

Abstract
Approximately 25–30% of the hemophilia A patients develop inhibitory antibodies against Factor VIII (FVIII) following protein-replacement therapy. This problem is also thought to occur following gene-replacement therapy. Recently, many approaches have been investigated to modulate FVIII-specific immune responses in either protein-replacement or gene therapy hemophilia A mouse models. Several promising protocols have been demonstrated to successfully prevent or modulate the formation of anti-FVIII antibodies, including methods to manipulate antigen presentation, development of less immunogenic FVIII proteins, or formulations or gene therapy protocols to evade immune responses, as well as immunomodulation strategies to target either T- and/or B-cell responses. Most of these successful protocols involve the induction of activated Treg cells to create a regulatory immune environment during tolerance induction. Innovative strategies to overcome pre-existing anti-FVIII immune responses and induce long-term tolerance in primed subjects still need to be developed.
Keywords: antibody, Factor VIII, gene therapy, hemophilia, immunomodulation, immunosuppressive regimen, inhibitor, protein-replacement therapy, tolerance induction, Treg
Hemophilia A is caused by a deficiency of blood-clotting Factor VIII (FVIII). FVIII participates as a coFin a critical initiation reaction of intrinsic Factor X activation in the coagulation cascade [1]. Hemophilia A is an X-linked recessive bleeding disorder and affects approximately one in 5000 males. Patients with FVIII deficiency due to FVIII mutations have lifelong bleeding tendencies, with variable severities. Affected individuals with more severe hemophilia are at risk of spontaneous bleeding episodes into the joints or muscles and, at times, into organs, including the brain. These episodes can be life-threatening, or lead to chronic problems, such as severe arthritis. Acute bleeding episodes treated with purified FVIII or IX concentrates from human plasma have often led to HIV or hepatitis B and C transmission. Recombinant factor concentrates are also now available. Owing to the short half-life of these factors, repeated infusions are required for major bleeding episodes. Even mildly deficient patients are at risk of life-threatening bleeding following moderately severe trauma.
A major ongoing problem in the clinical treatment of hemophilia A using factor-replacement therapy is a very high frequency in the formation of inhibitory antibodies against FVIII. This problem is also predicted to occur following strategies currently aimed at targeted genetic correction of this disease (and clearly occurs in animal models). Approximately 25% of hemophilia A patients develops antibodies after repeated infusion of FVIII protein [2,3]. Development of inhibitory antibodies significantly increases morbidity and lowers the quality of life of hemophilia A patients, and treatment of these patients is costly and very challenging [4-6]. At present, relatively few predictive criteria exist to identify the individual patients most likely to develop antibodies. The development of inhibitory antibodies correlates partially with the type of mutation within the FVIII gene, the mode of protein administration, the immunological state of the individual at the time of the infusion and, possibly, specific MHC class II types [7]. These observations indicate that factors influencing antibody formation are probably complex and incompletely defined.
Currently, protein-replacement therapy to treat hemophilia patients is very costly, and repeated infusions are required for both acute and prophylactic treatment. In addition, because of the risk of bleeding and the fact that their disease results from a single factor deficiency that can potentially be treated by a single gene addition or correction, hemophilic patients have been considered as an excellent candidate population for developing gene therapy approaches. Gene therapy has been explored as a promising treatment for hemophilia A through Phase I clinical trials [8-10]. However, to date, only transient, low-level FVIII protein expression has been achieved, owing to the development of immune responses against FVIII and/or associated gene-transfer vectors. In many preclinical experiments using immunocompetent hemophilia A mice and dogs, strong immune responses against FVIII following gene transfer have completely inhibited circulating FVIII activity and, thus, subverted the effect of gene therapy. Similar to immune responses induced by protein-replacement therapy, transgene-induced immune responses are primarily humoral responses. However, cytotoxic T lymphocytes (CTLs) can be induced in the presence of other strong signals, such as viral vector components, in the context of gene therapy applications. Administration of an E1/E3-deleted adenoviral vector encoding FVIII activated both cytotoxic and humoral responses in hemophilia mice [11,12]. However, infusion of adenoassociated vectors (AAV) carrying FVIII into mouse livers induced only high-titer anti-FVIII antibodies [13]. Inhibitory antibodies were also observed following gene transfer of a vesicular stomatitis virus (VSV)-G pseudo-typed, oncoretroviral vector encoding human B-domain deleted (BDD) FVIII [14,15], and a feline immunodeficiency virus (FIV)-based lentiviral-hFVIII vector [16] into hemophilia A mice. In a more recent case, naked gene transfer of FVIII into the liver using a hydrodynamics-based delivery method achieved initial high levels of hFVIII [17]. However, a robust humoral immune response against FVIII occurred 2 weeks post-treatment, and led to complete inhibition of circulating FVIII activity [18]. No evidence is observed for the induction of CTLs.
The hemophilia A murine model has been successfully used to mimic the immune response in hemophilia A patients treated with repeated infusions of FVIII protein [19]. These mice are genetically deficient in FVIII (through targeted disruption of exon 16 [or 17] of the FVIII gene). This strain expresses a nonfunctional, heavy-chain FVIII protein, with undetectable (<1%) FVIII activity of the normal protein product in the plasma [12]. The phenotype of these animals is similar to that of patients with severe hemophilia A, including significantly impaired hemostasis, severe bleeding after minor injuries and spontaneous bleeding. Anti-FVIII antibodies are reproducibly generated after multiple injections of hFVIII protein into hemophilia A mice [20,21]. Furthermore, as mentioned previously, nonviral gene transfer of a FVIII plasmid into hemophilia A mice induces strong humoral responses through predominantly Th2 signals [18]. The plasmid-treated mice with persistent, high-level inhibitory antibody against FVIII enables the analysis of immune responses specifically against neoantigen in the absence of other immunostimulatory effects of the delivery system. It represents a unique and useful model system for testing various immunomodulation strategies.
Immune tolerance induction protocols
Immune tolerance induction (ITI) protocols have been utilized since the 1970s in an effort to tolerize hemophilia patients to infused hFVIII. The strategy can not only eliminate anti-FVIII inhibitory antibodies, but also induce FVIII-specific tolerance in patients. However, a third of the patients that have undergone ITI failed to generate tolerance to FVIII. The success rate is dependent on the pretreatment and peak inhibitor titers of the patient, and possibly other factors, such as the type of FVIII used in the infusion. The protocols require long-term and repetitive infusions of FVIII, which are both very costly and practically challenging.
Although little is known about the mechanism how tolerance to FVIII is induced following successful ITI in hemophilia patients, studies in animal models demonstrated that ITI may inhibit the restimulation of FVIII-specific memory B cells, and their differentiation into antibody-secreting plasma cells, as an early event in the process of inducing tolerance [22]. The eradication of memory B cells may generate a deficiency of effective antigen-presenting cells required for the re-stimulation of FVIII-specific effector T cells, which may lead to the induction of Treg cells. This will create a regulatory environment to facilitate tolerance induction in the presence of antigen. ITI protocols have also been combined with long-term use of immunosuppressive agents [23,24] or immunoglobulin. Intravenous immunoglobulin was recently demonstrated to expand CD4+CD25+ Treg cells [25]. It has also been recently shown that natural Treg cells can be activated by IgG Fc-derived peptides (Tregitopes) [26]. The positive effect of tolerance induction by intravenous imunoglobulin may be closely related to the induction and activation of Treg cells.
New strategies of immunomodulation of FVIII inhibitors
Recently, many approaches have been investigated to modulate FVIII-specific immune responses in either protein-replacement or gene therapy hemophilia A mouse models.
Antigen presentation
Oral or nasal administration of FVIII domain or peptides
Using the similar concept as ITI by high-dose FVIII in human patients, efforts have been made to present antigens for a prolonged period using more convenient and economic methods in hemophilia A mouse models. New strategies include oral feeding of FVIII-C2 domain [27], nasal administration of FVIII peptide(s) [27], and infusion of lipopolysacchride (LPS)-activated B-cell blasts, transduced with a fusion IgG containing the C2 or A2 domains of FVIII (see ‘Immunosuppressive therapies targeting B cells’ section) [28]. Mucosal administration of the FVIII-C2 peptide decreased the titer of anti-FVIII-C2 inhibitory antibodies following challenge with FVIII-C2, and this tolerance can be adoptively transferred to naive mice. However, when the treated mice were challenged with the whole FVIII protein, the FVIII-C2-specific antibodies were decreased, but tolerance was not established towards FVIII protein.
Immature dendritic cells
Lillicrap and colleagues recently investigated FVIII-specific tolerance induction using canine FVIII-pulsed immature dendritic cells (cFVIII-iDCs) isolated from the bone marrow of hemophilia A mice, and incubated without (cFVIII-iDCs) or with (Andro-cFVIII-iDCs) the NF-κB pathway-blocking compound andrographolide [29]. The mice infused with cFVIII-iDCs and Andro-cFVIII-iDCs and challenged with cFVIII had 25 and 40% reduction of FVIII inhibitory antibody titers for long periods of time. Studies of cytokine release and T-cell phenotypes indicate that the mechanisms responsible for reducing immunologic responsiveness to cFVIII involve the expansion of CD4+Foxp3+ Treg cells after infusion of cFVIII-iDC, as well as an increase in the immunosuppressive cytokines IL-10 and TGF-β levels following infusion of Andro-cFVIII-iDCs. A similar study using cFVIII-iDCs also showed regulation of FVIII-specific immune responses in the presence of granulocyte–macrophage colony-stimulating factor and TGF-β in hemophilia A mice [30]. In this study, second challenge with recombinant FVIII produced no increase in anti-F.VIII antibody response. The immunomodulation effect was associated with high IL-10 and low IL-2 levels, splenic T-cell hyporesponsiveness to FVIII, and expansion of CD4+Foxp3high Treg cells.
Apoptotic fibroblast cells
Delivery of apoptotic syngeneic fibroblasts modified by FVIII expressing foamy vectors was shown to suppress FVIII inhibitor formation in both naive and primed hemophilia A mice [31]. Infusion of the FVIII-modified cells into hemophilia A mice with concomitant FVIII infusion reduced inhibitor responses to FVIII compared with much less suppression by infusion of the unmodified cells mediated by nonspecific anti-inflammatory signals. Secondary transfer of CD4+ T cells from the mice treated with FVIII-modified apoptotic cells also suppressed the immune response to FVIII in the recipient mice. In addition, CD4+CD25+ Treg cells isolated from the mice treated with apoptotic cells suppressed the proliferation of responder T cells stimulated by FVIII. This protocol may be useful for treating patients who develop high-titer inhibitors.
Evasion of immune responses to FVIII
Preparation of less immunogenic FVIII proteins or formulations
Immunogenicity of various preparations of FVIII protein has been targets of many studies. It is still under debate whether plasma-derived FVIII may be less immunogenic than recombinant FVIII [32,33]. It is hypothesized that, since plasma-derived FVIII contains many factors, including von Willebrand factor and other proteins in the preparation, these factors could compete with FVIII in the endocytosis pathway to antigen-presenting cells, or exert direct effect in altering the immune activation pathway, such as TGF-β. These influences may lead to reduced immunogenicity of plasma-derived FVIII.
Recently, efforts have also been made to generate longer acting, more active and/or less immunogenic FVIII formulation or variant molecules. It would, of course, be preferable if all three goals can be achieved in one variant protein. If not, at least any modifications on the FVIII molecule will not compromise any of the three properties (e.g., a longer acting or more active FVIII should not increase the immunogenicity of FVIII). The potential advantages and disadvantages of some of the modifications, including the addition of polyethylene glycol (PEG) polymers or polysialic acids or PEG-modified liposomes, and modification of FVIII molecules (FVIII variants), were reviewed by Saenko and Pipe [34]. Furthermore, a BDD-FVIII/N6 variant was found to produce lower anti-FVIII antibody titers than BDD-FVIII [35] when delivered by either an adenoviral vector [36] or a plasmid vector [37]. Modifications of immunodominant T-cell epitopes are also being investigated to reduce the immunogenicity of FVIII, while maintaining its full functional activity [38].
Neonatal gene transfer
Gene transfer has been utilized to induce tolerance by persistent transgene expression mimicking the repeated infusions in ITI protocols. However, although immune tolerance to Factor IX (FIX) has been successfully induced in adult animals by hepatic gene transfer using AAVs [39], immunosuppressive regimens are needed to ensure long-term expression of FVIII following AAV-mediated gene transfer in adult hemophilia A mice [40]. Some success have been achieved to produce persistent transgene expression following neonatal gene transfer of FVIII [15,41,42]. Induction of long-term FVIII tolerance could be achieved using a lentiviral vector encoding a B-domain-deleted canine FVIII gene, driven by a liver-specific promoter (HCR/α1-antitrypsin promoter) into neonatal hemophilia A mice. Tolerance to FVIII could be adoptively transferred to naive hemophilia-recipient mice, and FVIII-stimulated CD4+ T cells isolated from the spleens of tolerized mice expressed increased levels of IL-10 and decreased levels of IL-6 and IFN-γ. Induction of FVIII tolerance mediated by this protocol is found to be associated with a FVIII-expandable population of CD4+CD25+Foxp3+ Treg cells.
Ex vivo transduction of hematopoietic stem cells
Transplantation of retrovirally transduced hematopoietic-carrying FVIII gene produced undetectable levels of FVIII protein in the circulation but induced tolerance against FVIII in 50% of treated animals [43]. Reduced antibody responses were also observed in hemophilia A mice transplanted with bone marrow cells transduced with an optimized murine stem cell virus (MSCV)-based FVIII oncoretroviral vector [44]. Recent improvement in the transduction protocol, in combination with the use of a vector encoding a B-domain-deleted porcine FVIII cDNA, and immunosuppressive agents of CTLA4-immunoglobulin and anti-CD40L or anti-(murine)thymocyte serum (ATS), achieved therapeutic levels of FVIII long term in an hemophilia A mouse model [45]. A nonmyeloblative conditioning regimen involving busulfan was used for the establishment of a mixed chimerism. Such a strategy was also effective in mice with inhibitors to FVIII prior to hematopoietic stem cell (HSC) transplantation [46]. The observed tolerance to foreign antigens was likely to be mediated by the engraftment of transduced cells and the migration of transduced APCs to the recipient thymus, where they induced negative selection of donor-reactive T cells, prior to release into the circulation. A recent attempt to transplant sibling HSCs into a hemophilia patient induced tolerance only to donor antigens but not to FVIII [47]. A reduced-intensity conditioning regimen was used, and followed by bone marrow infusion and continuous FVIII administration during immune reconstitution. Inhibitory antibody titer rose at day 23 following the procedure, indicating that transplantation procedure cannot control the boosting of long-term memory effector cells. The success of the gene-modified HSC transplantation to induce long-term tolerance to FVIII in hemophilia A mice may be associated with their use of porcine FVIII and transient immunosuppression [46].
Specific site-targeted FVIII gene expression
Shi et al. developed transgenic mice harboring a human BDDFVIII gene under the control of the platelet-specific αIIb promoter [48]. The FVIII expressed was stored in platelets and released at the site of platelet activation. Although there is no FVIII detected in plasma of the transgenic mice, the hemophilia A phenotype was corrected, even in the presence of high inhibitory antibody titers, by a tail vein bleeding assay. The same group subsequently investigated the ex vivo gene therapy approach using a lentiviral vector carrying the FVIII gene driven by αIIb promoter [49]. Following the transfer of FVIII-modified bone marrow cells, FVIII expression was detected in platelet lysates. The treated mice survived tail-clipping assay and no anti-FVIII antibodies were detected over 11 months. Platelet-specific gene expression may be a way to evade the inhibitory antibody responses for treating hemophilia A. The current major obstacle for this protocol is the low-level transgene expression and functional activity, which need to be improved to ensure a therapeutic effect.
In addition, ex vivo gene therapy for hemophilia A was explored by implanting lentivirally transduced endothelial progenitor cells subcutaneously in a Matrigel scaffold [50]. Circulating blood outgrowth endothelial cells (BOECs) were isolated and transduced with a lentiviral vector encoding the canine FVIII transgene under the direction of a cytomegalovirus promoter or a thrombomodulin regulatory element. The hemophilia A mice were pretreated with tolerizing doses of FVIII (two intravenous injection of recombinant canine FVIII [20 units/kg per injection]) 5 and 2 days prior to the implant, or with transient immunosuppression (intraperitoneal injection of cyclophosphamide [20 mg/kg per injection] administered on the day of the implantation and then biweekly for 4 weeks). Therapeutic FVIII gene expression persisted for 27 weeks following transplantation before dropping to baseline levels owing to the loss of implanted cells. Interestingly, none of the treated mice developed inhibitory antibodies to FVIII. Another group used a nanocapsule system to direct FVIII gene into liver sinusoidal endothelial cells [51]. Sleeping Beauty transposon expressing the BDD-canine FVIII in cis with Sleeping Beauty transposase was encapsulated in hyaluronan nanocapsules, and infused intravenously into hemophilia A mice. Long-term (50 weeks) and high-level (equal to normal level) gene expression of FVIII was achieved, without apparent antibody formation along with improvement of hemophilia A phenotype in treated mice. Recently, induced pluripotent stem (iPS) cells were generated from murine tail-tip fibroblast cells by the ectopic expression of three transcription factors, Oct4, Sox2 and Klf4. These murine iPS cells were further differentiated to both FVIII-secreting endothelial cells and endothelial progenitor cells by using the embryoid body differentiation method, and injected directly to the liver of irradiated hemophilia A mice. The recipient mice produced 8–12% FVIII levels, and corrected the phenotype correction of murine hemophilia A for at least 3 months [52]. Moreover, lineage-negative murine bone marrow cells were isolated and transplanted directly into the host mouse livers, which were perturbed with acetaminophen to enhance the cell engraftment. The engrafted cells transdifferentiated into liver cells, including both hepatocytes and endothelial cells, and produced approximately 20% normal levels of FVIII activities, which persisted for more than 1 year without detection of anti-FVIII inhibitory antibodies [53].
Incorporation of miRNA site in FVIII gene-expression cassettes
miRNA is a newly discovered RNA group composed of approximately 21–23-nucleotide RNAs that negatively regulate gene expression at the post-transcriptional level. miRNAs act as a guide for the RNA-induced silencing complex (RISC) to repress translation of cellular transcripts. miRNAs have distinct expression profiles in different tissues. Recently, a lentiviral-green fluorescent protein (GFP) construct incorporating miRNA binding sites (miR-142–3p) specific for HSCs effectively suppressed transgene expression in APCs, whereas expression was maintained in nonhematopoietic cells without inducing transgene-specific immune responses [54]. Incorporation of a miRNA site may limit the FVIII gene expression to nonhematopoietic cells within the liver, and reduce FVIII presentation by local APCs, leading to reduced immune responses to FVIII.
Immunosuppressive therapy targeting T cells
Tolerance-induction protocols involving antigen presentation, and the methods to evade anti-FVIII immune responses, often are successful in only a fraction of the treated patients or animals and/ or in combination with immunosuppressive agents. Therefore, transient immunomodulation regimens to reduce or eliminate anti-FVIII inhibitory antibodies are highly desirable. Peripheral tolerance to a specific antigen can be induced through several different mechanisms to disrupt T-cell activation or T- and B-cell interaction, or inhibit T- or B-cell function (Figure 1). In addition, dominant tolerance can be established through active suppression of activated effector T cells by Treg cells.
Figure 1. Induction of peripheral tolerance can be achieved by several different methods.
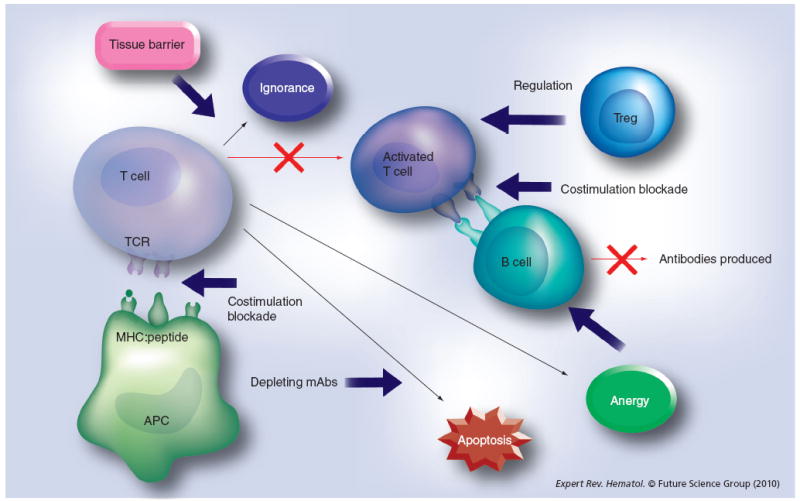
Depleting mAbs were used to deplete activated T cells to generate T-cell apoptosis. Blockade of costimulatory pathways can induce T-cell apoptosis or anergy. A dominant tolerance can be induced by suppression of activated T cells by Tregs.
APC: Antigen-presenting cell; mAb: Monoclonal antibody; TCR: T-cell receptor.
Common immunosuppressive drugs
Common immunosuppressive drugs nonspecifically targeting T-cell activation, clonal expansion or differentiation into effector T cells have been used to prevent antigen-specific responses. Cyclophosphamide has been used along with AAV- [40] or lentiviral-mediated gene transfer of FVIII [50] to suppress anti-FVIII responses. In hFVIII plasmid-transferred hemophilia A mice, the administration of cyclophosphamide following gene transfer of naked FVIII plasmid resulted in the prevention (two out of six animals) or delay (five out of six animals) of inhibitory antibody formation against FVIII [18]. In addition, ciclosporin A (CSA) suppresses the early stages of lymphocyte activation. Rapamycin selectively suppresses activated T cells and favors the expansion of Treg cells. Mycophenolate mophetil (MMF) is an antiproliferative agent for both B and T cells. In the FVIII plasmid, gene-therapy treated hemophilia A mice, and application of either single-agent, or combined therapy with MMF, CSA and rapamycin only resulted in delayed immune responses [55]. Inhibitory antibodies appeared quickly upon withdrawal of the drug(s). Although the transient treatment of MMF considerably suppressed the production of antigen-specific IgG1 and IgG2b antibodies, the effect, however, was not potent enough to prevent the induction of long-lasting inhibitory antibodies against FVIII.
Costimulatory blockade
Recently, mAbs have emerged as a new class of immunosuppressive agents that appear to be both more effective and selective in facilitating immune tolerance induction, and are generally very well tolerated by recipients. These agents targeting specific pathways also make them less toxic than nonspecific, traditional immunosuppressive agents, and can induce antigen-specific tolerance in the presence of antigens. Monoclonal antibodies can be used to block the costimulatory pathway to induce T-cell apoptosis or anergy. Multiple T-cell costimulatory pathways, including CD28 and B7 molecules (CD80, CD86), inducible costimulatory molecule (ICOS), ICOS-L, CD-40L and CD40, OX40 (CD134) and OX40-L, provide a functional network to ensure robust T-cell activation to mount immune responses against foreign antigens.
Several agents to interrupt costimulatory pathways have been used to induce tolerance to FVIII. CTLA4-immunoglobulin (CTLA4-Ig) is a soluble fusion protein of the extracellular domain of CTLA4 and the heavy-chain constant regions 2 and 3 of IgG1. CTLA4-Ig blocks the costimulatory interaction of B7 and CD28. It has been shown to transiently block the inhibitor formation in hemophilia A mice [56]. CTLA4-Fc inhibits the restimulation and differentiation of FVIII-specific memory B cells in the presence of FVIII [57]. In relation to CTLA4-Ig, the part of its action, at least in certain models, is through the induction of indoleamine 2,3-dioxygenase (IDO), a tryptophan-degrading enzyme. Codelivery of FVIII and IDO genes in a transposon system yielded long-term therapeutic FVIII expression and significantly reduced anti-FVIII antibody titers [58]. The attenuated antibody response was associated with increased plasma kynurenine (one of the IDO metabolites) levels, inhibition of T-cell infiltration, and increased apoptosis of T cells in the liver, leading to a localized state of general immunosuppression. This observation agrees with most other studies; however, there have been reports of IDO over-expression causing modulation of Treg cells populations in other model systems [59]. In vivo analysis of CD4+CD25+ Treg cells was not performed in this study to confirm if the immune tolerance observed is the result of upregulation of Treg cells.
Anti-CD40L blocks the interaction of CD40 and CD40-ligand (CD40L). Anti-murine CD40L mAb has been previously used to transiently block the inhibitory antibody formation in hemophilia A mice [56,60,61]. Similar to CTLA4-Fc, anti-CD40L has been shown to block the restimulation and differentiation of FVIII-specific memory B cells in the presence of FVIII antigen [22,57].
Dual blockade of CD40/CD40L and B7/CD28 pathways using a combination regimen of anti-CD40L and CTLA4-Ig haven been demonstrated to act synergistically to prevent antigen-specific immune responses and induce long-term tolerance to FVIII in FVIII plasmid-treated hemophilia A mice [55]. Following treatment with this combined immunotherapy regimen, only one out of nine mice developed low-titer antibodies that were transient and only partially blocked the functional activity of hFVIII in plasma after naked gene transfer of hFVIII plasmid. All nine animals, including the one with transient antibodies, produced persistent, therapeutic or supraphysiologic levels of hFVIII expression. When challenged with the T-dependent antigen, bacteriophage Φx174, tolerized mice exhibited normal primary and secondary antibody responses. Thus, transient inhibition of costimulatory molecules can promote a long-term immune tolerance that is specific for hFVIII, allowing long-term gene correction in immunocompetent hemophilia A mice. CTLA4-Ig has been shown to not induce long-term immune compromise in humans [62], and has recently been approved by the US FDA for clinical use. Clinical trials to test efficacy and safety of anti-CD40L have yielded controversial results. In particular, some trials induced thrombotic events or increased thrombotic risks in patients [63,64], whereas some did not [65]. Further study is needed to delineate functional and safety profiles of different monoclonal antibodies targeting different CD40L epitopes and functions. Currently, however, this agent is not available for clinical use.
Anti-inducible costimulatory molecule blocks the third costimulatory pathway of interaction between ICOS and ICOS-L. In plasmid-treated hemophilia A mice, inhibitory antibody formation was successfully prevented following nonviral gene transfer of FVIII and anti-ICOS treatment [66]. Blockade of the ICOS–ICOS-L pathway by anti-ICOS alone as a single agent was effective to eliminate immune responses. This observation is consistent with the earlier findings that agents that block T-help function were more effective (including MR1 alone) than CTLA4-Ig alone for prevention of the antibody response [55]. In established animal models of tolerance, the presence of antigen was also required to maintain the tolerant state. The minimum concentration of FVIII required to maintain hyporesponsiveness is in the range of 10-11–10-10 M in the circulation [67,68], a level readily achieved by the sustained hFVIII gene expression (10-9–10-8 M) in the gene transfer model. In anti-ICOS-treated hemophilia mice, tolerance was maintained along with high-level (100–300% of normal) constitutive hFVIII gene expression [66]. It was also found that:
Anti-ICOS treatment resulted in transient depletion of CD4+ T cells during initial treatment period;
Blockade of ICOS–ICOS-L costimulatory pathway by anti-ICOS induced elimination of T-effector cells in the short term and T memory cells in the long term;
Anti-ICOS treatment promoted upregulation of CD4+CD25+Foxp3+Treg cells in peripheral blood, spleen, and lymph nodes, in terms of both percentage and activity;
CD4+ T cells from anti-ICOS-treated mice did not proliferate in response to hFVIII stimulation, and produced high levels of regulatory cytokines, including IL-10 and TGF-β;
CD4+CD25+ Treg cells from tolerized mice adoptively transferred dominant tolerance in syngeneic hFVIII plasmid-treated hemophilia A mice and reduced the production of antibodies against FVIII.
These results indicated the involvement of antigen-specific Treg cells in tolerance induction following anti-ICOS treatment. Furthermore, anti-ICOS-treated mice tolerized to hFVIII generated normal primary and secondary antibody responses after immunization with the T-dependent antigen, bacteriophage Φx174, indicating maintenance of immune competency of the treated mice. Anti-ICOS, which is currently under development for clinical applications, represents a highly effective agent for modulating antigen-specific immune responses.
It is very interesting that administration of anti-ICOS mAbs over an extended period, effectively promoted tolerance to FVIII despite there being multiple other costimulatory pathways present. Our results were different form the results reported by Hausl et al. [57] that the interference with ICOS–ICOS-L interaction by anti-ICOS-L monoclonal antibody did not inhibit the restimulation of FVIII-specific memory B cells. This difference is likely owing to the fact the model used was one of an established disease and/or that the blocking antibodies anti-ICOS and anti-ICOS-L act differently. On conjugation, anti-ICOS-L may indeed initiate B-cell activation bypassing the ICOS–ICOS-L costimulatory pathway, similar to the effect of anti-CD40 antibody [69], which achieves B-cell activation by directly crosslinking CD40. Alternatively, anti-ICOS-L antibody may be less effective because memory B-cell responses are less T-cell dependent.
T-cell depleting antibody: anti-CD3
Depletion of T cells and, sometimes, in combination with B cells, can significantly reduce the number of effector T cells capable of mounting an immune response at the time of initial exposure to foreign antigens. Humanized anti-CD3 antibodies (OKT3γ1 Ala-Ala and ChAglyCD3) are non-mitogenic and are devoid of deleterious cytokine-releasing activity and serious side effects [70,71], and exhibit their therapeutic potency in several disease models. Five consecutive anti-CD3 treatments concomitant with FVIII plasmid injection prevented the formation of inhibitory antibodies against FVIII and achieved persistent, therapeutic levels of FVIII gene expression in treated hemophilia A mice [72]. Repeated plasmid gene transfer is applicable to tolerized mice without eliciting immune responses. In the initial several weeks following treatment, anti-CD3 depleted both CD4+ and CD8+ T cells to very low levels, but increased TGF-β levels in plasma and the frequency of both CD4+CD25+Foxp3+ and CD4+CD25+Foxp3+ Treg cells. Interestingly, prior depletion of CD4+CD25+ cells did not abrogate tolerance induction; however, adoptive transfer of CD4+ T cells from tolerized mice at 6 weeks after treatment protected recipient mice form anti-FVIII immune responses. These results demonstrate that FVIII-specific tolerance was induced by anti-CD3 treatment via a mechanism involving the increase in TGF-β levels and the generation of adaptive FVIII-specific CD4+Foxp3+ Treg cells in the periphery [72]. Depletion of effector T cells following anti-CD3 administration generated a suitable window for antigen-specific Treg cells to proliferate and be activated.
Waters et al. recently reported that anti-CD3 is also able to induce tolerance to FVIII after repeated injections of FVIII proteins in hemophilia A mice via a CD4+CD25+ Treg-dependent mechanism [73]. Their result is consistent with our study demonstrating that Treg cells are pivotal for establishing FVIII tolerance. However, the development and characterization of functional antigen-specific Treg cells are different in these two studies. We propose that inducible CD4+CD25-/lowFoxp3+ initiated the tolerance and then matured into CD4+CD25+Foxp3+ Treg cells for maintaining long-term tolerance. In their model, CD4+CD25+Foxp3+ Treg cells are important for both induction and maintenance of the tolerance. This difference may be the result of the different protocols of anti-CD3 treatment. Anti-human CD3 has been approved by the FDA for clinical use, and has been widely applied in transplantation and autoimmune models. The dosage and schedule used in both studies mentioned are comparable with those used in human trials. Anti-CD3 immunomodulation has the potential to be a safe and effective strategy to prevent FVIIII-specific immune responses after gene therapy or protein-replacement therapy. It was also demonstrated that anti-CD3 can reduce the titers of pre-existing inhibitors [72] and will be a candidate in combination therapy with other immunosuppressive agents for modulating anti-FVIII immune responses in hemophilia mice with established inhibitors.
Adoptive Treg cell therapy
A dominant tolerance can be induced by suppression of activated T cells by Treg cells. Recent advances in the understanding of how Treg cells suppress various immune responses may provide new opportunities to develop a novel strategy to prevent and/or eliminate inhibitory antibodies, and possibly induce long-term immune tolerance against specific antigens.
Development of tolerance is a dynamic process involving several mechanisms, including deletion of the alloreactive T-cell pool and immunoregulation [74-76]. A critical role of CD4+CD25+ Treg cells in the regulation and suppression of autoimmune and alloimmune responses has been demonstrated [77-81] . T-cell homeostasis was achieved by balancing the CD4+CD25+ Treg cells and effector T cells; tolerance induction can be accomplished by inducing a balance shift between Treg and effector T cells. CD4+CD25+Foxp3+ Treg cells originate in the thymus during ontogeny as fully differentiated suppressor cells and are referred to as natural Treg cells. Treg cells can also be induced from naive T cells in the periphery [82]. Some, but not all, of these peripherally induced adaptive Treg cells also express Foxp3. At present, there are several limitations that significantly limit the use of naturally occurring Treg cells [83]. First, only very small numbers of the Treg cells that can be obtained and their proliferative potential is very low. Second, it appears that only a minority of CD4+CD25+ T cells have strong suppressive activity, and this function is antigen nonspecific. These limitations have prompted efforts to identify novel mechanisms to generate ‘antigen-specific’ Treg cells.
Recently, CD4+ CD25+ Treg cell populations have been successfully induced and/or expanded, and shown to suppress autoimmune and alloimmune responses. In particular, supplementation of donor grafts with freshly isolated CD4+CD25+ Treg cells can downregulate the graft-versus-host disease (GVHD) lethality to various degrees [84-86]. Good manufacturing practice techniques have been developed to isolate and expand human Treg cells for use in human clinical trials in bone marrow transplant patients. Furthermore, antigen-specific FOXP3-transduced CD4+CD25+ Treg cells can control established Type 1 diabetes in mice [87].
To test if FVIII-specific Treg cells can modulate immune responses against FVIII, a hemophilia A mouse model in which all T cells overexpressed Foxp3 (HemA/Foxp3-Tg) [88] was developed (Figure 2) [89]. FVIII plasmid transfer did not induce antibody production in HemA/Foxp3-Tg mice. CD4+Foxp3+ T cells isolated from FVIII plasmid-treated HemA/Foxp3-Tg mice significantly suppressed proliferation of FVIII-stimulated CD4+ effector T cells. Adoptive transfer of Treg cells isolated from FVIII-exposed HemA/Foxp3-Tg mice produced significantly reduced antibody titers in Treg-recipient hemophilia A mice compared with untreated control mice after initial challenge with FVIII plasmid and second challenge 16 weeks following the first plasmid treatment [89]. Adoptively transferred Treg cells engrafted and distributed at 2–4% in the Treg compartment of the blood, lymph nodes and spleens of the recipient mice; however, the engraftment decreased to negligible levels over 8–12 weeks, indicating that very little adoptively transferred Treg cells were present when the second plasmid challenge was performed. By contrast, the adoptively transferred Treg cells induced significant activation of endogenous Treg cells in the recipient mice after adoptive transfer plus FVIII plasmid treatment. Thus, the Treg-mediated generation of new functional FVIII-specific CD4+Foxp3+ Treg cells via an infectious tolerance mechanism was responsible for their suppressive abilities to maintain long-term protective effect in vivo against FVIII-specific immune responses and limit recall responses induced by a second challenge. Nevertheless, adoptively transferred transgenic CD4+Foxp3+ T cells can only partially modulate the immune responses against FVIII following plasmid transfer into hemophilia A mice for prolonged periods of time. The insufficient regulation could be owing to insufficient Treg cells engrafted after adoptive transfer and inadequate antigen specificity. Improvements of the adoptive transfer therapy of Treg cells are currently under investigation.
Figure 2. Adoptive transfer of CD4+Foxp3+ T cells from Factor VIII plasmid-treated HemA/Foxp3-Tg mice can modulate anti-FVIII immune responses in recipient hemophilia A mice.
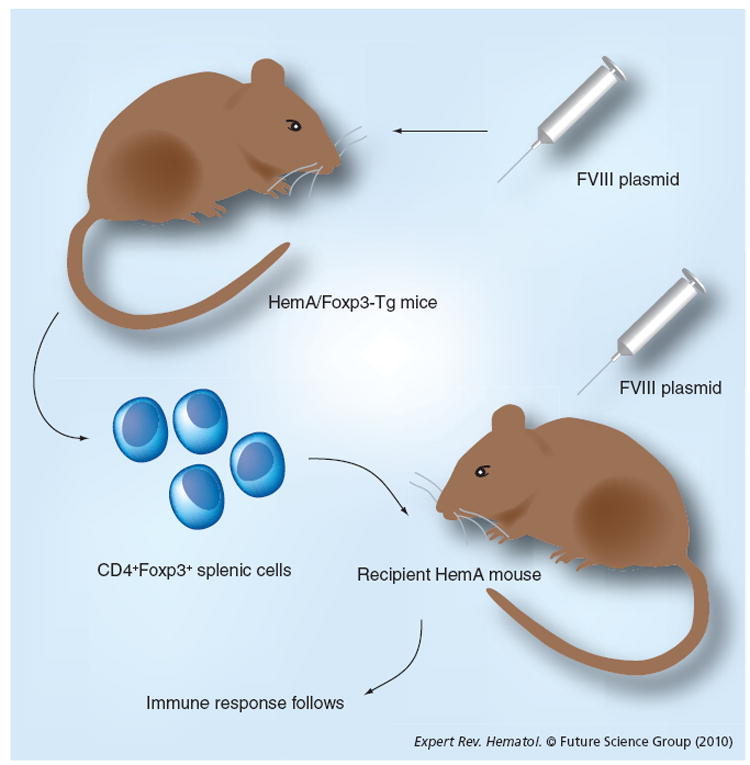
CD4+ T cells were isolated from the spleens of HemA/Foxp3-Tg mice 2 weeks post-FVIII plasmid treatment and adoptively transferred into naive HemA mice. Recipient mice, as well as a control group of naive hemophilia A mice, were subsequently challenged with FVIII plasmid injection 1 day following adoptive transfer.
Adoptive transfer therapy of Treg cells to modulate antigen-specific responses has many advantages over conventional treatments. Some of these benefits include protection through antigen-specific activity without general immunosuppression, the possibility of inducing physiological long-lasting regulation in vivo, and customized therapy for individual patient with limited or no side effects. For human applications, Treg cells will be isolated from a patient, enriched and/or selected, expanded ex vivo, and re-infused back into the patient. The major challenge lies on how to efficiently isolate and expand pure Treg cells with or without defined allospecificities ex vivo. Recently, ex vivo cell isolation and culture systems are becoming more advanced and efficient, ensuring a safe, reproducible, closed cell-processing system that can operate on a large scale [90]. A number of clinical studies using adoptive Treg cell therapy to induce transplantation tolerance are currently underway, and some have shown a positive association between circulating Treg cells and good graft function. Potential risks of adoptive Treg cell therapy include the possibility of expanded Treg cells reverting to pro-inflammatory effector cells and excessive Treg activity impacting the immune system and blunting the response to infectious agents. In the case of modulation of FVIII-specific immune responses, the transferred Treg cells will be transiently present at low percentages in the Treg cell compartment, and should have minimum effects on immune system, even in young children. Another potential approach is to generate Treg cells with added suicide-gene safety features to allow rapid removal of transferred cells [91].
In vivo expansion of Treg cells
As discussed, many successful approaches developed towards induction of tolerance to FVIII and other antigens involve suppressive function of Treg cells [92]. Adoptively transferred transgenic hemophilia A/Foxp3 Treg cells reduced anti-FVIII inhibitory antibody titers following FVIII plasmid-mediated gene transfer into hemophilia A mice. An in vivo approach for inducing selective expansion of Treg cells by injecting hemophilia A mice with IL-2 mixed with a particular IL-2 mAB (JES6–1) was used to modulate FVIII-specific immune responses [93]. The mice treated with IL-2–IL-2 mAb complex did not generate inhibitory antibodies against FVIII, and achieved therapeutic level of FVIII gene expression in FVIII plasmid-treated mice [94]. The treatment led to a marked five- to seven-fold increase in total numbers of Treg cells in the peripheral blood on the peak day (day 6 following the last IL-2–IL-2 mAb complex treatment), and these levels gradually returned to normal within the next 7–14 days. These short-lived expanded Treg cells are highly activated and display superior suppressive function. Little or no change in other cell populations was observed. Furthermore, IL-2–IL-2 mAb complexes can transiently reduce inhibitory antibody titers in mice with pre-existing inhibitory antibodies following gene transfer [94]. These results demonstrate the important role of Treg cells in suppressing anti-FVIII immune responses and the potential of developing Treg expansion therapy to induce long-term tolerance to FVIII.
Immunosuppressive therapy targeting B cells
Tolerogenic B-cell therapy
David Scott’s laboratory has investigated a B-cell therapy by using tolerogenic B cells, transduced with a retroviral vector carrying the cDNA encoding the A2 or C2 domains of FVIII fused to the heavy chain of murine IgG to induce FVIII tolerance [28,95]. Specific tolerance to A2 or C2 domain was induced by this protocol. Furthermore, a combination of A2-IgG- and C2-IgG-expressing B cells induced tolerance to the full-length FVIII protein. This therapy successfully prevented antibody production in naive mice without inhibitors or reduced the inhibitor titers in FVIII-primed hemophilia A mice (mice with pre-existing inhibitors) [28]. This tolerance induction protocol required MHC class II and B7 expression on the retrovirally transduced activated B cells [96,97], and was dependent on transfer to the endosomes and processing in an IFN-γ-inducible lysosomal thiol reductase-dependent fashion [28,95]. In addition, the tolerance was dependent on Treg cells; significant increase of Treg cells was detected in B-cell-transduced animals, and depletion of CD4+CD25+ Treg cells completely abolished the tolerance [98].
B-cell depleting antibody: anti-CD20
Rituximab is a humanized mAb against the pan-B-cell antigen CD20, and induces rapid in vivo depletion of mature human B lymphocytes. Limited reports described the use of rituximab in children [99-101] and in adults [24,100,102-105] with congenital hemophilia and inhibitors. Case reports of rituximab treatment in small numbers of congenital hemophilia patients with inhibitors have been inconclusive [106,107]. It was hypothesized that concurrent administration of high-dose FVIII is beneficial. A Phase II clinical trial of rituximab in the treatment of hemophilia A high-responding inhibitor formation is currently ongoing in the Transfusion Medicine Hemostasis Clinical Network (TMH-CTN) [108]. However, the effects of rituximab in combination with other immunomodulatory agents or immune tolerance therapy have not yet been tested, and such studies in humans will be challenging to perform.
Scott and colleagues recently studied the effect of anti-murine CD20 IgG1 to selectively deplete follicular B cells, while sparing marginal zone (MZ) B cells on the immune response to FVIII and its influence on an ITI protocol [109]. This is due to the hypothesis that MZ B cells may be tolerogenic APCs; thus, selective depletion of follicular B cell by anti-CD20 IgG1 may favor tolerance induction to human FVIII. Both groups of hemophilia A mice treated with either anti-CD20 IgG1 or a control IgG1 developed high-titer anti-FVIII antibodies following a protocol similar to ITI. However, the increase in anti-FVIII IgG levels was significantly lower than that in control IgG1-treated group.
In FVIII plasmid-treated hemophilia A mice, we found that administration of anti-murine CD20 IgG2a into hemophilia A mice significantly reduced CD19+ B cells in the blood, spleen and lymph nodes [110]. The depletion of B cells lasted for sustained periods of time, and gradually returned to normal at approximately 8 weeks following anti-CD20 treatment. Of the mice, 25% undergoing treatment with FVIII plasmid and anti-CD20 had persistent FVIII activity without detectable inhibitory anti-FVIII antibodies. The rest of the treated mice had delayed immune responses; however, all generated moderate-to-high titers of inhibitory antibodies, with FVIII activity decreasing to undetectable levels at 6–15 weeks. A reduction of inhibitory antibody titers was observed following anti-CD20 treatment; however, the titer rose back when anti-CD20 decreases over time. In these cases, depletion of B cells following gene transfer did not create a sufficient window for tolerance induction to occur in the presence of antigen. No significant change in the numbers or percentages of CD4+ T cells and CD4+CD25+Foxp3+Treg cells was observed during the experimental period. These results indicate that anti-CD20 can partially regulate anti-FVIII immune responses. It is suspected that anti-CD20 depleted most B cells; however, it failed to eliminate long-lived CD20- plasma and memory B cells. Anti-CD20 still represents a potential candidate in therapies combined with other immunomodulation agents.
Treg cell plays an important role in immunomodulation of inhibitors in hemophilia A
As described in this article, many successful protocols to modulate FVIII-specific immune responses involve increases in either or both of the percentages and total numbers of CD4+Foxp3+Treg cells in either protein-replacement or gene therapy settings. It is also important that these induced Treg cells are activated in order to exert their regulatory function to suppress FVIII-specific responses. Interestingly, immunomodulation strategies with such capacity can successfully prevent induction of anti-FVIII immune responses and induce long-term tolerance to FVIII (Table 1), whereas strategies targeting other cell populations without induction of Treg cells can only partially reduce the anti-FVIII titers following treatment, and no long-term tolerance can be established. These results indicate that a shift from an immune-activating environment to a regulatory environment by induction of activated Treg cells to suppress T-helper cell function is not only important in blocking the initial activation of antibody responses, but also facilitate the induction and maintenance of antigen-specific tolerance.
Table 1.
Changes in T-lymphocyte subpopulations in the spleens of mice under treatment with various immunomodulation protocols.
| Immunomodulation protocols† | Prevention of Factor VIII inhibitor formation | Modulation of pre-existing Factor VIII inhibitors | CD4+ T cells | Treg cells‡ in CD4+ T cells | Activated§ Factor VIII-specific Treg cells | Treg cells (total numbers) | Ref. |
|---|---|---|---|---|---|---|---|
| ITI (human protocol)¶ ± immunosuppressant or intravenous immunoglobulin | Yes | Yes | No report | Possible increase | Possible increase | Possible increase | [22-25] |
| Mucosal or oral exposure of Factor VIII domain or peptides | Partial | [27] | |||||
| Immature dendritic cells | Partial | No change | Increase | Increase | Increase | [29,30] | |
| Apoptotic fibroblast cells | Partial | Partial | Increase | [31] | |||
| Neonatal gene transfer | Yes | NA | No change | Increase | Increase | [15,41,42] | |
| Ex vivo HSC gene therapy + ATS | Yes | Yes | [43-46] | ||||
| Gene therapy targeting liver, platelet or endothelial cells ± immunosuppressant | Yes or partial | Yes or partial | [48-50] | ||||
| Cyclophosphamide | Partial | Decrease | [18,40,50] | ||||
| Cyclosporin | Transient | Decrease | [55] | ||||
| Rapamycin | Transient | Decrease | [55] | ||||
| MMF | Transient | Decrease | [55] | ||||
| CTLA4-Ig | Transient | Transient | [56,57] | ||||
| IDO | Partial | Decrease | [58] | ||||
| Anti-CD40-L | Transient | Transient | [22,56,57, 60,61] | ||||
| CTLA4-Ig + CD40-L | Yes | [55] | |||||
| Anti-inducible costimulatory molecule | Yes | Decrease | Increase | Increase | Slight increase | [66] | |
| Anti-CD3 | Yes | Partial | Decrease | Increase | Increase | No change | [72,73] |
| Adoptive Treg cell therapy | Partial | Transient | No change | Slight increase | Increase | Slight increase | [89] |
| IL-2–IL-2 monoclonal antibody complex | Yes | No change | Increase | Increase | Increase | [93] | |
| Tolerogenic B-cell therapy | Yes | Partial | No change | Increase | Increase | Increase | [28,95,98] |
| Antimurine CD20 IgG1 or IgG2a | Partial | Transient and partial | No change | No change | No change | No change | [109,110] |
All immunomodulation therapies were applied in the presence of either Factor VIII protein or gene expression.
Treg cells denotes CD4+CD25+Foxp3+ Treg cells.
Activated Treg cells are evaluated by expression of activation markers.
This is the human protocol for comparison purposes.
ATS: Anti-(murine)thymocyte serum; HSC: Hematopoietic stem cell; IDO: Indoleamine 2,3-dioxygenase; Ig: Immunoglobulin; ITI: Immune tolerance induction; MMF: Mycophenolate mophetil; NA: Not applicable.
Nevertheless, it appears to be much more challenging to modulate the pre-existing inhibitory antibody responses in mice primed with FVIII. Even with five- to seven-fold increases of Treg cells in FVIII primed mice treated with IL-2–IL-2 mAb complexes [94], anti-FVIII antibody titers were only partially and transiently reduced to lower levels, and the titers returned to higher levels when the Treg numbers returned to normal at approximately 7 days following treatment. In this case, the IL-2–IL-2 mAb treatment may not eliminate long-term memory B and/or T cells already established in primed mice; thus, antibody responses returns as soon as T-cell help becomes available again in the absence of suppression by Treg cells. It may require combination therapy targeting both memory cells and T-helper cells to treat pre-established inhibitory antibody responses, and induce long-term tolerance to FVIII.
Expert commentary
Approximately 25% of hemophilia patients develop antibodies after repeated infusions of FVIII protein. Treatments of bleeding episodes in hemophilia patients with inhibitors include the use of bypassing agents, or induction of tolerance by infusing massive doses of FVIII. However, high doses of FVIII are very costly, and the rate of success is not ensured. As described in this article, many effective strategies have been explored recently to prevent or reduce inhibitory antibody responses against FVIII. As discussed, most of the successful strategies involve the participation of functional antigen-specific Treg cells. Manipulation of antigen presentation or new FVIII variants/formulations and gene therapy protocols can, hopefully, prevent or reduce the antibody production in a fraction of the treated subjects. Some of these may need to combine with transient immunosuppressive protocols targeting B and/or T cells to ensure the success of the treatment.
Prevention therapy can be used in patients who have never been exposed to FVIII proteins – therefore, most likely, in very young patients. Safety precautions for regimens that can be used for patients at such young age will need to be considered. In addition, successful strategies should also meet the following criteria:
Require only transient exposure to immunosuppressive protocols;
Induce minimal cytoablation and other associated toxicity;
Exhibit minimal effects on immune competence;
Mediate FVIII-specific long-term tolerance.
Five-year view
It is anticipated that some of the preventive therapies will enter the phases of large animal studies and clinical trials in the near future. In the next step, further improvements and/or combination of effective regimens need to be investigated to modulate pre-existing anti-FVIII antibodies in hemophilia A mouse models. Currently, only ITI is successful in some patients to eradicate pre-existing inhibitory antibodies. None of the single immunomodulation protocols are able to eliminate pre-existing inhibitors and induce long-term tolerance to FVIII. A combination therapy that targets different pathways is most likely to succeed for this task. A strategy will need to be devised so that it can not only deplete memory B and/or T cells and/or long-lived plasma cells to eliminate pre-existing immune responses, but also induce expansion of functional antigen-specific Treg cells to maintain long-term T-cell hyporesponsiveness, and tolerance to FVIII. It is anticipated that combined therapies targeting distinct pathways will permit use of more-limited dosages of each individual agent, thereby providing improved safety while more effectively modulating pre-existing inhibitory immune responses.
Key issues.
Approximately 25–30% of hemophilia A patients develop inhibitory antibodies against Factor VIII (FVIII) or against Factor IX following protein-replacement therapy.
This problem is also anticipated to occur following gene-replacement therapy.
Immune tolerance induction is currently the only effective therapy to induce tolerance to FVIII in hemophilia patients. However, this procedure is both costly and inconvenient.
The protocols that can modulate anti-FVIII immune responses include methods to manipulate antigen presentation, development of less immunogenic FVIII proteins or formulations or gene therapy protocols to evade immune responses, as well as immunomodulation strategies to target either T- and/or B-cell responses.
Many transient immunomodulation therapies have been successfully designed and tested to prevent the formation of inhibitory antibodies against FVIII.
Most of the successful prevention protocols involve the expansion of suppressive CD4+CD25+Foxp3+ Treg cells.
Strategies to eliminate pre-existing anti-FVIII antibodies and induce long-term tolerance to FVIII still need to be developed.
Acknowledgments
Carol Miao is supported by the grants from NIH/NHLBI (R01 HL082600, R01 HL069049 and R21/R33 HL089038) and a special project grant from the Bayer Hemophilia Foundation.
Footnotes
Financial & competing interests disclosure The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
-
•
of interest
-
••
of considerable interest
- 1.Davie EW, Fujikawa K, Kisiel W. The coagulation cascade: initiation, maintenance, and regulation. Biochemistry. 1991;30:10363–10370. doi: 10.1021/bi00107a001. [DOI] [PubMed] [Google Scholar]
- 2.Hoyer LW, Scandella D. Factor VIII inhibitors: structure and function in autoantibody and hemophilia A patients. Semin Hematol. 1994;31(2 Suppl. 4):1–5. [PubMed] [Google Scholar]
- 3.Briet E, Mauser-Bunschoten EP. Revision consensus hemophilia: treatment and responsibility. Nederlandse Vereniging van Hemophilia Patients. Ned Tijdschr Geneeskd. 1997;141(52):2566–2571. [PubMed] [Google Scholar]
- 4.Darby SC, Keeling DM, Spooner RJ, et al. The incidence of Factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977–1999. J Thromb Haemost. 2004;2(7):1047–1054. doi: 10.1046/j.1538-7836.2004.00710.x. [DOI] [PubMed] [Google Scholar]
- 5.Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of Factor VIII and factor IX inhibitors in haemophiliacs. Lancet. 1992;339(8793):594–598. doi: 10.1016/0140-6736(92)90874-3. [DOI] [PubMed] [Google Scholar]
- 6.Lusher JM, Arkin S, Abildgaard CF, Schwartz RS. Recombinant Factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. Kogenate Previously Untreated Patient Study Group. N Engl J Med. 1993;328(7):453–459. doi: 10.1056/NEJM199302183280701. [DOI] [PubMed] [Google Scholar]
- 7.Jacquemin M, Vantomme V, Buhot C, et al. CD4+ T-cell clones specific for wild-type Factor VIII: a molecular mechanism responsible for a higher incidence of inhibitor formation in mild/moderate hemophilia A. Blood. 2003;101(4):1351–1358. doi: 10.1182/blood-2002-05-1369. [DOI] [PubMed] [Google Scholar]
- 8.Roth DA, Tawa NE, Jr, O’Brien JM, Treco DA, Selden RF. Nonviral transfer of the gene encoding coagulation Factor VIII in patients with severe hemophilia A. N Engl J Med. 2001;344(23):1735–1742. doi: 10.1056/NEJM200106073442301. [DOI] [PubMed] [Google Scholar]
- 9.Powell JS, Ragni MV, White GC, 2nd, et al. Phase 1 trial of FVIII gene transfer for severe hemophilia A using a retroviral construct administered by peripheral intravenous infusion. Blood. 2003;102(6):2038–2045. doi: 10.1182/blood-2003-01-0167. [DOI] [PubMed] [Google Scholar]
- 10.White GC., 2nd Gene therapy in hemophilia: clinical trials update. Thromb Haemost. 2001;86(1):172–177. [PubMed] [Google Scholar]
- 11.Andrews JL, Kadan MJ, Gorziglia MI, Kaleko M, Connelly S. Generation and characterization of E1/E2a/E3/E4-deficient adenoviral vectors encoding human Factor VIII. Mol Ther. 2001;3(3):329–336. doi: 10.1006/mthe.2001.0264. [DOI] [PubMed] [Google Scholar]
- 12.Sarkar R, Gao GP, Chirmule N, Tazelaar J, Kazazian HHJ. Partial corretion of murine hemophilia A with neo-antigenic murine Factor VIII. Hum Gene Ther. 2000;11:881–894. doi: 10.1089/10430340050015491. [DOI] [PubMed] [Google Scholar]
- 13.Chao H, Mao L, Bruce AT, Walsh CE. Sustained expression of human Factor VIII in mice using a parvovirus-based vector. Blood. 2000;95(5):1594–1599. [PubMed] [Google Scholar]
- 14.Greengard JS, Jolly PJ. Animal testing of retroviral-mediated gene therapy for Factor VIII deficiency. Thromb Haemost. 1999;82(2):555–561. [PubMed] [Google Scholar]
- 15.VandenDriessche T, Vanslembrouck V, Goovaerts I, et al. Long-term expression of human coagulation Factor VIII and correction of hemophilia A after in vivo retroviral gene transfer in Factor VIII-deficient mice. Proc Natl Acad Sci USA. 1999;96(18):10379–10384. doi: 10.1073/pnas.96.18.10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stein CS, Kang Y, Sauter SL, et al. In vivo treatment of hemophilia A and mucopolysaccharidosis type VII using nonprimate lentiviral vectors. Mol Ther. 2001;3(6):850–856. doi: 10.1006/mthe.2001.0325. [DOI] [PubMed] [Google Scholar]
- 17.Miao CH, Ye X, Thompson AR. Highlevel Factor VIII gene expression in vivo achieved by nonviral liver-specific gene therapy vectors. Hum Gene Ther. 2003;14(14):1297–1305. doi: 10.1089/104303403322319381. [DOI] [PubMed] [Google Scholar]
- 18.Ye P, Thompson AR, Sarkar R, et al. Naked DNA transfer of Factor VIII induced transgene-specific, species-independent immune response in hemophilia A mice. Mol Ther. 2004;10(1):117–126. doi: 10.1016/j.ymthe.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Bi L, Lawler AM, Antonarakis SE, et al. Targeted disruption of the mouse Factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10(1):119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 20.Qian J, Collins M, Sharpe AH, Hoyer LW. Prevention and treatment of Factor VIII inhibitors in murine hemophilia A. Blood. 2000;95(4):1324–1329. [PubMed] [Google Scholar]
- 21.Wu H, Reding M, Qian J, et al. Mechanism of the immune response to human Factor VIII in murine hemophilia A. Thromb Haemost. 2001;85(1):125–133. [PubMed] [Google Scholar]
- 22.Hausl C, Ahmad RU, Sasgary M, et al. High-dose Factor VIII inhibits Factor VIII-specific memory B cells in hemophilia A with Factor VIII inhibitors. Blood. 2005;106(10):3415–3422. doi: 10.1182/blood-2005-03-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berntorp E, Astermark J, Carlborg E. Immune tolerance induction and the treatment of hemophilia. Malmo protocol update. Haematologica. 2000;85(10 Suppl):48–50. discussion 50–51. [PubMed] [Google Scholar]
- 24.Carlborg E, Astermark J, Lethagen S, Ljung R, Berntorp E. The Malmo model for immune tolerance induction: impact of previous treatment on outcome. Haemophilia. 2000;6(6):639–642. doi: 10.1046/j.1365-2516.2000.00436.x. [DOI] [PubMed] [Google Scholar]
- 25.Ephrem A, Chamat S, Miquel C, et al. Expansion of CD4+CD25+ regulatory T cells by intravenous immunoglobulin: a critical factor in controlling experimental autoimmune encephalomyelitis. Blood. 2008;111(2):715–722. doi: 10.1182/blood-2007-03-079947. [DOI] [PubMed] [Google Scholar]
- 26.De Groot AS, Moise L, McMurry JA, et al. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes”. Blood. 2008;112(8):3303–3311. doi: 10.1182/blood-2008-02-138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rawle FE, Pratt KP, Labelle A, et al. Induction of partial immune tolerance to Factor VIII through prior mucosal exposure to the Factor VIII C2 domain. J Thromb Haemost. 2006;4(10):2172–2179. doi: 10.1111/j.1538-7836.2006.02118.x. [DOI] [PubMed] [Google Scholar]
- 28.Lei TC, Scott DW. Induction of tolerance to Factor VIII inhibitors by gene therapy with immunodominant A2 and C2 domains presented by B cells as Ig fusion proteins. Blood. 2005;105(12):4865–4870. doi: 10.1182/blood-2004-11-4274.. •Shows a successful tolerance induction protocol using a tolerogenic B-cell gene therapy protocol.
- 29.Qadura M, Othman M, Waters B, et al. Reduction of the immune response to Factor VIII mediated through tolerogenic Factor VIII presentation by immature dendritic cells. J Thromb Haemost. 2008;6(12):2095–2104. doi: 10.1111/j.1538-7836.2008.03165.x. [DOI] [PubMed] [Google Scholar]
- 30.Ragni MV, Wu W, Liang X, et al. Factor VIII-pulsed dendritic cells reduce anti-Factor VIII antibody formation in the hemophilia A mouse model. Exp Hematol. 2009;37(6):744–754. doi: 10.1016/j.exphem.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su RJ, Epp A, Latchman Y, et al. Suppression of FVIII inhibitor formation in hemophilic mice by delivery of transgene modified apoptotic fibroblasts. Mol Ther. 2008;18(1):214–222. doi: 10.1038/mt.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goudemand J, Rothschild C, Demiguel V, et al. Influence of the type of Factor VIII concentrate on the incidence of Factor VIII inhibitors in previously untreated patients with severe hemophilia A. Blood. 2006;107(1):46–51. doi: 10.1182/blood-2005-04-1371. [DOI] [PubMed] [Google Scholar]
- 33.Waters B, Lillicrap D. The molecular mechanisms of immunomodulation and tolerance induction to Factor VIII. J Thromb Haemost. 2009;7(9):1446–1456. doi: 10.1111/j.1538-7836.2009.03538.x.. •Discusses various mechanisms involved in immunomodulation methods used along with protein-replacement therapy.
- 34.Saenko EL, Pipe SW. Strategies towards a longer acting Factor VIII. Haemophilia. 2006;12(Suppl. 3):42–51. doi: 10.1111/j.1365-2516.2006.01260.x. [DOI] [PubMed] [Google Scholar]
- 35.Pipe SW. Functional roles of the Factor VIII B domain. Haemophilia. 2009;15(6):1187–1196. doi: 10.1111/j.1365-2516.2009.02026.x.. •Discusses the advantages and disadvantages of many different Factor VIII variants.
- 36.Cerullo V, Seiler MP, Mane V, et al. Correction of murine hemophilia A and immunological differences of Factor VIII variants delivered by helper-dependent adenoviral vectors. Mol Ther. 2007;15(12):2080–2087. doi: 10.1038/sj.mt.6300308. [DOI] [PubMed] [Google Scholar]
- 37.Jirovska D, Ye P, Pipe SW, Miao CH. Reduction of inhibitory anti-FVIII actor antibody titer by using a domain variant FVIII/N6 cDNA for nonviral gene therapy in hemophilia A mice. Presented at: 50th Annual Meeting of American Society of Hematology; San Francisco, CA, USA. 6–10 December 2008. [Google Scholar]
- 38.Ettinger RA, Lieverman JA, Boigiano DC, Thompson AR, Pratt KP. Reduced immunogenicity of FVIII peptides by rational modifications of an immunodominant T cell epitope. Presented at: 51st Annual Meeting of American Society of Hematology; New Orleans, LA, USA. 4–7 December 2009. [Google Scholar]
- 39.Mingozzi F, Liu YL, Dobrzynski E, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest. 2003;111(9):1347–1356. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarkar R, Mucci M, Addya S, et al. Long-term efficacy of adeno-associated virus serotypes 8 and 9 in hemophilia A dogs and mice. Hum Gene Ther. 2006;17(4):427–439. doi: 10.1089/hum.2006.17.427. [DOI] [PubMed] [Google Scholar]
- 41.Liu L, Mah C, Fletcher BS. Sustained FVIII expression and phenotypic correction of hemophilia A in neonatal mice using an endothelial-targeted sleeping beauty transposon. Mol Ther. 2006;13(5):1006–1015. doi: 10.1016/j.ymthe.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 42.Matsui H, Shibata M, Brown B, et al. A murine model for induction of long-term immunologic tolerance to Factor VIII does not require persistent detectable levels of plasma Factor VIII and involves contributions from Foxp3+ T regulatory cells. Blood. 2009;114(3):677–685. doi: 10.1182/blood-2009-03-202267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Evans GL, Morgan RA. Genetic induction of immune tolerance to human clotting Factor VIII in a mouse model for hemophilia A. Proc Natl Acad Sci USA. 1998;95(10):5734–5739. doi: 10.1073/pnas.95.10.5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moayeri M, Hawley TS, Hawley RG. Correction of murine hemophilia A by hematopoietic stem cell gene therapy. Mol Ther. 2005;12(6):1034–1042. doi: 10.1016/j.ymthe.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ide LM, Gangadharan B, Chiang KY, Doering CB, Spencer HT. Hematopoietic stem-cell gene therapy of hemophilia A incorporating a porcine Factor VIII transgene and nonmyeloablative conditioning regimens. Blood. 2007;110(8):2855–2863. doi: 10.1182/blood-2007-04-082602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doering CB, Gangadharan B, Dukart HZ, Spencer HT. Hematopoietic stem cells encoding porcine Factor VIII induce pro-coagulant activity in hemophilia A mice with pre-existing Factor VIII immunity. Mol Ther. 2007;15(6):1093–1099. doi: 10.1038/sj.mt.6300146. [DOI] [PubMed] [Google Scholar]
- 47.Uprichard J, Dazzi F, Apperley JF, Laffan MA. Haemopoietic stem cell transplantation induces tolerance to donor antigens but not to foreign FVIII peptides. Haemophilia. 2010;16(1):143–147. doi: 10.1111/j.1365-2516.2009.02099.x. [DOI] [PubMed] [Google Scholar]
- 48.Shi Q, Wilcox DA, Fahs SA, et al. Factor VIII ectopically targeted to platelets is therapeutic in hemophilia A with high-titer inhibitory antibodies. J Clin Invest. 2006;116(7):1974–1982. doi: 10.1172/JCI28416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi Q, Wilcox DA, Fahs SA, et al. Lentivirus-mediated platelet-derived Factor VIII gene therapy in murine haemophilia A. J Thromb Haemost. 2007;5(2):352–361. doi: 10.1111/j.1538-7836.2007.02346.x. [DOI] [PubMed] [Google Scholar]
- 50.Matsui H, Shibata M, Brown B, et al. Ex vivo gene therapy for hemophilia A that enhances safe delivery and sustained in vivo Factor VIII expression from lentivirally engineered endothelial progenitors. Stem Cells. 2007;25(10):2660–2669. doi: 10.1634/stemcells.2006-0699. [DOI] [PubMed] [Google Scholar]
- 51.Kren BT, Unger GM, Sjeklocha L, et al. Nanocapsule-delivered Sleeping Beauty mediates therapeutic Factor VIII expression in liver sinusoidal endothelial cells of hemophilia A mice. J Clin Invest. 2009;119(7):2086–2099. doi: 10.1172/JCI34332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu D, Alipio Z, Fink LM, et al. Phenotypic correction of murine hemophilia A using an iPS cell-based therapy. Proc Natl Acad Sci USA. 2009;106(3):808–813. doi: 10.1073/pnas.0812090106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yadav N, Kanjirakkuzhiyil S, Kumar S, et al. The therapeutic effect of bone marrow-derived liver cells in the phenotypic correction of murine hemophilia A. Blood. 2009;114(20):4552–4561. doi: 10.1182/blood-2009-02-202788. [DOI] [PubMed] [Google Scholar]
- 54.Brown BD, Venneri MA, Zingale A, Sergi Sergi L, Naldini L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med. 2006;12(5):585–591. doi: 10.1038/nm1398. [DOI] [PubMed] [Google Scholar]
- 55.Miao CH, Ye P, Thompson AR, Rawlings DJ, Ochs HD. Immunomodulation of transgene responses following naked DNA transfer of human Factor VIII into hemophilia A mice. Blood. 2006;108(1):19–27. doi: 10.1182/blood-2005-11-4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qian J, Burkly LC, Smith EP, et al. Role of CD154 in the secondary immune response: the reduction of pre- existing splenic germinal centers and anti-Factor VIII inhibitor titer. Eur J Immunol. 2000;30(9):2548–2554. doi: 10.1002/1521-4141(200009)30:9<2548::AID-IMMU2548>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 57.Hausl C, Ahmad RU, Schwarz HP, et al. Preventing restimulation of memory B cells in hemophilia A: a potential new strategy for the treatment of antibody-dependent immune disorders. Blood. 2004;104(1):115–122. doi: 10.1182/blood-2003-07-2456. [DOI] [PubMed] [Google Scholar]
- 58.Liu L, Liu H, Mah C, Fletcher BS. Indoleamine 2,3-dioxygenase attenuates inhibitor development in gene-therapy-treated hemophilia A mice. Gene Ther. 2009;16(6):724–733. doi: 10.1038/gt.2009.13. [DOI] [PubMed] [Google Scholar]
- 59.Yu G, Dai H, Chen J, et al. Gene delivery of indoleamine 2,3-dioxygenase prolongs cardiac allograft survival by shaping the types of T-cell responses. J Gene Med. 2008;10(7):754–761. doi: 10.1002/jgm.1201. [DOI] [PubMed] [Google Scholar]
- 60.Reipert BM, Sasgary M, Ahmad RU, et al. Blockade of CD40/CD40 ligand interactions prevents induction of Factor VIII inhibitors in hemophilic mice but does not induce lasting immune tolerance. Thromb Haemost. 2001;86(6):1345–1352. [PubMed] [Google Scholar]
- 61.Rossi G, Sarkar J, Scandella D. Long-term induction of immune tolerance after blockade of CD40–CD40L interaction in a mouse model of hemophilia A. Blood. 2001;97(9):2750–2757. doi: 10.1182/blood.v97.9.2750. [DOI] [PubMed] [Google Scholar]
- 62.Abrams JR, Lebwohl MG, Guzzo CA, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest. 1999;103(9):1243–1252. doi: 10.1172/JCI5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boumpas DT, Furie R, Manzi S, et al. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003;48(3):719–727. doi: 10.1002/art.10856. [DOI] [PubMed] [Google Scholar]
- 64.Kawai T. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. 2000;6:114. doi: 10.1038/72162. [DOI] [PubMed] [Google Scholar]
- 65.Davis JC, Jr, Totoritis MC, Rosenberg J, Sklenar TA, Wofsy D. Phase I clinical trial of a monoclonal antibody against CD40-ligand (IDEC-131) in patients with systemic lupus erythematosus. J Rheumatol. 2001;28(1):95–101. [PubMed] [Google Scholar]
- 66.Peng B, Ye P, Blazar BR, et al. Transient blockade of the inducible costimulator pathway generates long-term tolerance to Factor VIII after nonviral gene transfer into hemophilia A mice. Blood. 2008;112(5):1662–1672. doi: 10.1182/blood-2008-01-128413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scott D. The Nature of Immunologic Tolerance. RG Landes Co; TX, USA: 1994. [Google Scholar]
- 68.Smith R. Immunologic tolerance in non-living antignes. Adv Immunol. 1961;1:67. [Google Scholar]
- 69.Heath AW, Chang R, Harada N, et al. Antibodies to murine CD40 stimulate normal B lymphocytes but inhibit proliferation of B lymphoma cells. Cell Immunol. 1993;152(2):468–480. doi: 10.1006/cimm.1993.1305. [DOI] [PubMed] [Google Scholar]
- 70.Woodle ES, Xu D, Zivin RA, et al. Phase I trial of a humanized, Fc receptor nonbinding OKT3 antibody, huOKT3γ1(Ala-Ala) in the treatment of acute renal allograft rejection. Transplantation. 1999;68(5):608–616. doi: 10.1097/00007890-199909150-00003. [DOI] [PubMed] [Google Scholar]
- 71.Friend PJ, Hale G, Chatenoud L, et al. Phase I study of an engineered aglycosylated humanized CD3 antibody in renal transplant rejection. Transplantation. 1999;68(11):1632–1637. doi: 10.1097/00007890-199912150-00005. [DOI] [PubMed] [Google Scholar]
- 72.Peng B, Ye P, Rawlings DJ, Ochs HD, Miao CH. Anti-CD3 antibodies modulate anti-Factor VIII immune responses in hemophilia A mice after Factor VIII plasmid-mediated gene therapy. Blood. 2009;114(20):4373–4382. doi: 10.1182/blood-2009-05-217315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Waters B, Qadura M, Burnett E, et al. Anti-CD3 prevents Factor VIII inhibitor development in hemophilia A mice by a regulatory CD4+CD25+-dependent mechanism and by shifting cytokine production to favor a Th1 response. Blood. 2009;113(1):193–203. doi: 10.1182/blood-2008-04-151597. [DOI] [PubMed] [Google Scholar]
- 74.Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3(3):199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 75.Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9(3):239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 77.Salomon B, Lenschow DJ, Rhee L, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12(4):431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 78.Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+) CD4(+) regulatory cells that control intestinal inflammation. J Exp Med. 2000;192(2):295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood. 2002;99(10):3493–3499. doi: 10.1182/blood.v99.10.3493. [DOI] [PubMed] [Google Scholar]
- 80.Edinger M, Hoffmann P, Ermann J, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003;9(9):1144–1150. doi: 10.1038/nm915. [DOI] [PubMed] [Google Scholar]
- 81.Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med. 2002;196(3):389–399. doi: 10.1084/jem.20020399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rβ-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 2002;17(2):167–178. doi: 10.1016/s1074-7613(02)00367-9. [DOI] [PubMed] [Google Scholar]
- 83.Vigouroux S, Yvon E, Biagi E, Brenner MK. Antigen-induced regulatory T cells. Blood. 2004;104(1):26–33. doi: 10.1182/blood-2004-01-0182. [DOI] [PubMed] [Google Scholar]
- 84.Taylor PA, Panoskaltsis-Mortari A, Swedin JM, et al. L-Selectin(hi) but not the L-selectin(lo) CD4+25+ T-regulatory cells are potent inhibitors of GVHD and BM graft rejection. Blood. 2004;104(12):3804–3812. doi: 10.1182/blood-2004-05-1850. [DOI] [PubMed] [Google Scholar]
- 85.Rifle G, Herve P. Regulatory (suppressor) T cells in peripheral allograft tolerance and graft-versus-host reaction. Transplantation. 2004;77(1 Suppl):S5. doi: 10.1097/01.TP.0000107184.18562.FC. [DOI] [PubMed] [Google Scholar]
- 86.Clark FJ, Gregg R, Piper K, et al. Chronic graft-versus-host disease is associated with increased numbers of peripheral blood CD4+CD25high regulatory T cells. Blood. 2004;103(6):2410–2416. doi: 10.1182/blood-2003-06-2073. [DOI] [PubMed] [Google Scholar]
- 87.Jaeckel E, von Boehmer H, Manns MP. Antigen-specific FoxP3-transduced T-cells can control established type 1 diabetes. Diabetes. 2005;54(2):306–310. doi: 10.2337/diabetes.54.2.306. [DOI] [PubMed] [Google Scholar]
- 88.Kasprowicz DJ, Smallwood PS, Tyznik AJ, Ziegler SF. Scurfin (FoxP3) controls T-dependent immune responses in vivo through regulation of CD4+ T cell effector function. J Immunol. 2003;171(3):1216–1223. doi: 10.4049/jimmunol.171.3.1216. [DOI] [PubMed] [Google Scholar]
- 89.Miao CH, Harmeling BR, Ziegler SF, et al. CD4+FOXP3+ regulatory T cells confer long-term regulation of Factor VIII-specific immune responses in plasmid-mediated gene therapy-treated hemophilia mice. Blood. 2009;114(19):4034–4044. doi: 10.1182/blood-2009-06-228155.. ••Adoptive transfer of CD4+Foxp3+ Treg cells isolated from transgenic hemophilia A/Foxp3 mice modulated short-term and long-term anti-FVIII responses in a plasmid-mediated gene transfer model.
- 90.Tran CA, Burton L, Russom D, et al. Manufacturing of large numbers of patient-specific T cells for adoptive immunotherapy: an approach to improving product safety, composition, and production capacity. J Immunother. 2007;30(6):644–654. doi: 10.1097/CJI.0b013e318052e1f4. [DOI] [PubMed] [Google Scholar]
- 91.Guillot-Delost M, Cherai M, Hamel Y, et al. Clinical-grade preparation of human natural regulatory T-cells encoding the thymidine kinase suicide gene as a safety gene. J Gene Med. 2008;10(8):834–846. doi: 10.1002/jgm.1220. [DOI] [PubMed] [Google Scholar]
- 92.Cao O, Loduca PA, Herzog RW. Role of regulatory T cells in tolerance to coagulation factors. J Thromb Haemost. 2009;7(Suppl. 1):88–91. doi: 10.1111/j.1538-7836.2009.03417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chai JG, Coe D, Chen D, et al. In vitro expansion improves in vivo regulation by CD4+CD25+ regulatory T cells. J Immunol. 2008;180(2):858–869. doi: 10.4049/jimmunol.180.2.858. [DOI] [PubMed] [Google Scholar]
- 94.Liu CL, Ye P, Y B, Peng B, Miao CH. In vivo expansion of Treg cells with IL2–mAb complexes prevents and modulates anti-Factor VIII immune responses in hemophilia A mice after Factor VIII plasmid-mediated gene therapy. Presented at: 13th Annual Meeting of American Society of Gene and Cell Therapy; Washington, DC, USA. 19–22 May 2010.. ••Showed that expansion of activated Treg cells has profound suppressive effect on anti-Factor VIII immune responses, which induced long-term tolerance to Factor VIII in a prevention model and modulated immune responses in antigen-primed mice with pre-existing antibodies.
- 95.Su Y, Carey G, Maric M, Scott DW. B cells induce tolerance by presenting endogenous peptide-IgG on MHC class II molecules via an IFN-γ-inducible lysosomal thiol reductase-dependent pathway. J Immunol. 2008;181(2):1153–1160. doi: 10.4049/jimmunol.181.2.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.El-Amine M, Melo M, Kang Y, et al. Mechanisms of tolerance induction by a gene-transferred peptide-IgG fusion protein expressed in B lineage cells. J Immunol. 2000;165(10):5631–5636. doi: 10.4049/jimmunol.165.10.5631. [DOI] [PubMed] [Google Scholar]
- 97.Litzinger MT, Su Y, Lei TC, Soukhareva N, Scott DW. Mechanisms of gene therapy for tolerance: B7 signaling is required for peptide–IgG gene-transferred tolerance induction. J Immunol. 2005;175(2):780–787. doi: 10.4049/jimmunol.175.2.780. [DOI] [PubMed] [Google Scholar]
- 98.Skupsky J, Su Y, Scott DW. Tolerance induced by B-cell gene therapy: expansion and increased activity of antigen-specific Tregs. Presented at: 51st Annual Meeting of American Society of Hematology; New Orleans, LA, USA. 4–7 December 2009.. •Demonstrates the induction of tolerance by tolerogenic B-cell gene therapy via the mechanism involving expansion and activation of functional Treg cells.
- 99.Mathias M, Khair K, Hann I, Liesner R. Rituximab in the treatment of alloimmune Factor VIII and IX antibodies in two children with severe haemophilia. Br J Haematol. 2004;125(3):366–368. doi: 10.1111/j.1365-2141.2004.04916.x. [DOI] [PubMed] [Google Scholar]
- 100.Carcao M, St Louis J, Poon MC, et al. Rituximab for congenital haemophiliacs with inhibitors: a Canadian experience. Haemophilia. 2006;12(1):7–18. doi: 10.1111/j.1365-2516.2005.01170.x. [DOI] [PubMed] [Google Scholar]
- 101.Curtin J, Misra A, Teo J, Webster B, Lammi A. Use of Rituximab as an laternarative strategy for the management of difficult high titre inhibitors in children with haemophilia A. Haemophilia. 2004;10(Suppl. 3) 12 PO 30. [Google Scholar]
- 102.Krause M, Betz C, Scharrer I, Krause M. Rituximab – a challenge for the therapy of inhibitors to Factor VIII. J Throm Haemost. 2003;1(Suppl. 1):P1615. [Google Scholar]
- 103.Pruthi RK, Schmidt KA, Slaby JA, Hook CC, Nchols WL. Rituximab treatment of FVIII inhibitors in congenital haemophilia A (cHA) J Thromb Haemost. 2005;3(Suppl. 1):P0652. [Google Scholar]
- 104.Dunkley S, Lindeman R. Successful treatment of a high titre Factor VIII inhibitor with rituximab alone. J Thromb Haemost. 2005;3(Suppl. 1):P0631. [Google Scholar]
- 105.Wiestner A, Cho HJ, Asch AS, et al. Rituximab in the treatment of acquired Factor VIII inhibitors. Blood. 2002;100(9):3426–3428. doi: 10.1182/blood-2002-03-0765. [DOI] [PubMed] [Google Scholar]
- 106.Biss TT, Velangi MR, Hanley JP. Failure of rituximab to induce immune tolerance in a boy with severe haemophilia A and an alloimmune Factor VIII antibody: a case report and review of the literature. Haemophilia. 2006;12(3):280–284. doi: 10.1111/j.1365-2516.2006.01212.x. [DOI] [PubMed] [Google Scholar]
- 107.Fox RA, Neufeld EJ, Bennett CM. Rituximab for adolescents with haemophilia and high titre inhibitors. Haemophilia. 2006;12(3):218–222. doi: 10.1111/j.1365-2516.2006.01215.x. [DOI] [PubMed] [Google Scholar]
- 108.Giulino LB, Bussel JB, Neufeld EJ. Treatment with rituximab in benign and malignant hematologic disorders in children. J Pediatr. 2007;150(4):338–344. 344e1. doi: 10.1016/j.jpeds.2006.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang AA, Skupsky J, Scott DW. Affect of B-cell depletion on inhibitor antibody formation and immune tolerance injction of human Factor VIII in hemophilia A mice. Presented at: 51st Annual Meeting of American Society of Hematology; New Orleans, LA, USA. 4–7 December 2009. [Google Scholar]
- 110.Ye P, Peng B, Kehry M, Rawlings DJ, Miao CH. Depletion of B cells by anti-CD20 partially regulates anti-FVIII antibody production in the nonviral gene therapy model. Presented at: 13th Annual Meeting of American Society of Gene and Cell Therapy; Washington, DC, USA. 19–22 May 2010. [Google Scholar]