidence detector necessary for STDP induction (Nishiyama et al. 2000; Shouval et al. 2002). Such a situation may not be the sole STDP mechanism since experiments and models have suggested the involvement of multiple coincidence detectors to induce bidirectional STDP (Karmarkar & Buonomano, 2002; Karmarkar et al. 2002; Sjöström et al. 2003; Bender et al. 2006; Nevian & Sakmann, 2006; Tzounopoulos et al. 2007). Our results support the model of multiple coincidence detectors controlling corticostriatal MSN STDP: t-LTP relies on postsynaptic NMDARs, while t-LTD involves PLCβ, IP3Rs and retrograde endocannabinoid signalling (Fig. 8). Strikingly, similar coincidence detectors determine the corticostriatal anti-Hebbian STDP and the Hebbian STDP reported in the basal dendrites of layer 2/3 pyramidal neurons of the somatosensory cortex (Bender et al. 2006; Nevian & Sakmann, 2006). In addition, t-LTP and t-LTD observed in cartwheel cells of the dorsal cochlear nucleus, which follow anti-Hebbian learning rules (Tzounopoulos et al. 2004), rely also on NMDA receptors and endocannabinoid signalling, respectively (Tzounopoulos et al. 2007).
t-LTP induction requires NMDARs as coincidence detector
A pre–post pairing generally triggers t-LTP in various mammalian brain structures (Dan & Poo, 2006; Caporale & Dan, 2008). In NMDAR activation-dependent t-LTP, it is assumed that the bAP facilitates Mg2+ unblocking of NMDARs and allows the calcium influx into the postsynaptic cell (Nevian & Sakmann, 2004; Johnston & Narayanan, 2008; Sjöström et al. 2008). A t-LTP induced by a post–pre sequence at corticostriatal synapses (Fino et al. 2005) could be explained by the fact that the postsynaptic element could be still depolarized and the Mg2+ still relieved when the presynaptic release of glutamate hits the MSN. Indeed, the depolarization of 30 ms duration and the postsynaptic AP might cause dendritic plateau potentials that result in a prolonged local depolarization. Therefore, such prolonged depolarization could efficiently interact with the EPSP and promote t-LTP. Contrarily to a 30 ms sub- or suprathreshold depolarization (Fino et al. 2005, 2009b), we show here that a very brief suprathreshold depolarization (5 ms) was not sufficient to remove the Mg2+ block of the NMDARs and induced a t-LTP. Similarly, 100 ms depolarizing somatic current injections in pyramidal cells permit rescue of a low frequency t-LTD, which could not be induced with 5 ms depolarization (Sjöström et al. 2001).
A complex interplay between depolarization and synaptic activation and spikes is required to induce t-LTP at corticostriatal synapses. Indeed, a single AP (triggered by a very short, 5 ms depolarization) did not induce t-LTP (this study), a subthreshold depolarization lasting for 30 ms induced a bidirectional plasticity (Fino et al. 2009b), and an AP evoked on top of a depolarization of 30 ms duration induced t-LTP (the present study). These observations are in accordance with corticostriatal physiology. MSNs act as coincidence detectors of cortical activity and have the ability to extract relevant information from the background noise. MSNs, very hyperpolarized at rest, display a low level of spontaneous activity explained by non-linear electrical membrane properties (Nisenbaum et al. 1994). These properties allow an efficient filtering of the small and uncorrelated synaptic events. Consequently, cortical inputs do not systematically lead to an AP in MSNs but to a wide range of postsynaptic depolarizations, which mostly remain subthreshold (Stern et al. 1998). Pairing presynaptic activation with postsynaptic subthreshold depolarization had previously been shown to elicit LTP (with 1 min duration postsynaptic depolarization) (Artola et al. 1990) or LTD (with 250 ms duration postsynaptic depolarization) (Sjöström et al. 2004) in cortex. Such subthreshold signals paired with cortical activity can lead to subthreshold depolarization-dependent plasticity (Fino et al. 2009b). We chose a 30 ms postsynaptic subthreshold depolarization to mimic corticostriatal subthreshold summation of EPSPs induced by cortical or thalamic activity (as observed in in vivo studies). Indeed, because of non-linear membrane properties, when a spike is evoked in a MSN (by a cortical activity), it requires a progressive depolarization, from −80 mV to −45 mV built by successive EPSP summation, lasting 20–50 ms. It appears that such a process is much slower than in cortex or in hippocampus where neurons act as integrators (but not as coincidence detectors as in MSNs). Moreover, subthreshold depolarization backpropagates very efficiently into the dendritic tree (Carter & Sabatini, 2004). Carter & Sabatini (2004) showed that a backpropagating AP evoked from a holding potential of −50 mV induced a smaller Ca2+ change in a dendritic spine than one evoked from −80 mV (a value close to the MSN RMP); this indicates that the duration and the amplitude of depolarization necessary to bring the MSN from its RMP to the AP threshold is the determinant for the activation of Ca2+-dependent pathways, and therefore very important for the induction of long-term plasticity. Accordingly, it is not surprising that a short (5 ms) depolarization failed to induce a t-LTP at MSN synapses.
Retrograde endocannabinoid signalling for the induction of t-LTD involves multiple coincidence detectors: PLCβ and IP3Rs
Corticostriatal LTD can be induced by various protocols: Hebbian high-frequency stimulation (HFS) (Calabresi et al. 1992), non-Hebbian and Hebbian low-frequency stimulation (LFS) (Fino et al. 2005), Hebbian medium-frequency stimulation (MFS) (Ronesi & Lovinger, 2005; Kreitzer & Malenka, 2005), STDP (Fino et al. 2005, 2008, 2009a; Pawlak & Kerr, 2008; Shen et al. 2008) or brief (30 ms) subthreshold depolarization-dependent plasticity (Fino et al. 2009b). Corticostriatal HFS- and MFS-induced LTD involve a retrograde endocannabinoid signalling (Kreitzer & Malenka, 2008; Di Filippo et al. 2009; but see controversies reviewed in Kano et al. 2009). Here we show that corticostriatal t-LTD also depends on retrograde endocannabinoid signalling. The action of endocannabinoids constitutes the last step of a signalling cascade involving activation of mGluR5/M1Rs/VSCCs, and then PLCβ and IP3Rs (Fig. 8).
t-LTD relies on the successive activation of at least two coincidence detectors: PLCβ and IP3Rs. To be activated, PLCβ needs calcium (originating from VSCC opening and IP3R-gated stores) with Gq/11 protein-coupled receptor activation. Coincident signalling through mGluR5/M1R and VSCCs leads to PLCβ activation (Kim et al. 2002; Fukudome et al. 2004; Hashimotodani et al. 2005, 2008; Maejima et al. 2005; Narushima et al. 2007) and promotes endocannabinoid synthesis and release (Hashimotodani et al. 2005). It has been suggested that PLCβ was also the coincidence detector for t-LTD in cortical synapses since blockade of group-I mGluR prevented the induction of t-LTD (Bender et al. 2006; Nevian & Sakmann, 2006).
IP3R acts as a coincidence detector because it requires both calcium and IP3 binding for its activation to induce calcium release (Bezprozvanny et al. 1991; Finch et al. 1991). Consequently, IP3R needs a PLCβ activation, leading to IP3 production, and a postsynaptic activity mediating a calcium entry. This calcium entry could be due to the bAP and/or the depolarization induced by postsynaptic AMPA receptor activation (Sjöström et al. 2008). The coincidence detectors (PLCβ and IP3Rs) controlling t-LTD are organized in series. Indeed, IP3R is clearly dependent on the IP3 production by PLCβ. Moreover, the calcium necessary for PLCβ activity arises not only from VSCCs, but also from IP3R-gated stores indicating an additional level of interactions between coincidence detectors. This semi-regenerative activation of PLCβ, demonstrated in striatal astrocytes (Venance et al. 1997), allows prolonged PLCβ activity and consequently sustained levels of calcium beneficial for endocannabinoid synthesis and release.
The two major endocannabinoids, anandamide and 2-AG, are produced through distinct pathways. Anandamide production involves N-acyltransferase and phospholipase D, whereas 2-AG is produced by PLCβ and DGLα (Piomelli, 2003). PLCβ and IP3Rs tightly control the retrograde 2-AG signalling. Indeed, PLCβ catalyses PIP2 into DAG, and IP3Rs (as well as VSCCs) allow a calcium increase necessary for the DGLα activity and 2-AG release. By blocking specifically PLCβ (with i-U73122) or DGLα (with i-THL), we demonstrate a critical involvement of 2-AG in the corticostriatal t-LTD. As previously observed in endocannabinoid-mediated short-term plasticity, 2-AG signalling depends on activation of mGluR5 or M1Rs, which are Gq/11 protein-coupled receptors to PLCβ (Chevaleyre et al. 2006; Kano et al. 2009). In addition, activation of group-I mGluRs triggers biosynthesis of 2-AG, but not anandamide, in a calcium-dependent manner (Jung et al. 2005). In the striatum, mGluR5, M1Rs, PLCβ1 and DGLα are widely distributed on the somatodendritic surface of MSNs and are physically apposed to presynaptic CB1Rs (Narushima et al. 2007; Uchigashima et al. 2007; Fukaya et al. 2008).
Interestingly, it has been reported that anandamide is the endocannabinoid molecule involved in corticostriatal LTD induced by HFS or MFS (Adermark & Lovinger, 2007). Therefore, depending on the corticostriatal paired activities (Hebbian HFS, MFS versus STDP), the identity of the endocannabinoid involved in LTD may vary. These discrepancies could be explained by different levels of intracellular calcium reached after HFS or STDP pairings. Different intracellular calcium concentration could then favour the synthesis of either anandamide or 2-AG.
Distinct coincidence detectors underlie t-LTP and t-LTD
A growing body of evidence indicates that a single coincidence detector model cannot explain STDP at different synapses (Karmarkar & Buonomano, 2002; Sjöström et al. 2003; Bender et al. 2006; Nevian & Sakmann, 2006; Tzounopoulos et al. 2007). Here, we report the involvement of distinct pathways for t-LTP and t-LTD at corticostriatal synapses, requiring distinct coincidence detectors (Fig. 8). The induction of both t-LTP and t-LTD was equally sensitive to the inhibition of T- and L-type VSCCs and to postsynaptic loading of BAPTA. Importantly, besides these two elements, the entire signalling cascades leading to t-LTP or t-LTD involved different receptors or enzymes. Indeed, t-LTP requires postsynaptic NMDARs whereas t-LTD does not. t-LTD requires the retrograde endocannabinoid signalling via the activation Gq/11 protein-coupled receptors (mGluR5 or M1Rs) and PLCβ activity. Moreover, t-LTP is not modified after the blockage of CB1Rs. A similar mechanism has been proposed for STDP in somatosensory cortex (Bender et al. 2006; Nevian & Sakmann, 2006) or in the dorsal cochlear nucleus (Tzounopoulos et al. 2007) as well as for short- or long-term depression induced by HFS in hippocampus (Varma et al. 2001; Fukudome et al. 2004; Hashimotodani et al. 2005, 2008), striatum (Gerdeman et al. 2002; Robbe et al. 2002; Kreitzer & Malenka, 2005; Lafourcade et al. 2007; Narushima et al. 2007) or cerebellum (Maejima et al. 2005). Interestingly, PLCβ inhibition does not impair t-LTP induction or maintenance but seems to influence its magnitude. This indicates that if two distinct pathways rule the induction of either t-LTP or t-LTD, some elements can be shared considering the strength of the evoked plasticity.
The striatum is engaged in procedural learning and memory (Graybiel, 2005; Yin & Knowlton, 2006), which are thought to involve long-term synaptic efficacy changes (Martin et al. 2000; Martin & Morris, 2002; Lynch, 2004). Here, we showed that distinct coincidence detectors are involved depending on the spike-timing dependence of the plasticity (t-LTP versus t-LTD). These various coincidence detectors constitute targets for dopamine, a major neuromodulator of striatum, which contributes to goal-directed operant learning. The dopaminergic control on corticostriatal plasticity induced by either HFS or MFS has been established, but the effects remain debated (reviewed in Kreitzer & Malenka, 2008; Kano et al. 2009). However, a recent study also focusing on STDP showed that D1/D5 receptor activation is critically required for corticostriatal t-LTP and t-LTD (Pawlak & Kerr, 2008). According to the results presented in our study, the impact of dopamine should be addressed by considering the different coincidence detector underlying corticostriatal STDP and taking into account that MSNs belong to the direct or indirect basal ganglia pathway.
Acknowledgments
This work was supported by the INSERM, ANR Mecarec, ANR Mobil and College de France.
Glossary
Abbreviations
- 2-AG
2-arachidonoylglycerol
- AMPAR
AMPA receptor
- AP
action potential
- bAP
back-propagating action potential
- CB1R
type-1 cannabinoid receptor
- DAG
diacylglycerol
- DGLα
diacylglycerol lipase
- EPSC
excitatory postsynaptic current
- HFS
high-frequency stimulation
- 5-HTR
serotoninergic receptor
- IP3
inositol trisphosphate
- IP3R
inositol trisphosphate receptor
- LFS
low-frequency stimulation
- LTD
long-term depression
- LTP
long-term potentiation
- M1R
type-1 muscarinic receptor
- mGluR
metabotropic glutamate receptor
- MFS
medium-frequency stimulation
- MSN
medium-sized spiny neuron
- NMDAR
NMDA receptor
- PIP2
phosphatidylinositol bisphosphate
- PLCβ
phospholipase Cβ
- STDP
spike timing-dependent plasticity
- t-LTP
spike-timing dependent long-term potentiation
- t-LTD
spike-timing dependent long-term depression
- THL
tetrahydrolipstatin
- VSCC
voltage sensitive calcium channel
Author contributions
All authors approved the final version of the manuscript for publication. Conception and design of the experiments: E.F., V.P. and L.V. Collection, analysis and interpretation of data: E.F., V.P., Y.C., T.M.-H. and L.V. Drafting the article and revising it critically for important intellectual content: E.F., V.P., J.-M.D. and L.V.
Author's present address
E. Fino: Department of Biological Science, Columbia University, New York, NY 10027, USA.
References
- Adermark L, Lovinger DM. Retrograde endocannabinoid signaling at striatal synapses requires a regulated postsynaptic release step. Proc Natl Acad Sci U S A. 2007;104:20564–20569. doi: 10.1073/pnas.0706873104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artola A, Bröcher S, Singer W. Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature. 1990;347:69–72. doi: 10.1038/347069a0. [DOI] [PubMed] [Google Scholar]
- Bell CC, Han VZ, Sugawara Y, Grant K. Synaptic plasticity in a cerebellum-like structure depends on temporal order. Nature. 1997;387:278–281. doi: 10.1038/387278a0. [DOI] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensorey cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Misgeld U, Dodt HU. Intrinsic membrane properties of neostriatal neurons can account for their low level of spontaneous activity. Neuroscience. 1987;20:293–303. doi: 10.1016/0306-4522(87)90021-2. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporale N, Dan Y. Spike timing-dependent plasticity: a Hebbian learning rule. Annu Rev Neurosci. 2008;31:25–46. doi: 10.1146/annurev.neuro.31.060407.125639. [DOI] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendrites spines of striatal spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- Debanne D, Gahwiler BH, Thompson SM. Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol. 1998;507:237–247. doi: 10.1111/j.1469-7793.1998.237bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debarbieux F, Brunton J, Charpak S. Effect of bicuculine on thalamic activity: a direct blockade of IAHP in reticularis neurons. J Neurophysiol. 1998;79:2911–2918. doi: 10.1152/jn.1998.79.6.2911. [DOI] [PubMed] [Google Scholar]
- Di Filippo M, Picconi B, Tantucci M, Ghiglieri V, Bagetta V, Sgobio C, Tozzi A, Parnetti L, Calabresi P. Short-term and long-term plasticity at corticostriatal synapses: implications for learning and memory. Behav Brain Res. 2009;119:108–118. doi: 10.1016/j.bbr.2008.09.025. [DOI] [PubMed] [Google Scholar]
- Di Matteo V, Pierucci M, Esposito E, Crescimanno G, Benigno A, Di Giovanni G. Serotonin modulation of the basal ganglia circuitry: therapeutic implication for Parkinson's disease and other motor disorders. Exp Brain Res. 2008;172:423–463. doi: 10.1016/S0079-6123(08)00921-7. [DOI] [PubMed] [Google Scholar]
- Finch EA, Turner TJ, Goldin SM. Calcium as a coagonist of inositol 1,4,5-triphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L. Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci. 2005;25:11279–11287. doi: 10.1523/JNEUROSCI.4476-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Deniau JM, Venance L. Cell-specific spike-timing-dependent plasticity in GABAergic and cholineregic interneurons in corticostriatal rat brain slices. J Physiol. 2008;586:265–282. doi: 10.1113/jphysiol.2007.144501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Paille V, Deniau JM, Venance L. Asymetric spike-timing dependent plasticity of striatal nitric oxide-synthase interneurons. Neuroscience. 2009a;160:744–754. doi: 10.1016/j.neuroscience.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Fino E, Deniau JM, Venance L. Brief subthreshold events can act as Hebbian signals for long-term plasticity. PLoS ONE. 2009b;4:e6557. doi: 10.1371/journal.pone.0006557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund T, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signalling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Uchigashima M, Nomura S, Hasegawa Y, Kibuchi H, Watanabe M. Predominant expression of phospholipase Cβ1 in telencephalic principal neurons and cerebellar interneurons, and its close association with related signaling molecules in somato-dendritic neuronal elements. Eur J Neurosci. 2008;28:1744–1759. doi: 10.1111/j.1460-9568.2008.06495.x. [DOI] [PubMed] [Google Scholar]
- Fukudome Y, Ohno-Shosaku T, Matsui M, Omori Y, Fukaya M, Tsubokawa H, Taketo MM, Watanabe M, Manabe T, Kano M. Two distinct classes of muscarinic action on hippocampal inhibitory synapses: M2-mediated direct suppression and M1-M3-mediated indirect suppression through endocannabinoid signaling. Eur J Neurosci. 2004;19:2682–2692. doi: 10.1111/j.0953-816X.2004.03384.x. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Lovinger DM. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol. 2001;85:468–471. doi: 10.1152/jn.2001.85.1.468. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Graybiel AM. The basal ganglia: learning new tricks and loving it. Curr Opin Neurobiol. 2005;15:638–644. doi: 10.1016/j.conb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi, Shin H-S, Kano M. Phospholipase Cβ serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Maejima T, Fukami K, Kano M. Pharmacological evidence for the involvement of diacylglycerol lipase in depolarization-induced endocannabinoid release. Neuropharmacology. 2008;54:58–67. doi: 10.1016/j.neuropharm.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, de Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res. 1991;547:267–274. doi: 10.1016/0006-8993(91)90970-7. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Gutekunst C, Rees HD, Heilman CJ, Levey AI. Distribution of m1-m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;17:3334–3342. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Lo SW, Hsu KS. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J Physiol. 2001;532:731–748. doi: 10.1111/j.1469-7793.2001.0731e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Narayanan R. Active dendrites: colorful wings of the mysterious butterflies. Trends Neurosci. 2008;31:309–316. doi: 10.1016/j.tins.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Jung KM, Mangieri R, Stapleton C, Kim J, Fegley D, Wallace M, Mackie K, Piomelli D. Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Mol Pharmacol. 2005;68:1196–1202. doi: 10.1124/mol.105.013961. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosahu T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Karmarkar UR, Buonomano DV. A model of spike-timing dependent plasticity: one or two coincidence detectors. J Neurophysiol. 2002;88:507–513. doi: 10.1152/jn.2002.88.1.507. [DOI] [PubMed] [Google Scholar]
- Karmarkar UR, Najarian MT, Buonomano DV. Mechanisms and significance of spike-timing dependent plasticity. Biol Cybern. 2002;87:373–382. doi: 10.1007/s00422-002-0351-0. [DOI] [PubMed] [Google Scholar]
- Katona I, Urban GM, Wallace M, Ledent C, Jung KM, Piomelli D, Mackie K, Freund TF. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26:5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- Kim J, Isokawa M, Ledent C, Alger BE. Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci. 2002;22:10182–10191. doi: 10.1523/JNEUROSCI.22-23-10182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, Manzoni O. Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS ONE. 2007;8:e709. doi: 10.1371/journal.pone.0000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Kampa BM, Stuart GJ. Learning rules for spike timing-dependent plasticity depend on dendritic synapse location. J Neurosci. 2006;26:10420–10429. doi: 10.1523/JNEUROSCI.2650-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Maejima T, Oka S, Hashimotodani Y, Ohno-Shosaku T, Aiba A, Wu D, Waku K, Sugiura T, Kano M. Synaptically driven endocannabinoid release requires Ca2+-assisted metabotropic glutamate receptor subtype 1 to phospholipase Cβ4 signaling cascade in the cerebellum. J Neurosci. 2005;25:6826–6835. doi: 10.1523/JNEUROSCI.0945-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Plasticity of dendritic function. Curr Opin Neurobiol. 2005;15:334–342. doi: 10.1016/j.conb.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Morris RG. New life in an old idea: the synaptic plasticity and memory hypothesis revisited. Hippocampus. 2002;12:609–636. doi: 10.1002/hipo.10107. [DOI] [PubMed] [Google Scholar]
- Narushima M, Uschigashima M, Fukaya M, Matsui M, Manabe T, Hashimoto K, Watanabe M, Kano M. Tonic enhancement of endocannabinoid-mediated retrograde suppression of inhibition by cholinergic interneuron activity in the striatum. J Neurosci. 2007;27:496–506. doi: 10.1523/JNEUROSCI.4644-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Single spine Ca2+ signals evoked by coincident EPSPs and backpropagating action potentials in spiny stellate cells of layer 4 in the juvenile rat somatosensory barrel cortex. J Neurosci. 2004;24:1689–1699. doi: 10.1523/JNEUROSCI.3332-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisenbaum ES, Xu ZC, Wilson CJ. Contribution of a slowly inactivating potassium current to the transition to firing of neostriatal spiny projections neurons. J Neurosci. 1994;15:4449–4463. doi: 10.1152/jn.1994.71.3.1174. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- Pawlak V, Kerr JND. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signaling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi J, Lovinger DM. Induction of striatal long-term synaptic depression by moderate frequency activation of cortical afferents in rat. J Physiol. 2005;562:245–256. doi: 10.1113/jphysiol.2004.068460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouval HZ, Bear MF, Cooper LN. A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc Natl Acad Sci U S A. 2002;99:10831–10836. doi: 10.1073/pnas.152343099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron. 2001;32:1149–1164. doi: 10.1016/s0896-6273(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Endocannabinoid-dependent neocortical layer-5 LTD in the absence of postsynaptic spiking. J Neurophysiol. 2004;92:3338–3343. doi: 10.1152/jn.00376.2004. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Häusser M. A cooperative switch determines the sign of synaptic plasticity in distal dendrites of neocortical pyramidal neurons. Neuron. 2006;51:227–238. doi: 10.1016/j.neuron.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A, Häusser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogeneous cannabinoid that modulates long-term potentiation. Nature. 1997;338:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Stern EA, Jaeger D, Wilson CJ. Membrane potential synchrony of simultaneously recorded striatal spiny neurons in vivo. Nature. 1998;394:475–478. doi: 10.1038/28848. [DOI] [PubMed] [Google Scholar]
- Testa CM, Standaert DG, Young AB, Penney JB., Jr Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci. 1994;14:3005–3018. doi: 10.1523/JNEUROSCI.14-05-03005.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounopoulos T, Kim Y, Oertel D, Trussell LO. Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat Neurosci. 2004;7:719–725. doi: 10.1038/nn1272. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Rubio ME, Keen JE, Trussell LO. Coactivation of pre- and postsynaptic signaling mechanisms determines cell-specific spike-timing-dependent plasticity. Neuron. 2007;54:291–301. doi: 10.1016/j.neuron.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M, Watanabe M. Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci. 2007;27:3663–3676. doi: 10.1523/JNEUROSCI.0448-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci. 2001;21:RC188. doi: 10.1523/JNEUROSCI.21-24-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venance L, Stella N, Glowinski J, Giaume C. Mechanism involved in initiation and propagation of receptor induced intercellular calcium signaling in cultured rat astrocytes. J Neurosci. 1997;17:1981–1992. doi: 10.1523/JNEUROSCI.17-06-01981.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venance L, Glowinski J, Giaume C. Electrical and chemical transmission between striatal GABAergic output neurones in rat brain slices. J Physiol. 2004;559:215–230. doi: 10.1113/jphysiol.2004.065672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickens JR. Synaptic plasticity in the basal ganglia. Behav Brain Res. 2009;199:119–128. doi: 10.1016/j.bbr.2008.10.030. [DOI] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- Yin HH, Mulcare SP, Hilario MRF, Clouse E, Holloway T, Davis MI, Hansson AC, Lovinger DM, Costa RM. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat Neurosci. 2009;12:333–341. doi: 10.1038/nn.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]