

Abstract
Acute intermittent hypoxia (AIH) elicits long-term increases in respiratory and sympathetic outflow (long-term facilitation, LTF). It is still unclear whether sympathetic LTF is totally dependent on changes in respiration, even though respiratory drive modulates sympathetic nerve activity (SNA). In urethane-anaesthetized, vagotomized mechanically ventilated Sprague–Dawley rats, we investigated the effect of ten 45 s episodes of 10% O2–90% N2 on splanchnic sympathetic nerve activity (sSNA) and phrenic nerve activity (PNA). We then tested whether or not hypoxic sympathetic chemoreceptor and baroreceptor reflexes were changed 60 min after AIH. We found that 17 animals manifested a sustained increase of sSNA (+51.2 ± 4.7%) 60 min after AIH, but only 10 of these rats also expressed phrenic LTF compared with the time controls (rats not exposed to hypoxia, n = 5). Inspiratory triggered averages of integrated sSNA showed respiratory modulation of SNA regardless of whether or not phrenic LTF had developed. The hypoxic chemoreceptor reflex was enhanced by 60 min after the development of AIH (peak change from 76.9 ± 13.9 to 159.5 ± 24.9%). Finally, sympathetic baroreceptor reflex sensitivity increased after sympathetic LTF was established (Gainmax from 1.79 ± 0.18 to 2.60 ± 0.28% mmHg−1). Our findings indicate that respiratory–sympathetic coupling does contribute to sympathetic LTF, but that an additional tonic increase of sympathetic tone is also present that is independent of the level of PNA. Sympathetic LTF is not linked to the change in baroreflex function, since the baroreflex appears to be enhanced rather than impaired, but does play an important role in the enhancement of the hypoxic chemoreflex.
Introduction
Obstructive sleep apnoea (OSA) is associated with repeated episodes of apnoea and hypoxaemia, causing marked increases in muscle sympathetic nerve activity (MSNA) primarily due to stimulation of carotid body and central chemoreceptors (Dempsey et al. 2010). Patients with OSA also present with impaired baroreflex function (Carlson et al. 1996) and cardiorespiratory sensitivity to hypoxia (Imadojemu et al. 2007). These changes in autonomic control may reflect the central nerve plasticity analogous to those previously described for respiratory control (Mitchell & Johnson, 2003). Several approaches have been used to mimic the neurocirculatory changes associated with sleep apnoea in the laboratory. One of the most frequently studied experimental models is acute intermittent hypoxia (AIH) (Mahamed & Mitchell, 2007).
AIH is capable of inducing phrenic long-term facilitation (pLTF) (Mitchell & Johnson, 2003), expressed as a ‘progressive and sustained increase in phrenic motor output that is independent of changes in chemoafferent input’ (Mitchell et al. 2001). Phrenic LTF is thought to be serotonin dependent, because pretreatment with methysergide, a serotonergic antagonist, blocks its development (Bach & Mitchell, 1996). It is well known that respiration markedly modulates the sympathetic nervous system (Adrian & Bronk, 1932; Miyawaki et al. 2002b), leading to the idea that respiratory LTF can induce sympathetic LTF (sLTF) (Zoccal et al. 2008). Dick et al. (2007) were the first to report that AIH can elicit both phrenic LTF and sympathetic LTF and that the SNA was not simply tonic, but correlated with respiratory bursts. They concluded that the AIH-induced increase in sympathetic activity is co-ordinated and caused by interactions between the respiratory and sympathetic control systems. However, the exact mechanism of AIH induced sympathoexcitation remains poorly understood.
Previous research has demonstrated that arterial chemoreflex abnormalities contribute to the development of hypertension in general, and numerous studies have hypothesized that altered chemoreflex control of SNA contributes to the development of hypertension in OSA patients (Narkiewicz et al. 1999). Leuenberger et al. (2007) demonstrated that short-term intermittent hypoxia enhances sympathetic responses to hypoxia in humans. They concluded that alteration of peripheral chemoreflex function is a potential mechanism leading to AIH-induced sympathoexcitation. Animal studies revealed that chronic intermittent hypoxia (CIH) can elicit chemosensory plasticity, which is expressed as increased basal discharge, hypoxic sensitivity and the capacity to express sensory long-term facilitation (Peng et al. 2003). However, it is not clear whether the hypoxic sympathetic chemoreflex is enhanced after AIH in animals.
Baroreflex impairment is associated with hypertension (Vitela et al. 2005), and baroreflex control appears to be impaired in OSA (Bonsignore et al. 2002). Treatment of OSA with continuous night-time positive airway pressure increases cardiovagal baroreflex function during sleep and wakefulness (Bonsignore et al. 2002). Some animal studies suggest that the impairment of baroreflex control is accompanied by an increase in blood pressure and sympathetic nerve activity after CIH exposure (Lai et al. 2006). Others indicated that the sympathetic-mediated hypertension observed in rats exposed to CIH is not secondary to a reduction in baroreflex gain (Zoccal et al. 2009). Evidence of the modulation of baroreflex function after AIH is scarce. It is still unclear if modulation of baroreflex contributes to the sympathetic LTF.
Therefore, the purpose of the present study is to address the following questions. (1) Does AIH elicit a sustained increase in resting sympathetic tone (i.e. sympathetic long-term facilitation)? (2) Does the increase in sympathetic tone rely on an alteration of respiratory drive or is it solely due to a change in tonic activity? (3) Is baroreflex and hypoxic chemoreflex function altered in the presence of AIH? To answer these questions, we recorded splanchnic sympathetic nerve activity (sSNA), phrenic nerve activity (PNA), mean arterial pressure (MPA) and heart rate (HR) during and 60 min after AIH. We also evaluated the peripheral chemoreflex and baroreflex function before and 60 min after AIH. We hypothesized that AIH would elicit a robust increase in tonic sympathetic resting tone, which is modulated by the respiratory drive but not dependent on it.
Methods
Ethical approval
This study was approved by the Animal Care and Ethics Committee of Macquarie University. Experiments were conducted on adult male Sprague–Dawley rats (350–600 g; Animal Resource Centre, Canning Vale, WA, Australia) in accordance with the Australian Code of Practice for the Care and Use of Animals as endorsed by the National Health and Medical Research Council of Australia.
General preparation
General surgical preparation was carried out as previously described (Abbott & Pilowsky, 2009). Briefly, rats were initially anaesthetized with a bolus of 10% urethane (10% w/v in saline, 1.1–1.3 g kg−1i.p.). Additional doses of urethane (30 mg in a 10% solution) were delivered intravenously as required to maintain adequate levels of anaesthesia. Depth of anaesthesia was assessed by checking for an absence of the withdrawal reflex and/or arterial blood pressure changes after a hindpaw pinch. The left and right femoral arteries and the right femoral vein were cannulated for arterial blood gas sampling (VetStat, IDEXX, Westbrook, ME, USA), measurement of arterial blood pressure, and administration of drugs and fluids, respectively. Heart rate (HR) was derived from ECG. The left phrenic nerve was approached dorsolaterally, isolated, tied with 5/0 silk thread, cut and recorded. The left greater splanchnic nerve was dissected using a retro-peritoneal approach. Animals were bilaterally vagotomized. Neurograms were amplified (×2000 for PNA; ×5000 for sSNA, CWE Inc., Ardmore, PA, USA), band pass filtered (0.2–3 kHz), sampled at 5 kHz (1401plus, CED Ltd, Cambridge, UK) and recorded on computer using Spike2 software (v. 7, CED Ltd). The trachea was cannulated, and the animals were subject to neuromuscular block (pancuronium; 0.8 mg i.v. induction, then 0.4 mg h−1i.v. maintenance, Astra Pharmaceuticals Pty Ltd, Sydney, NSW, Australia). Mechanical ventilation was instituted, with room air enriched with 100% oxygen throughout the experiment (pump frequency, 65–90 min−1; tidal volume, 2.5–4 ml; Ugo Basile, Italy). Core temperature was measured with a rectal probe and maintained at ∼37°C with a homeothermic blanket (Harvard Apparatus, Holliston, MA, USA). Animals were infused with a 50:50 mixture of lactated Ringer solution and 5% glucose (5 ml kg−1 h−1). At the end of experiments, animals were euthanatized with injection of 10% KCl.
Experimental protocol
To established baseline nerve activity, the  level was corrected to 40 ± 2 mmHg by adjusting the ventilator pump frequency and tidal volume; arterial blood pH was corrected to 7.35–7.45 by bolus injection of 5% sodium bicarbonate if metabolic acidosis was apparent. Sympathetic baroreflex function curves (n = 8) were generated by sequential intravenous injection of sodium nitroprusside (10 μg kg−1) and phenylephrine (10 μg kg−1) 30 min after stabilization at baseline. The animals were then stabilized for at least 10 min and then exposed to acute intermittent hypoxia (AIH), consisting of 10 exposures to a hypoxic gas mixture (10% O2 in N2, 45 s per episode) separated by 5 min intervals of ventilation with 100% O2. Arterial blood gas measurements were made at baseline and at 15, 30, 45 and 60 min post-hypoxia to ensure that values remained constant (
level was corrected to 40 ± 2 mmHg by adjusting the ventilator pump frequency and tidal volume; arterial blood pH was corrected to 7.35–7.45 by bolus injection of 5% sodium bicarbonate if metabolic acidosis was apparent. Sympathetic baroreflex function curves (n = 8) were generated by sequential intravenous injection of sodium nitroprusside (10 μg kg−1) and phenylephrine (10 μg kg−1) 30 min after stabilization at baseline. The animals were then stabilized for at least 10 min and then exposed to acute intermittent hypoxia (AIH), consisting of 10 exposures to a hypoxic gas mixture (10% O2 in N2, 45 s per episode) separated by 5 min intervals of ventilation with 100% O2. Arterial blood gas measurements were made at baseline and at 15, 30, 45 and 60 min post-hypoxia to ensure that values remained constant (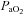 > 140 mmHg,
> 140 mmHg,  ± 1 mmHg from baseline value). Arterial
± 1 mmHg from baseline value). Arterial  was corrected to the target range by adjusting ventilator frequency as necessary. After recording for more than 60 min following the last hypoxic episode, the sympathetic baroreflex function was assessed again and peripheral chemoreceptors were stimulated by ventilating the animal with 10% O2 in N2 for 45 s. Another group of animals that were not exposed to AIH (n = 5) served as time controls.
was corrected to the target range by adjusting ventilator frequency as necessary. After recording for more than 60 min following the last hypoxic episode, the sympathetic baroreflex function was assessed again and peripheral chemoreceptors were stimulated by ventilating the animal with 10% O2 in N2 for 45 s. Another group of animals that were not exposed to AIH (n = 5) served as time controls.
Data analysis
For averaging purposes, sSNA was rectified and smoothed (t = 2 s); sSNA was normalized against baseline as 100% and death level as zero. PNA was rectified and smoothed (t = 0.1 s). Variables determined include the amplitude of integrated phrenic activity (μV), phrenic nerve burst frequency (cycles min−1) and minute phrenic activity (amplitude × burst frequency). For phrenic LTF, changes of phrenic amplitude and minute phrenic activity were normalized as a percentage of the baseline (%baseline). Changes from baseline in burst frequency used absolute units (cycles min−1). PNA and sSNA were analysed over a 1 min period after the blood sampling. To evaluate cardiorespiratory coupling, cycle-triggered averages (CTA) of sSNA were triggered from the onset of the inspiration of the phrenic cycle. The phrenic cycle, and corresponding sSNA, was divided into three phases: inspiratory (I), post-inspiratory (PI), and expiratory (E) (Fig. 1). The amplitude of sSNA from −200 ms to 0 ms before the onset of the phrenic nerve discharge, which shows the most constant amplitude during the respiratory cycle, was taken as the basal level (Miyawaki et al. 2002a). The PI-peak and amplitude of the PI-peak of sSNA were expressed as a percentage change from the baseline.
Figure 1. Data analysis of cycle triggered average of sSNA (lower) based on PNA (upper).
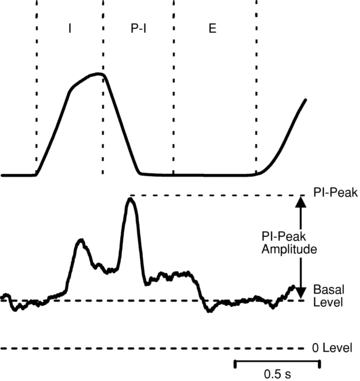
Typically, the predominant patterns include a peak in sSNA coincident with the phrenic burst (inspiratory peak, I-peak) and another peak immediately before the cessation of phrenic burst (post-inspiratory peak, PI-peak). The value of sSNA from −200 ms to 0 ms before the onset of the phrenic nerve discharge was taken as the basal level. The value of sSNA at the apex in the post-inspiratory phase was taken as PI-peak. The PI-peak amplitude is the difference of PI-peak and basal value. The basal level, PI-peak and amplitude of PI-peak of sSNA were expressed as a percentage change from the baseline. The change of the I-peak of sSNA was not included in the analysis.
To analyse the data from the baroreflex function tests, mean arterial blood pressure (MAP) was divided into 1 s consecutive bins and the average sSNA during each bin was determined; successive values were tabulated and graphed, with MAP as the abscissa and sSNA as the ordinate. Each data set was then analysed using GraphPad Prism (v. 5.0; GraphPad Software Inc., La Jolla, CA, USA) to determine the sigmoidal curve of best fit (Kent et al. 1972), which is described by the following equation:
where y is sSNA, A1 is the y range (y at the top plateau – y at the bottom plateau), A2 is the gain coefficient, A3 is the value of x at the midpoint (which is also the point of maximum gain), and A4 is y at the bottom plateau. The peak gain of each curve was determined by obtaining the maximal value of the first derivative across the full range of MAP. The range of sSNA was calculated as the difference between the values at the upper and lower plateaus of the curve. The threshold and saturation values for MAP were defined as the values of MAP that corresponded to the points where y was 5% (of the y range) below and above the upper and lower plateaus, respectively. Only curves where the correlation coefficient (R2) was greater than 0.9 were included in the analysis.
Since the increase in PNA is progressive, phrenic LTF in this study was defined as an increase in the average amplitude of phrenic output from 45 to 60 min that is more than 20% compared with the 15 min before AIH. For LTF, both the change from baseline and the differences between groups in the post-hypoxic phrenic amplitude (also burst frequency and minute phrenic activity) and sSNA were statistically analysed using a two-way ANOVA with repeated measures, followed by post hoc analysis using multiple comparisons and Bonferroni's correction (Graphpad Prism, v. 5.0). Student's t test for paired data was used to analyse the comparative results of baroreflex and peripheral chemoreflex before and 60 min after AIH. For blood gas values, one-way ANOVA was used to determine if there were differences between and within groups. P < 0.05 was considered significant. All values are expressed as mean ±s.e.m.
Results
Effects of AIH on phrenic nerve and splanchnic sympathetic nerve activity
Following AIH all 17 animals exhibited enhancement of sSNA (Fig. 2), while 10 out of 17 exhibited both phrenic (pLTF) and sympathetic LTF (sLTF) after AIH (Fig. 2A). Therefore, the animals were further divided into two groups according to whether the pLTF was induced or not (‘pLTF+sLTF’ group, n = 10 Fig. 2A; ‘Only sLTF’ group, n = 7 Fig. 2B; also Fig. 3A and B and Supplementary figure).
Figure 2. Representative neurograms from rats exposed to AIH.
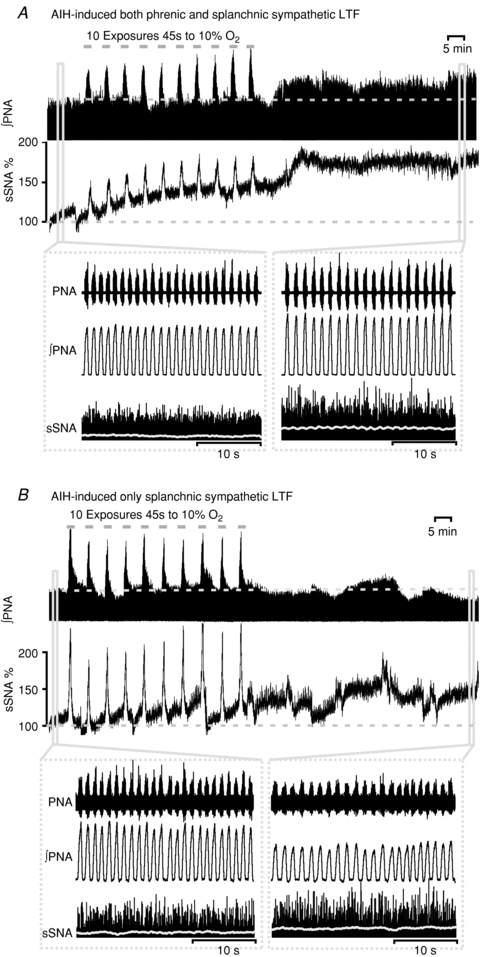
A, acute intermittent hypoxia (10% O2 in N2 45 s × 10, grey bars) with 5 min 100% O2 intervals elicited a significant increase in integrated phrenic nerve activity (phrenic long-term facilitation, pLTF) as long as the increase in normalized splanchnic sympathetic nerve activity (sympathetic long-term facilitation, sLTF) for at least 60 min post-hypoxia. Inset: expanded 30 s traces of phrenic and sympathetic nerve activity at baseline and 60 min following the last hypoxic exposure. Traces from top to bottom represent raw recording of PNA, integrated PNA and rectified sSNA. Normalized integrated sSNA (grey) is superimposed over rectified sSNA. Both of the PNA and sSNA fired more robustly after the LTF is established. Note that the sSNA showed a step-like increase after each hypoxia exposure during the AIH. B, AIH only elicited the sLTF without pLTF. Inset: same as A, but only the sSNA was firing more robustly 60 min after hypoxia. Note that the fluctuation of integrated sSNA is because of the arterial blood sampling and arterial line flush. Arterial blood (see also Table 1) was sampled and assessed for pH, 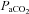 and HCO3− at baseline and 15, 30, 45 and 60 min after the last hypoxic exposure. Also note that during exposure to intermittent hypoxia sSNA gradually increased from the initial hypoxic episode to the final hypoxic episode, referred to as progressive augmentation.
and HCO3− at baseline and 15, 30, 45 and 60 min after the last hypoxic exposure. Also note that during exposure to intermittent hypoxia sSNA gradually increased from the initial hypoxic episode to the final hypoxic episode, referred to as progressive augmentation.
Figure 3. The effect of AIH on phrenic (pLTF) and sympathetic (sLTF) long-term facilitation.

A, average value of integrated and normalized splanchnic sympathetic nerve activity. B, average changes from baseline in peak amplitude of integrated phrenic nerve activity, normalized as a percentage of the baseline (%baseline). C, changes from baseline in minute phrenic nerve activity, normalized as a percentage of the baseline (%baseline). Data are expressed as means ±s.e.m.*P < 0.05, **P < 0.001.
Blood gases
Baseline 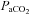 was not significantly different (P = 0.2) between the three groups: ‘pLTF+sLTF’ (40.3 ± 1.2 mmHg, n = 10), ‘only sLTF’ group (39.6 ± 0.5 mmHg, n = 7) and the time control group (41.2 ± 0.4, n = 5). The baseline pH was slightly higher in the ‘only sLTF’ group than in the ‘pLTF+sLTF’ group (7.40 ± 0.01 vs. 7.36 ± 0.01, P < 0.05), but there was no difference in the HCO3− levels between the groups (22.4 ± 0.5 vs. 21.3 ± 0.7 mmol l−1, P = 0.2). The baseline pH levels of all animals were within the physiological range (7.35–7.45) (Table 1). The mean
was not significantly different (P = 0.2) between the three groups: ‘pLTF+sLTF’ (40.3 ± 1.2 mmHg, n = 10), ‘only sLTF’ group (39.6 ± 0.5 mmHg, n = 7) and the time control group (41.2 ± 0.4, n = 5). The baseline pH was slightly higher in the ‘only sLTF’ group than in the ‘pLTF+sLTF’ group (7.40 ± 0.01 vs. 7.36 ± 0.01, P < 0.05), but there was no difference in the HCO3− levels between the groups (22.4 ± 0.5 vs. 21.3 ± 0.7 mmol l−1, P = 0.2). The baseline pH levels of all animals were within the physiological range (7.35–7.45) (Table 1). The mean  values at 15, 30, 45 and 60 min post-hypoxia were not significantly different from baseline (Table 1), indicating a consistent isocapnic condition throughout the experiment.
values at 15, 30, 45 and 60 min post-hypoxia were not significantly different from baseline (Table 1), indicating a consistent isocapnic condition throughout the experiment. 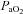 remained above 140 mmHg in all samples examined before and after AIH indicating a consistent hyperoxic condition before and after AIH.
remained above 140 mmHg in all samples examined before and after AIH indicating a consistent hyperoxic condition before and after AIH.
Table 1.
Blood gas values and mean blood pressure before (i.e. baseline) and 15, 30, 45 and 60 min after acute intermittent hypoxia
| Group | pH |
 (mmHg) (mmHg) |
HCO3− (mmol l−1) | MAP (mmHg) | |
|---|---|---|---|---|---|
| Baseline | pLTF+sLTF | 7.36 ± 0.01 | 40.3 ± 1.2 | 21.3 ± 0.7 | 103 ± 7 |
| Only sLTF | 7.40 ± 0.01* | 39.6 ± 0.5 | 22.4 ± 0.5 | 103 ± 7 | |
| Time control | 7.38 ± 0.01 | 41.2 ± 0.4 | 22.3 ± 0.5 | 110 ± 6 | |
| 15 min | pLTF+sLTF | 7.37 ± 0.01 | 40.7 ± 0.9 | 21.6 ± 0.1 | 99 ± 8 |
| Only sLTF | 7.40 ± 0.01 | 39.0 ± 0.7 | 22.3 ± 0.4 | 92 ± 7 | |
| Time control | — | — | — | 109 ± 6 | |
| 30 min | pLTF+sLTF | 7.38 ± 0.01 | 40.0 ± 0.9 | 21.9 ± 0.4 | 99 ± 8 |
| Only sLTF | 7.39 ± 0.01 | 39.4 ± 0.9 | 22.1 ± 0.5 | 100 ± 8 | |
| Time control | 7.37 ± 0.01 | 40.4 ± 0.8 | 21.6 ± 0.5 | 104 ± 6 | |
| 45 min | pLTF+sLTF | 7.37 ± 0.01 | 40.1 ± 1.2 | 21.4 ± 0.4 | 100 ± 8 |
| Only sLTF | 7.38 ± 0.01 | 40.8 ± 0.5 | 22.5 ± 0.5 | 100 ± 10 | |
| Time control | — | — | — | 105 ± 5 | |
| 60 min | pLTF+sLTF | 7.37 ± 0.01 | 40.0 ± 1.2 | 21.1 ± 0.4 | 99 ± 7 |
| Only sLTF | 7.39 ± 0.01 | 39.4 ± 0.6 | 22.1 ± 0.7 | 93 ± 7 | |
| Time control | 7.38 ± 0.01 | 40.0 ± 1.7 | 21.3 ± 0.8 | 102 ± 7 | |
| Δ(60 min – baseline) | pLTF+sLTF | 0.01 | −0.3 | −0.14 | −6 |
| Only sLTF | −0.01 | −0.2 | −0.3 | −10 | |
| Time control | −0.01 | −0.2 | −1.0 | −8 |
Values are means ± s.e.m. There was no difference within or between groups in pH, 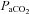 , HCO3− and MAP, except the baseline of pH between the ‘pLTF+sLTF’ and ‘Only sLTF’ groups. Also provided is the calculated mean difference between baseline and 60 min.
, HCO3− and MAP, except the baseline of pH between the ‘pLTF+sLTF’ and ‘Only sLTF’ groups. Also provided is the calculated mean difference between baseline and 60 min.
P < 0.05 compared with ‘pLTF+sLTF’ group.
Hypoxic phrenic responses (HPRs)
The mean HPRs, measured as peak phrenic amplitude changes from baseline during AIH, was not significantly different (P = 0.6) between the ‘pLTF+sLTF’ group (68.0 ± 16.0% change from baseline) and the ‘only sLTF’ group (78.7 ± 8.3%) (Fig. 2). The mean HPRs in peak burst frequency (‘pLTF+sLTF’ group, 64 ± 3 cycles min−1vs. ‘only pLTF’ group, 68 ± 6 cycles min−1) and post-hypoxia frequency decline (‘pLTF+sLTF’ group, 22 ± 2 cycles min−1vs.‘only sLTF’ group, 17 ± 3 cycles min−1) were also not significantly different between groups (both P > 0.2). These data suggested that no HPRs difference was detected between the ‘pLTF+sLTF’ and ‘only sLTF’ groups.
Sympathetic long-term facilitation (sLTF)
Two-way ANOVA showed a significant interaction effect (P < 0.001) between group factor (3 levels: ‘pLTF+sLTF’, ‘only sLTF’ and time control group) and time factor (5 levels: baseline, 15, 30, 45, 60 min post-hypoxia) in sSNA data. The post-hypoxia sSNA was significantly increased in both the AIH groups from baseline at each time point (all P < 0.05; Fig. 3A). In the ‘pLTF+sLTF’ group, the sSNA significantly increased at each time point compared with time control (131 ± 8 vs. 103 ± 2% at 15 min post-hypoxia, P < 0.05; 146 ± 6 vs. 107 ± 4% at 30 min post-hypoxia, P < 0.001; 157 ± 7 vs. 100 ± 6% at 45 min post-hypoxia, P < 0.001; 156 ± 7 vs. 101 ± 7% at 60 min post-hypoxia, P < 0.001, Fig. 3A). Maximum facilitation was generally achieved within 5–10 min following AIH, and subsequently no difference was found between each hypoxic time point until the termination of the experiment after 60 min. In the ‘only sLTF’ group, the sSNA also significantly increased at each time point compared with time control (139 ± 6% at 15 min post-hypoxia, P < 0.01; 145 ± 6% at 30 min post-hypoxia, P < 0.001; 144 ± 7% at 45 min post-hypoxia, P < 0.001; 144 ± 5% at 60 min post-hypoxia, P < 0.001, Fig. 3A).
Phrenic long-term facilitation (pLTF)
Two-way ANOVA showed a significant interaction effect (P < 0.001) between group factor (3 levels: ‘pLTF+sLTF’, ‘only sLTF’ and time control group) and time factor (5 levels: baseline, 15, 30, 45, 60 min post-hypoxia) in phrenic amplitude data. As stated in Methods, the post-hypoxia phrenic amplitude value was not different between the time controls and in the AIH rats where pLTF did not occur; phrenic amplitude did not rise more than 20% from baseline in AIH/non-pLTF rats (Fig. 3B). In the ‘pLTF+sLTF’ group (n = 10), the AIH-induced LTF of phrenic amplitude was significantly elevated at 30, 45 and 60 min, but not at 15 min when compared with the time control group (all P < 0.001, Fig. 3B). In the ‘only sLTF’ group (n = 7), the post-hypoxia phrenic amplitude was not different from time control group at any time point (Fig. 3B). The minute phrenic activity in the ‘pLTF+sLTF’ group was also increased at 30, 45 and 60 min time points compared with the time control group (all P < 0.05, Fig. 3C). No LTF of phrenic burst frequency was induced in the current AIH protocol.
Effects of AIH on cardiorespiratory coupling change
Two-way ANOVA showed significant interaction effects in basal level (P < 0.001) and PI-peaks (P < 0.001) between groups (3 levels: ‘pLTF+sLTF’, ‘only sLTF’ and time control group) and over time (5 levels: baseline, 15, 30, 45, 60 min post-hypoxia). Both the basal level and PI-peak present in sSNA were increased 60 min post-hypoxia regardless of the presence or absence of phrenic LTF (Fig. 4A and B). In the ‘pLTF+sLTF’ group, the basal value and PI-peak increased to 28.0 ± 4.2% and 43.9 ± 6.2% from baseline, respectively, 60 min after the last hypoxic exposure compared with time control (Fig. 4C; both P < 0.001). In the ‘only sLTF’ group, the basal value and PI-peak increased to 23.1 ± 2.60% and 31.4 ± 4.4%, respectively (Fig. 4C; both P < 0.05). In the two-way ANOVA, there was a significant interaction factor (P < 0.05) and time factor (P < 0.001) in amplitude of PI-peak, but there was no significance in group factor (P > 0.05). The increase in amplitude of PI-peak was only observed in ‘pLTF+sLTF’ group at 45 and 60 min post hypoxia (81.4 ± 18.3% and 88.3 ± 16.1%, P < 0.05). These results suggest that the central respiratory drive continued to modulate the sympathetic activity after the establishment of the sympathetic LTF.
Figure 4. Effect of AIH on respiratory modulation of sSNA.
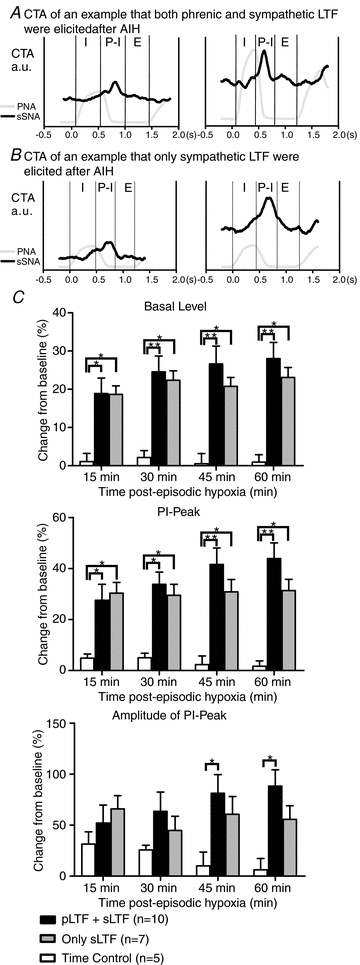
A, sSNA was modulated with respiration before AIH (left), and was lowest during the inspiratory phase (I) and highest during the post-inspiratory phase (PI). Sixty minutes after AIH, both PNA and sSNA had increased, but sSNA remained correlated with respiration, reaching its peak in PI phase. B, sSNA was modulated with respiration before AIH (left) as shown in A. Sixty minutes after AIH, only sSNA had increased, but the modulation stayed the same. C, grouped change in of sSNA basal value, PI peak and PI amplitude (Fig. 1) at 15, 30, 45 and 60 min after the last hypoxic exposure. *P < 0.05 compared with the time control; **P < 0.001 compared with time control.
Effects of AIH on cardiovascular response to hypoxia
After the establishment of sympathetic LTF, a subgroup of animals were exposed again to brief isocapnic hypoxia (10% O2–90% N2 for 45 s) to assess if there was any change in the peripheral chemoreflex change following AIH. In eight animals in the ‘pLTF+sLTF’ group, isocapnic hypoxia increased the amplitude of the sympathetic response by 202% (from 92 ± 22% to 186 ± 38%, P < 0.05; Fig. 5A and B). The hypotensive response during the hypoxia exposure was not changed (−30 ± 6 mmHg to −35 ± 13 mmHg, P > 0.05; Fig. 5A and B). No change in HR was observed (Fig. 5A and B). The sympathetic chemoreflex was also enhanced in the ‘only sLTF’ group by 217% (n = 6; from 57 ± 8% to 125 ± 18%, P < 0.05), but no changes in MAP or HR were found (Fig. 5D and E). The peak amplitude of PNA, but not the peak phrenic discharge frequency (PNF), during hypoxia 60 min after AIH, was increased in the ‘pLTF+sLTF’ group (PNA: 168 ± 16 vs. 217 ± 20% of control baseline, P < 0.001; PNF: 63 ± 3 vs. 64 ± 3 cycles min−1, P = 0.9; Fig. 5C). Similarly in the ‘only sLTF’ group the peak amplitude of PNA, but not the peak phrenic discharge frequency, was increased (PNA: 179 ± 8 vs. 194 ± 7% of control baseline, P < 0.05; PNF: 68 ± 6 vs. 70 ± 6 cycles min−1, P = 0.6; Fig. 5F). No changes in post-hypoxia frequency decline (PHFD) were observed (Fig. 5C and F). The results suggest that the peripheral chemoreflex was facilitated after sympathetic and phrenic LTF.
Figure 5. Response to brief hypoxia is enhanced following establishment of LTF.

A, an experimental recording from an animal which exhibited both phrenic and sympathetic LTF after AIH. Left, the first two 45 s hypoxic exposures during AIH. Right: another two 45 s hypoxic exposures 60 min after AIH. Smoothed and normalized sSNA (grey) is superimposed over rectified sSNA. B, peak change from baseline in MAP, HR and sSNA caused by hypoxia (n = 8) from the animals which exhibited both phrenic and sympathetic LTF after AIH. C, grouped absolute values of peak PNF and peak PNA during hypoxia, and also post-hypoxia frequency decline (PHFD). (n = 8). D, an experimental recording from an animal which only exhibited sympathetic LTF after AIH. E, peak change from baseline in MAP, HR and sSNA caused by hypoxia (n = 6) from the animals which only exhibited sympathetic LTF after AIH. F, grouped absolute values of peak PNF and peak PNA during hypoxia, along with PHFD (n = 6). *P < 0.05, **P < 0.01.
Effects of AIH on modulation of baroreceptors reflex
The MAP–sSNA function curves (n = 8) at 60 min after AIH were not shifted vertically or horizontally compared to baseline, but the range of sSNA was significantly expanded (Fig. 6). The upper plateau, range of sSNA and maximal gain of the MAP–sSNA curves were significantly increased (Fig. 6); however, there were no changes in lower plateau, threshold level, midpoint or saturation level (Table 2). The R2 values for each of the baroreflex function curves were identical before and after AIH (0.97 ± 0.01).
Figure 6. AIH enhances the gain and range of the sympathetic baroreceptor reflex.
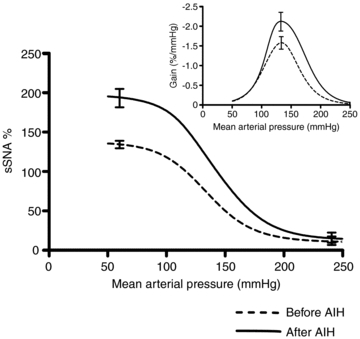
Averaged sigmoidal function curves derived from baroreflex tests of baseline (before AIH, dashed line) and 60 min after the last hypoxic episode (after AIH, straight line). The gain of the curves was determined by first derivative across the full range of MAP. For each curve upper plateau, lower plateau and maximum gain represent averages for 8 experiments. Error bars represent s.e.m. for upper plateau, lower plateau and maximum gain for averaged baroreflex function curves.
Table 2.
Parameters describing baroreflex control of sSNA before and 60 min after AIH
| Lower plateau (%) | Upper plateau (%) | Rang of sSNA (%) | Threshold leve (mmHg) | Midpoint (mmHg) | Saturation level (mmHg) | Max gain (% mmHg−1) | |
|---|---|---|---|---|---|---|---|
| Before | 10 ± 6 | 138 ± 6 | 128 ± 6 | 78 ± 7 | 135 ± 4 | 191 ± 9 | −1.79 ± 0.18 |
| After | 13 ± 7 | 198 ± 12** | 185 ± 10** | 85 ± 6 | 142 ± 5 | 198 ± 9 | −2.60 ± 0.28* |
Values are means ±s.e.m. (n = 8).
P < 0.05
P < 0.01 compared with before AIH exposure.
Discussion
The major novel findings of this study are as follows: (1) acute intermittent hypoxia (ten 45 s hypoxic episodes) induced sympathetic LTF that was independent of phrenic LTF, indicating that the effect on sSNA can be independent of the effect on PNA; (2) after the establishment of sLTF, the PNA related modulation of sSNA is increased, whether or not there is evidence for LTF of PNA; (3) the peripheral chemoreflex (45 s of 10% O2 in nitrogen) responses in both sSNA and PNA were enhanced following AIH, and (4) baroreceptor reflex function was facilitated following AIH.
Technical considerations
The baseline level of PNA and sSNA in this study was established under hyperoxic and normocapnic conditions ( > 140 mmHg,
> 140 mmHg, 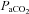 40 ± 2 mmHg) by adjusting ventilation (tidal volume and pump frequency), without determination of apnoeic and recruitment threshold. This is different from the approach used to establish baseline PNA in other studies (Bach & Mitchell, 1996; Dick et al. 2007). In early studies, it was suggested that hypocapnia might restrain the manifestation of LTF (Olson et al. 2001). Consequently, the apnoeic threshold was determined, and carbon dioxide elevated and maintained just above this threshold during and following exposure to AIH to ensure the manifestation of LTF. In a previous meta-analysis, Baker-Herman & Mitchell (2008) found that phrenic amplitude LTF is correlated with phrenic burst amplitude during hypoxia. Since arterial blood gas values and short-term hypoxic responses at baseline were not different between the ‘pLTF+sLTF’ and ‘only sLTF’ groups, it is unlikely that the different ways of determination of baseline activity from other studies will cause the absence of pLTF after AIH. The consistent isocapnic and metabolic condition (Table 1) during all the experiments indicates that no extra central chemoreflex or metabolic factors may cause alteration of central respiratory and presympathetic drive. Since the animals were mechanically ventilated with room air enriched with 100% oxygen, the
40 ± 2 mmHg) by adjusting ventilation (tidal volume and pump frequency), without determination of apnoeic and recruitment threshold. This is different from the approach used to establish baseline PNA in other studies (Bach & Mitchell, 1996; Dick et al. 2007). In early studies, it was suggested that hypocapnia might restrain the manifestation of LTF (Olson et al. 2001). Consequently, the apnoeic threshold was determined, and carbon dioxide elevated and maintained just above this threshold during and following exposure to AIH to ensure the manifestation of LTF. In a previous meta-analysis, Baker-Herman & Mitchell (2008) found that phrenic amplitude LTF is correlated with phrenic burst amplitude during hypoxia. Since arterial blood gas values and short-term hypoxic responses at baseline were not different between the ‘pLTF+sLTF’ and ‘only sLTF’ groups, it is unlikely that the different ways of determination of baseline activity from other studies will cause the absence of pLTF after AIH. The consistent isocapnic and metabolic condition (Table 1) during all the experiments indicates that no extra central chemoreflex or metabolic factors may cause alteration of central respiratory and presympathetic drive. Since the animals were mechanically ventilated with room air enriched with 100% oxygen, the 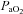 remains stable at well above 140 mmHg except for the hypoxia exposures, which is sufficient to prevent the firing of carotid chemoreceptors (Lopez-Barneo et al. 2001). Therefore, it is unlikely that the sustained sympathetic activation after hypoxic exposure was caused by persistent chemoreceptor stimulation.
remains stable at well above 140 mmHg except for the hypoxia exposures, which is sufficient to prevent the firing of carotid chemoreceptors (Lopez-Barneo et al. 2001). Therefore, it is unlikely that the sustained sympathetic activation after hypoxic exposure was caused by persistent chemoreceptor stimulation.
Blood pressure tended to decrease slightly but not significantly, rather than increasing during the study (Bach & Mitchell, 1996; Dick et al. 2007). This may be due to multiple arterial blood samplings and the long period of recording. Hypotension can result in increased nerve activity because of decreased brain blood flow, increasing CNS tissue 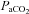 at a fixed level of
at a fixed level of  , indirectly causing the LTF following episodic hypoxia. Therefore, MAP was maintained within 10 mmHg (Table 1) of baseline at all times with continuous i.v. infusion of fluids.
, indirectly causing the LTF following episodic hypoxia. Therefore, MAP was maintained within 10 mmHg (Table 1) of baseline at all times with continuous i.v. infusion of fluids.
Tonic sSNA excitation after AIH
We demonstrated in the present study that 10 episodes of 45 s hypoxia are capable of eliciting a tonic sSNA excitation that is not necessarily dependent on central respiratory drive. Sustained elevation of sympathetic nerve activity induced by short-term exposure to AIH occurs in both animals (Dick et al. 2007) and humans (Cutler et al. 2004; Leuenberger et al. 2005). Here, we used the same AIH protocol and recorded from the same sympathetic bed as Dick et al. (2007), while no sLTF without pLTF was reported in their study. They hypothesized that the sustained increase in sympathetic activity is strongly influenced by respiratory LTF. We speculated that the disparity between our data and theirs is likely to be due to the frequency of adjustment of arterial blood gas. In Dick et al.'s study, the arterial blood gas was only adjusted 60 min post-hypoxia when the data were analysed, so the possibility of poikilocapnia during the period cannot be ruled out. It has been proposed that strengthened respiratory–sympathetic coupling contributes to sympathoexcitation after intermittent hypoxia (Dick et al. 2007; Zoccal et al. 2008). Nevertheless, in the same experimental protocol, the frequency of sLFT is much higher than the pLTF (100%vs. 65% of all the experiments) in the present study. Our findings indicate that sympathetic LTF does not necessarily require augmentation of phrenic motor output; in fact, the augmentation of the respiratory modulation sSNA and the enhanced sympathetic tone appear to be unrelated.
The mechanism for the tonic increase of sympathetic tone is not well known. We speculate that the memory-like effect of sLTF occurs at the level of the barosensitive bulbospinal neurones in the rostral ventrolateral medulla (RVLM) neurones, as well as the caudal ventrolateral medulla (CVLM) neurones, which are responsible for generating sympathetic ‘tone’ (Pilowsky & Goodchild, 2002; Pilowsky et al. 2009), and that it is caused by modulation of central processing of afferent inputs from peripheral chemoreceptors. First of all, chemoreceptor information can reach the presympathetic RVLM neurones without an intervening relay within the respiratory network. The sympathetic chemoreflex originates from the caudal aspect of the nucleus tractus solitarius (NTS) and requires the activation of RVLM barosensitive neurones (Guyenet, 2000). The peripheral chemoreceptor reflex presumably involves a monosynaptic, and or polysynaptic, pathway from the NTS to the RVLM barosensitive neurones, and indirect connections to these cells via the respiratory pattern generator (Guyenet, 2000). Carotid body denervation abrogates the reflex, preventing all of the cardiorespiratory responses and the release of glutamate into the caudal NTS (Mizusawa et al. 1994). In anaesthetized animals, elimination of all central respiratory activity abolished respiratory drive but did not attenuate the sympathetic response to stimulation of peripheral chemoreceptors (Koshiya & Guyenet, 1996). Increases in cervical preganglionic sympathetic nerve activity can be elicited by peripheral chemoreceptor stimulation even when PNA is abolished by hyperventilation to hypocapnia (Huang et al. 1988). Secondly, several Fos studies of presympathetic neurones in the RVLM following hypoxia also support the idea that neurones in the RVLM are important mediators of the central effects of hypoxia. Increased levels of Fos in RVLM were observed after relatively brief (60 min) and longer term (8 h day−1 for 30 days) exposures to hypoxia (Greenberg et al. 1999b). Increased Fos expression in the RVLM was also found after 10 min of electrical stimulation of the carotid sinus nerves (Erickson & Millhorn, 1994). Similar increases also occur following exposure to hypercapnia; however, high levels of inspired CO2 fraction (13%) seem to be required for Fos expression (Sato et al. 1992). Thirdly, although neurones in the RVLM play a role in integrating inputs to provide a final excitatory output to sympathetic preganglionic neurones, the neurones in the CVLM play an opposite but equally important role to inhibit sympathetic discharge. Individual CVLM neurones have basal patterns of respiratory-related activity (Mandel & Schreihofer, 2006). Those that display peak activity during expiration tend to be inhibited during hypoxia (Mandel & Schreihofer, 2009). Thus it is plausible that the tonic increase of SNA can be caused by disinhibition of RVLM neurones resulting from the removal of excitatory input to CVLM neural population. Last but not least, both animal and human studies demonstrated that the enhancement of chemoreflex induced by recurrent hypoxia contributed to the sympathoexcitation. In CIH, bilateral carotid body denervation abolished the increase in SNA and chemoreflex sensitivity (Fletcher et al. 1992). Intermittent hypoxia sensitizes the carotid body chemoreceptors to hypoxia and causes the chemoreceptor afferents to be tonically active even when the blood oxygen concentration is normal (Prabhakar et al. 2005). The present study also showed the enhancement of sympathetic chemoreflex 60 min after AIH (Fig. 5A and D). However, whether carotid body denervation can prevent the effect remains to be determined. Narkiewicz et al. (1999) demonstrated that untreated OSA is associated with selective potentiation of peripheral chemoreflex sensitivity and MSNA decreased in untreated OSA patients during administration of 100% O2. In short, the role of RVLM presympathetic neurones and modification of peripheral chemo-sensitivity requires further investigation, but on the basis of the findings here, is likely to be important.
Effect of AIH on the cardio-respiratory coupling
As expected, respiratory drive still modulated sympathetic tone regardless of whether LTF was induced in PNA or not; however both the resting sympathetic tone and PI-peak were significantly elevated (Fig. 4). We found no statistical difference in the amplitude of the PI-peak between the ‘only sLTF’ and the time control group (Fig. 4C). We speculate that the increase in both the basal level of sSNA and the PI-peak may prevent the increase of amplitude of PI-peak to a significant level. Respiration is well known to modulate SNA (Adrian & Bronk, 1932; Miyawaki et al. 2002b). Using the working-heart–brainstem preparation (WHBP), Zoccal et al. (2008) reported that rats submitted to CIH showed an additional ramping pattern of sympathetic activity coincident with the late expiratory phase and exhibited enhanced/forced expiratory activity by abdominal motor activity. Simms et al. (2009), also using the WHBP, demonstrated that in juvenile spontaneously hypertensive rats at an age prior to the development of hypertension there was already an amplified respiratory modulation of sympathetic activity. Our data support the proposal that an enhanced respiratory–sympathetic coupling in rats has the potential for sympathoexcitation, at least in the ‘pLTF+sLTF’ group of the present study. However, several points need to be mentioned. (1) The mechanism by which phrenic LTF occurs is considered to be strengthening of the postsynaptic membrane of phrenic motoneurones (Mahamed & Mitchell, 2007). It remains to be determined if a similar mechanism is operating at the level of sympathetic preganglionic neurones to cause the increase in sympathetic tone observed here; possible effects on presympathetic neurones in the RVLM also remain to be evaluated. (2) Phrenic LTF can be abrogated by treatments that affect phrenic motoneurones; however, to our knowledge evidence for plasticity of central respiratory output, with the exception of hypoglossal motoneurones, is not available. (3) Although it is reported that neurones in the ventral and dorsal respiratory group and raphé have increased firing rates by carotid chemoreceptor stimulation, no phrenic LTF lasting over 1–2 h was reported in these studies (Morris et al. 2000; Morris et al. 2001). (4) Finally, although there are reports of connections between neurones that generate respiration, the precise nature of the connection between respiratory rhythm and pattern generating neurones, and neurones that regulate sympathetic outflow is unknown (Sun et al. 1997).
Modulation of cardiovascular and respiratory reflex to hypoxia
The other principal finding of our study is that acute intermittent hypoxia produced by 10 episodes of isocapnic hypoxia results in enhanced respiratory and sympathetic neural responses to acute hypoxia, whether pLTF occurs or not. Our data show for the first time that the sympathetic hypoxic chemoreflex is augmented following acute intermittent hypoxia (Fig. 6). These findings agree with previous reports that CIH enhanced sympathetic responses to hypoxia in anaesthetized rats (Greenberg et al. 1999a; Braga et al. 2006), and conscious rats (Huang et al. 2009). Data from human studies strongly supported the idea that short-term intermittent exposure to hypoxia can facilitate the reflex response to hypoxia (Cutler et al. 2004; Leuenberger et al. 2007). The enhancement of phrenic amplitude chemoreflex was also observed. Interestingly, with phrenic LTF, the peak phrenic amplitude during hypoxia increased 129% after AIH (from 168 ± 16 to 217 ± 20% of control baseline, P < 0.001); but if phrenic LTF had not occurred, the peak phrenic amplitude increased by only 108% (from 179 ± 8.3 to 194 ± 7% of control baseline, P < 0.05). It is unclear whether the sensitization of the respiratory chemoreflex is associated with pLTF. No phrenic frequency change during hypoxia was found when comparing any of the groups at baseline to any time after AIH. The finding that no frequency LTF is elicited suggests that AIH has little effect on the central respiratory generator. Our work agrees with previous reports that the reflex response to hypoxia is sensitized in animals after AIH (Fuller, 2005) and CIH (Ling et al. 2001), and in healthy human after AIH (Lusina et al. 2006).
The mechanism underlying the enhanced peripheral chemoreflex responses following intermittent hypoxia is not clear. Two main possibilities should be considered and are likely to represent neural plasticity. First, intermittent hypoxia may lead to altered activity of neurochemical circuits at the level of the brainstem. The caudal NTS (i.e. commissural NTS, SolC) is the central site of termination of afferent neurones whose peripheral axons are found in the carotid body, providing important homeostatic feedback on the  (as well as the CO2/pH) status of the arterial blood (Guyenet, 2000; Lahiri et al. 2006). Plasticity in synaptic transmission in SolC could occur on a short- and/or long-term time scale. Mifflin (1997) demonstrated in intact rats that 2 min of 100–300 Hz stimulation of the carotid sinus, aortic or vagus nerve augmented monosynaptic and polysynaptic EPSPs and action potential discharge. De Paula et al. (2007) also demonstrated that exposure to CIH for 7 days alters the responses of NTS neurones to exogenous application of the inotropic excitatory amino acid agonists AMPA and NMDA. The changes occur in NTS neurones receiving arterial chemoreceptor afferent inputs and ATP-sensitive potassium channels may play an important role in this long-term potentiation (Zhang et al. 2008). Secondly, such an effect could be due to functional alterations in arterial chemoreceptors. Peng et al. (2003) reported that following (prolonged) intermittent but not continuous hypoxia the discharge frequency of chemoreceptor afferents is increased, an effect that is mediated by reactive oxygen species formed during recurrent cycles of hypoxia–reoxygenation (Prabhakar et al. 2005). However, most of the studies were conducted in prolonged intermittent hypoxic conditions. It is not clear whether the functional or histological changes may already exist at an early stage, following intermittent hypoxia.
(as well as the CO2/pH) status of the arterial blood (Guyenet, 2000; Lahiri et al. 2006). Plasticity in synaptic transmission in SolC could occur on a short- and/or long-term time scale. Mifflin (1997) demonstrated in intact rats that 2 min of 100–300 Hz stimulation of the carotid sinus, aortic or vagus nerve augmented monosynaptic and polysynaptic EPSPs and action potential discharge. De Paula et al. (2007) also demonstrated that exposure to CIH for 7 days alters the responses of NTS neurones to exogenous application of the inotropic excitatory amino acid agonists AMPA and NMDA. The changes occur in NTS neurones receiving arterial chemoreceptor afferent inputs and ATP-sensitive potassium channels may play an important role in this long-term potentiation (Zhang et al. 2008). Secondly, such an effect could be due to functional alterations in arterial chemoreceptors. Peng et al. (2003) reported that following (prolonged) intermittent but not continuous hypoxia the discharge frequency of chemoreceptor afferents is increased, an effect that is mediated by reactive oxygen species formed during recurrent cycles of hypoxia–reoxygenation (Prabhakar et al. 2005). However, most of the studies were conducted in prolonged intermittent hypoxic conditions. It is not clear whether the functional or histological changes may already exist at an early stage, following intermittent hypoxia.
Phrenic post-hypoxia frequency decline (PHFD) was attenuated after repeated exposure to brief periods of hypoxia, which were separated by 30 min or more (Bach et al. 1999), while unexpectedly no changes in PHFD were determined in any animals exposed to AIH. This may be due to different durations and degrees of hypoxic exposure. Ilyinsky et al. (2003) demonstrated that CIH abolished the PHFD, while the time of increased phrenic burst frequency and amplitude after hypoxia was prolonged because the inhibition of PHFD. The abolition of PHFD was also reported by Ling et al. (2001). The mechanism of PHFD is still unclear. It is reported that the actions of inhibitory α2 receptors on both brainstem respiratory neurones (Bach et al. 1999) and pontine respiratory group participate in the response (Dick & Coles, 2000). It still remains to be determined if AIH has any functional or histological effects on brainstem or pontine respiratory neurones.
Modulation of baroreflex function after AIH
Baroreflex control of cardiovagal and sympathetic outflow is reported to be impaired in OSA (Bonsignore et al. 2002). Moreover, in healthy humans, baroreflex function is impaired acutely by exposure to hypoxia (Cooper et al. 2004). Our present results suggest that the baroreflex function is sensitized, due to the increase of maximal gain without change of the working range. Our data are consistent with the findings of Soukhova-O’Hare et al. (2006), who used a similar experimental approach, and demonstrated that the gain of the baroreflex sympatho-excitatory response increased approximately twofold after AIH, while the gain of sympatho-inhibitory responses to SNP was not affected by AIH. Interestingly, the change of gain in baroreflex function could be abolished by neonatal CIH exposure. Soukhova-O’Hare et al. (2006) concluded that postnatal intermittent hypoxia causes a long-lasting impairment in chemoreceptor and baroreceptor control of SNA. Therefore, we speculate that the facilitation of the sympathetic baroreflex acts to compensate for the sympathoexcitation that follows AIH. This idea is supported by the finding in humans that short-term exposure to hypoxic apnoea does not impair cardiovagal or sympathetic baroreflex sensitivity (Monahan et al. 2006).
The mechanism leading to sensitization of the sympathetic baroreflex function is unknown. We postulate that the synthesis of proteins induced by AIH is essential to cause the change. Phrenic long-term facilitation requires protein synthesis at the spinal level (Baker-Herman & Mitchell, 2002). Intrathecal blockade of brain-derived neurotrophic factor (BDNF) synthesis prevents LTF evoked in PNA (Baker-Herman et al. 2004). It is reported that BDNF is a target-derived survival factor for arterial baroreceptors’ primary sensory neurones (Martin et al. 2009). If AIH also evokes synthesis and release of BDNF in the brainstem, then BDNF may play a similar role at multiple levels of the neuraxis and mediate increased short-term between raphé neurones. Further studies are required to determine if BDNF or other neurotrophins play a role in the modification of baroreflex function during the establishment of AIH.
In conclusion, we find that acute intermittent hypoxia induces both phrenic and sympathetic long-term facilitation and that an additional effect on sympathetic tone is also present. Cardio-respiratory coupling may be a potential mechanism contributing to the enhancement of SNA, but the enhancement of the chemoreflex, especially the peripheral chemoreflex, is likely to play a more important role in the sympathoexcitation. Further work is needed to elucidate the mechanisms and anatomical substrates for this effect. Our findings provide important insights into the acute adaptations of chemoreflex control systems of both the respiratory and sympathetic responses to AIH, and, perhaps more importantly, the early pathophysiology of OSA. Furthermore, our findings reveal an increase in baroreflex sensitivity following the increase in resting sympathetic tone.
Acknowledgments
T. Xing is supported by a Macquarie Research Excellence Scholarship. This work was supported by the National Health and Medical Research Council of Australia (457069, 457080 and 604002), Garnett Passe, and Rodney Williams Memorial Foundation and Macquarie University. The Authors wish to thank Drs Q-J. Sun and Simon McMullan for their assistance and advice.
Glossary
Abbreviations
- AIH
acute intermittent hypoxia
- CIH
chronic intermittent hypoxia
- CVLM
caudal ventrolateral medulla
- LTF
long-term facilitation
- pLTF
phrenic long-term facilitation
- sLTF
sympathetic long-term facilitation
- MSNA
muscle sympathetic nerve activity
- NTS
nucleus tractus solitarius
- OSA
obstructive sleep apnoea
- PHFD
post-hypoxia frequency decline
- PNA
phrenic nerve activity
- RVLM
rostral ventrolateral medulla
- SNA
sympathetic nerve activity
- sSNA
splanchnic sympathetic nerve activity
Author contributions
Dr T. Xing and P.M. Pilowsky conceived the experiment, designed the protocol and prepared the manuscript. Dr T. Xing conducted all the experiments and data analysis. Dr P.M. Pilowsky was the principle investigator in the study. Both authors approved the final version for publication. This study was conducted in Australian School of Advanced Medicine, Macquarie University, Sydney, Australia.
Supplemental material
Supplementary figure
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Abbott SBG, Pilowsky PM. Galanin microinjection into rostral ventrolateral medulla of the rat is hypotensive and attenuates sympathetic chemoreflex. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1019–1026. doi: 10.1152/ajpregu.90885.2008. [DOI] [PubMed] [Google Scholar]
- Adrian ED, Bronk DW. Discharges in mammalian sympathetic nerves. J Physiol. 1932;74:115–133. doi: 10.1113/jphysiol.1932.sp002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach KB, Kinkead R, Mitchell GS. Post-hypoxia frequency decline in rats: Sensitivity to repeated hypoxia and α2-adrenoreceptor antagonism. Brain Res. 1999;817:25–33. doi: 10.1016/s0006-8993(98)01181-0. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Determinants of frequency long-term facilitation following acute intermittent hypoxia in vagotomized rats. Respir Physiol Neurobiol. 2008;162:8–17. doi: 10.1016/j.resp.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsignore MR, Parati G, Insalaco G, Marrone O, Castiglioni P, Romano S, Di Rienzo M, Mancia G, Bonsignore G. Continuous positive airway pressure treatment improves baroreflex control of heart rate during sleep in severe obstructive sleep apnea syndrome. Am J Respir Crit Care Med. 2002;166:279–286. doi: 10.1164/rccm.2107117. [DOI] [PubMed] [Google Scholar]
- Braga VA, Soriano RN, Machado BH. Sympathoexcitatory response to peripheral chemoreflex activation is enhanced in juvenile rats exposed to chronic intermittent hypoxia. Exp Physiol. 2006;91:1025–1031. doi: 10.1113/expphysiol.2006.034868. [DOI] [PubMed] [Google Scholar]
- Carlson JT, Hedner JA, Sellgren J, Elam M, Wallin BG. Depressed baroreflex sensitivity in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 1996;154:1490–1496. doi: 10.1164/ajrccm.154.5.8912770. [DOI] [PubMed] [Google Scholar]
- Cooper VL, Bowker CM, Pearson SB, Elliott MW, Hainsworth R. Effects of simulated obstructive sleep apnoea on the human carotid baroreceptor-vascular resistance reflex. J Physiol. 2004;557:1055–1065. doi: 10.1113/jphysiol.2004.062513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler MJ, Swift NM, Keller DM, Wasmund WL, Burk JR, Smith ML. Periods of intermittent hypoxic apnea can alter chemoreflex control of sympathetic nerve activity in humans. Am J Physiol Heart Circ Physiol. 2004;287:H2054–2060. doi: 10.1152/ajpheart.00377.2004. [DOI] [PubMed] [Google Scholar]
- De Paula PM, Tolstykh G, Mifflin S. Chronic intermittent hypoxia alters NMDA and AMPA-evoked currents in NTS neurons receiving carotid body chemoreceptor inputs. Am J Physiol Regul Integr Comp Physiol. 2007;292:R2259–2265. doi: 10.1152/ajpregu.00760.2006. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. 2010;90:47–112. doi: 10.1152/physrev.00043.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick TE, Coles SK. Ventrolateral pons mediates short-term depression of respiratory frequency after brief hypoxia. Respir Physiol. 2000;121:87–100. doi: 10.1016/s0034-5687(00)00121-3. [DOI] [PubMed] [Google Scholar]
- Dick TE, Hsieh YH, Wang N, Prabhakar N. Acute intermittent hypoxia increases both phrenic and sympathetic nerve activities in the rat. Exp Physiol. 2007;92:87–97. doi: 10.1113/expphysiol.2006.035758. [DOI] [PubMed] [Google Scholar]
- Erickson JT, Millhorn DE. Hypoxia and electrical stimulation of the carotid sinus nerve induce Fos-like immunoreactivity within catecholaminergic and serotoninergic neurons of the rat brainstem. J Comp Neurol. 1994;348:161–182. doi: 10.1002/cne.903480202. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Behm R, Miller CC, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol. 1992;72:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- Fuller DD. Episodic hypoxia induces long-term facilitation of neural drive to tongue protrudor and retractor muscles. J Appl Physiol. 2005;98:1761–1767. doi: 10.1152/japplphysiol.01142.2004. [DOI] [PubMed] [Google Scholar]
- Greenberg HE, Sica A, Batson D, Scharf SM. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol. 1999a;86:298–305. doi: 10.1152/jappl.1999.86.1.298. [DOI] [PubMed] [Google Scholar]
- Greenberg HE, Sica AL, Scharf SM, Ruggiero DA. Expression of c-fos in the rat brainstem after chronic intermittent hypoxia. Brain Res. 1999b;816:638–645. doi: 10.1016/s0006-8993(98)01222-0. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. Neural structures that mediate sympathoexcitation during hypoxia. Respir Physiol. 2000;121:147–162. doi: 10.1016/s0034-5687(00)00125-0. [DOI] [PubMed] [Google Scholar]
- Huang J, Lusina S, Xie T, Ji E, Xiang S, Liu Y, Weiss JW. Sympathetic response to chemostimulation in conscious rats exposed to chronic intermittent hypoxia. Respir Physiol Neurobiol. 2009;166:102–106. doi: 10.1016/j.resp.2009.02.010. [DOI] [PubMed] [Google Scholar]
- Huang W, Lahiri S, Mokashi A, Sherpa AK. Relationship between sympathetic and phrenic nerve responses to peripheral chemoreflex in the cat. J Auton Nerv Sys. 1988;25:95–105. doi: 10.1016/0165-1838(88)90014-8. [DOI] [PubMed] [Google Scholar]
- Ilyinsky O, Tolstykh G, Mifflin S. Chronic hypoxia abolishes posthypoxia frequency decline in the anesthetized rat. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1322–1330. doi: 10.1152/ajpregu.00033.2003. [DOI] [PubMed] [Google Scholar]
- Imadojemu VA, Mawji Z, Kunselman A, Gray KS, Hogeman CS, Leuenberger UA. Sympathetic chemoreflex responses in obstructive sleep apnea and effects of continuous positive airway pressure therapy. Chest. 2007;131:1406–1413. doi: 10.1378/chest.06-2580. [DOI] [PubMed] [Google Scholar]
- Kent BB, Drane JW, Blumenstein B, Manning JW. A mathematical model to assess changes in the baroreceptor reflex. Cardiology. 1972;57:295–310. doi: 10.1159/000169528. [DOI] [PubMed] [Google Scholar]
- Koshiya N, Guyenet PG. Tonic sympathetic chemoreflex after blockade of respiratory rhythmogenesis in the rat. J Physiol. 1996;491:859–869. doi: 10.1113/jphysiol.1996.sp021263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S, Roy A, Baby SM, Hoshi T, Semenza GL, Prabhakar NR. Oxygen sensing in the body. Prog Biophys Mol Biol. 2006;91:249–286. doi: 10.1016/j.pbiomolbio.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Lai CJ, Yang CCH, Hsu YY, Lin YN, Kuo TBJ. Enhanced sympathetic outflow and decreased baroreflex sensitivity are associated with intermittent hypoxia-induced systemic hypertension in conscious rats. J Appl Physiol. 2006;100:1974–1982. doi: 10.1152/japplphysiol.01051.2005. [DOI] [PubMed] [Google Scholar]
- Leuenberger UA, Brubaker D, Quraishi S, Hogeman CS, Imadojemu VA, Gray KS. Effects of intermittent hypoxia on sympathetic activity and blood pressure in humans. Auton Neurosci. 2005;121:87–93. doi: 10.1016/j.autneu.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Leuenberger UA, Hogeman CS, Quraishi S, Linton-Frazier L, Gray KS. Short-term intermittent hypoxia enhances sympathetic responses to continuous hypoxia in humans. J Appl Physiol. 2007;103:835–842. doi: 10.1152/japplphysiol.00036.2007. [DOI] [PubMed] [Google Scholar]
- Ling L, Fuller DD, Bach KB, Kinkead R, Olson EB, Jr, Mitchell GS. Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. J Neurosci. 2001;21:5381–5388. doi: 10.1523/JNEUROSCI.21-14-05381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Ortega-Saenz P. Cellular mechanism of oxygen sensing. Annu Rev Physiol. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- Lusina SJC, Kennedy PM, Inglis JT, McKenzie DC, Ayas NT, Sheel AW. Long-term intermittent hypoxia increases sympathetic activity and chemosensitivity during acute hypoxia in humans. J Physiol. 2006;575:961–970. doi: 10.1113/jphysiol.2006.114660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol. 2007;92:27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- Mandel DA, Schreihofer AM. Central respiratory modulation of barosensitive neurones in rat caudal ventrolateral medulla. J Physiol. 2006;572:881–896. doi: 10.1113/jphysiol.2005.103622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel DA, Schreihofer AM. Modulation of the sympathetic response to acute hypoxia by the caudal ventrolateral medulla in rats. J Physiol. 2009;587:461–475. doi: 10.1113/jphysiol.2008.161760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JL, Jenkins VK, Hsieh HY, Balkowiec A. Brain-derived neurotrophic factor in arterial baroreceptor pathways: Implications for activity-dependent plasticity at baroafferent synapses. J Neurochem. 2009;108:450–464. doi: 10.1111/j.1471-4159.2008.05781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifflin SW. Short-term potentiation of carotid sinus nerve inputs to neurons in the nucleus of the solitary tract. Respir Physiol. 1997;110:229–236. doi: 10.1016/s0034-5687(97)00087-x. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Invited review: Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol. 2003;94:358–374. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- Miyawaki T, Goodchild AK, Pilowsky PM. Activation of mu-opioid receptors in rat ventrolateral medulla selectively blocks baroreceptor reflexes while activation of delta opioid receptors blocks somato-sympathetic reflexes. Neuroscience. 2002a;109:133–144. doi: 10.1016/s0306-4522(01)00439-0. [DOI] [PubMed] [Google Scholar]
- Miyawaki T, Goodchild AK, Pilowsky PM. Evidence for a tonic GABA-ergic inhibition of excitatory respiratory-related afferents to presympathetic neurons in the rostral ventrolateral medulla. Brain Res. 2002b;924:56–62. doi: 10.1016/s0006-8993(01)03025-6. [DOI] [PubMed] [Google Scholar]
- Mizusawa A, Ogawa H, Kikuchi Y, Hida W, Kurosawa H, Okabe S, Takishima T, Shirato K. In vivo release of glutamate in nucleus tractus solitarii of the rat during hypoxia. J Physiol. 1994;478:55–66. doi: 10.1113/jphysiol.1994.sp020229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan KD, Leuenberger UA, Ray CA. Effect of repetitive hypoxic apnoeas on baroreflex function in humans. J Physiol. 2006;574:605–613. doi: 10.1113/jphysiol.2006.108977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KF, Baekey DM, Shannon R, Lindsey BG. Respiratory neural activity during long-term facilitation. Respir Physiol. 2000;121:119–133. doi: 10.1016/s0034-5687(00)00123-7. [DOI] [PubMed] [Google Scholar]
- Morris KF, Shannon R, Lindsey BG. Changes in cat medullary neurone firing rates and synchrony following induction of respiratory long-term facilitation. J Physiol. 2001;532:483–497. doi: 10.1111/j.1469-7793.2001.0483f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkiewicz K, Van De Borne PJH, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- Olson EB, Jr, Bohne CJ, Dwinell MR, Podolsky A, Vidruk EH, Fuller DD, Powell FL, Mitchel GS. Ventilatory long-term facilitation in unanesthetized rats. J Appl Physiol. 2001;91:709–716. doi: 10.1152/jappl.2001.91.2.709. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: Implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilowsky PM, Goodchild AK. Baroreceptor reflex pathways and neurotransmitters: 10 years on. J Hypertens. 2002;20:1675–1688. doi: 10.1097/00004872-200209000-00002. [DOI] [PubMed] [Google Scholar]
- Pilowsky PM, Lung MS, Spirovski D, McMullan S. Differential regulation of the central neural cardiorespiratory system by metabotropic neurotransmitters. Philos Trans R Soc Lond B Biol Sci. 2009;364:2537–2552. doi: 10.1098/rstb.2009.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Jacono FJ, Kumar GK, Dick TE. Cardiovascular alterations by chronic intermittent hypoxia: Importance of carotid body chemoreflexes. Clin Exp Pharmacol Physiol. 2005;32:447–449. doi: 10.1111/j.1440-1681.2005.04209.x. [DOI] [PubMed] [Google Scholar]
- Sato M, Severinghaus JW, Basbaum AI. Medullary CO2 chemoreceptor neuron identification by c-fos immunocytochemistry. J Appl Physiol. 1992;73:96–100. doi: 10.1152/jappl.1992.73.1.96. [DOI] [PubMed] [Google Scholar]
- Simms AE, Paton JFR, Pickering AE, Allen AM. Amplified respiratory-sympathetic coupling in the spontaneously hypertensive rat: Does it contribute to hypertension? J Physiol. 2009;587:597–610. doi: 10.1113/jphysiol.2008.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukhova-O’Hare GK, Roberts AM, Gozal D. Impaired control of renal sympathetic nerve activity following neonatal intermittent hypoxia in rats. Neurosci Lett. 2006;399:181–185. doi: 10.1016/j.neulet.2006.01.054. [DOI] [PubMed] [Google Scholar]
- Sun QJ, Minson J, Llewellyn-Smith IJ, Arnolda L, Chalmers J, Pilowsky P. Botzinger neurons project towards bulbospinal neurons in the rostral ventrolateral medulla of the rat. J Comp Neurol. 1997;388:23–31. doi: 10.1002/(sici)1096-9861(19971110)388:1<23::aid-cne2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Vitela M, Herrera-Rosales M, Haywood JR, Mifflin SW. Baroreflex regulation of renal sympathetic nerve activity and heart rate in renal wrap hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R856–862. doi: 10.1152/ajpregu.00620.2004. [DOI] [PubMed] [Google Scholar]
- Zhang W, Carreño FR, Cunningham JT, Mifflin SW. Chronic sustained and intermittent hypoxia reduce function of ATP-sensitive potassium channels in nucleus of the solitary tract. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1555–1562. doi: 10.1152/ajpregu.90390.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccal DB, Bonagamba LGH, Paton JFR, Machado BH. Sympathetic-mediated hypertension of awake juvenile rats submitted to chronic intermittent hypoxia is not linked to baroreflex dysfunction. Exp Physiol. 2009;94:972–983. doi: 10.1113/expphysiol.2009.048306. [DOI] [PubMed] [Google Scholar]
- Zoccal DB, Simms AE, Bonagamba LGH, Braga VA, Pickering AE, Paton JFR, Machado BH. Increased sympathetic outflow in juvenile rats submitted to chronic intermittent hypoxia correlates with enhanced expiratory activity. J Physiol. 2008;586:3253–3265. doi: 10.1113/jphysiol.2008.154187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.