

Abstract
It is well known that using random RNA/DNA sequences for SELEX experiments will generally yield low-complexity structures. Early experimental results suggest that having a structurally diverse library, which, for instance, includes high-order junctions, may prove useful in finding new functional motifs. Here, we develop two computational methods to generate sequences that exhibit higher structural complexity and can be used to increase the overall structural diversity of initial pools for in vitro selection experiments. Random Filtering selectively increases the number of five-way junctions in RNA/DNA pools, and Genetic Filtering designs RNA/DNA pools to a specified structure distribution, whether uniform or otherwise. We show that using our computationally designed DNA pool greatly improves access to highly complex sequence structures for SELEX experiments (without losing our ability to select for common one-way and two-way junction sequences).
Keywords: in vitro selection, random pool, aptamer pool design, RNA/DNA secondary structure, genetic algorithm, ATP aptamer
INTRODUCTION
Background
Aptamers are target-binding nucleic acid molecules that can bind with high affinity and selectivity to a range of molecules including antibiotics, proteins, viruses, catalytic RNA molecules, and most recently, whole cells (Wilson and Szostak 1999; Hermann and Patel 2000; Hodgson and Suga 2004; Shangguan et al. 2006). Methods for the evolution of aptamers were first established by Gold and Szostak (Ellington and Szostak 1990; Tuerk and Gold 1990). To date, over 3400 aptamers that bind to a wide variety of ligands have been selected using a biochemical process known as Systematic Evolution of Ligands by Exponential Enrichment (SELEX) (Lee et al. 2004; Thodima et al. 2006). SELEX involves the repetitive partitioning of a library of random nucleic acid sequences on the basis of selective binding to the desired target, followed by amplification by PCR.
Recent in vitro selection experiments have led to the identification of novel aptamers for biotechnological purposes such as functional genomics (Famulok and Verma 2002) and biosensing (Soukup and Breaker 1999). Other recent applications of aptamers include control of gene expression and medical diagnosis (Breaker 2004; Isaacs et al. 2006). As the applications of aptamers grow, the need for design, identification, and selection of novel and improved aptamers becomes even more apparent.
Problem
In RNA and DNA, junctions are common secondary structural elements that connect different helical segments (Lilley et al. 1995). RNA junctions are important for the structural and catalytic properties of RNAs. It has been shown that they are involved in a variety of different functional roles in nucleic acids, including the self-cleaving catalytic properties of the hammerhead ribozyme (Scott et al. 1996), promotion of functional folded states of the hairpin ribozyme (Wilson et al. 2005), recognition of the binding pocket domain by purine riboswitches (Batey et al. 2004; Serganov et al. 2004), and translation initiation of the hepatitis C virus at the internal ribosome entry site (Kieft et al. 2002).
It is well known that in vitro random RNA/DNA pools are not structurally diverse and heavily favor simple topological structures such as stem–loops structures due to incomplete and insufficient random sequence sampling (Gevertz et al. 2005). Analysis of existing aptamers has revealed that the majority of oligonucleotide sequences generated from random in vitro selection experiments have simple structures with low degrees of complexity and that highly complex structures are far less abundant (Khoo et al. 2002; Zinnen et al. 2002; Laserson et al. 2004). This lack of structural diversity in random pools may explain why complex structure motifs such as high-order junctions are rare in known synthetic aptamers. To further demonstrate this, we folded the 2793 known aptamers from the Ellington Lab aptamer database (Lee et al. 2004) by ViennaRNA (Hofacker et al. 1994; Hofacker 2003) and then counted the number of junctions of these known aptamers using the CountJunctions algorithm described in the Supplemental Material. The results showed that among these known aptamers, <1% contain four-way and five-way junction structures (see Supplemental Fig. S1). However, natural aptamers (e.g., riboswitches) more commonly contain high-order junctions. For example, the lysine riboswitch involves three-helical and two-helical bundles joined by a five-way junction (Serganov et al. 2008). Almost all other riboswitches can be classified into one of two types based on their high-junction number. Type I riboswitches contain a fundamental three-way junction architecture. In Type II riboswitches, such as the SAM-I binding riboswitch, the aptamer domain is arranged in a four-way junction structure (Schwalbe et al. 2007).
It has recently been shown that increasing the structural diversity of the starting oligonucleotide pool can enhance the possibility of finding novel aptamers with improved activity (Carothers et al. 2004, 2006). In a recent investigation, five-way junctions were found to provide the scaffold for the formation of functionally more active catalytic deoxyribozymes (Chiuman and Li 2006). It was shown that the structures with 5Js were evolved from pre-existing and less complex structures with three-way junctions, and that the more complex structures had a significantly higher rate of activity.
Consequently, novel approaches have been used that aim to increase the structural complexity of starting pools. It has been shown that structural complexity is largely dependent on sequence length (Sabeti et al. 1997). However, RNA/DNA pools for SELEX are usually restricted to sequences of at most 100 nucleotides (100 nt) in length due to the difficulty involved in the synthesis above this length. This 100-nt starting sequence typically includes 15-nt primer sites at both the 3′ and 5′ ends, which reduces the total number of variable positions from 100 to 70. Thus, effective methods should make use of primer sites as part of their strategy for predicting high-complexity structures. One approach to increase the structural complexity is to keep the structure constant and introduce random segments in the proximity of the existing structure (Jaeger et al. 1999; Ohuchi et al. 2002; Lau et al. 2004; Yoshioka et al. 2004). Other approaches involve changing sequence length and composition (Knight et al. 2005; Legiewicz et al. 2006). Davis and Szostak (2002) isolated a high-affinity GTP aptamer from a partially structured RNA library, one-half of which consisted of fully random sequences and the other half of a random region with a designed stem–loop in the middle. Subsequent analysis of the obtained aptamers revealed that most of those with highest affinity originated from the partially structured portion of their pool. They also observed that the highest affinity aptamers had fairly complex structures (Carothers et al. 2004) and concluded from their experiments that more complex RNA structures are required for greater GTP-binding activity.
However, the above observations are based on ad-hoc pool designs for individual experiments and targets. Furthermore, these ad-hoc pools with more complex structures were generated by inserting particular stem–loops that were known a priori to be relevant for the given target (Davis and Szostak 2002). Such prior knowledge is not available in many cases. Hence, it is necessary to develop more general pool design approaches that can be used for targets without such prior knowledge. Recently, Schlick and coworkers (Kim et al. 2007a,b, 2010) developed a computational approach for designing structured RNA pools by modeling the pool generation process using mixing matrices. However, the scope of this approach was limited and the approach is not computationally efficient (see Discussion).
Contributions
We have studied the design of RNA/DNA pools for in vitro selection experiments and developed two new methods to improve their structural complexity and diversity:
A computational method called Random Filtering to selectively increase the number of five-way junctions in RNA/DNA pools.
A computational method called Genetic Filtering to design RNA/DNA pools with any desired structure distribution, including a uniform structure distribution, i.e., 20% 1-way (1J), 2-way (2J), 3-way (3J), 4-way (4J), and 5-way (5J) junctions each.
Our first aim was to increase the structural complexity of RNA/DNA pools. Since RNA/DNA secondary structures can be described using unlabeled tree graphs (Gan et al. 2003), we used the degree of branching, i.e., the number of junctions, to measure their structural complexity. In this approach, a tree graph with a higher order junction has a greater structural complexity. Random Filtering led to a significant enrichment in the number of highly complex structures in DNA pools. For example, pools were generated where >10% of the sequences are five-way junctions and >15% of the sequences are four-way junctions as compared with nearly zero five-way junctions and around 0.2% four-way junctions in a typical random DNA pool. We found very similar results for 100-nt DNA pools where 30 nt are fixed primer-binding regions.
Our second aim was to engineer a RNA/DNA pool with a uniform distribution over a range of possible structural complexities to maximize the diversity of sequences available in the starting pool for in vitro selection (Gevertz et al. 2005). For a given set of primer sites determined by the respective SELEX experiment, our Genetic Filtering method can provide a roadmap for creating a RNA/DNA pool with nearly uniform structure distribution (i.e., 20% of each 1J, 2J, 3J, 4J, and 5J structures).
We tested the hypothesis that our designed pools can increase the chance of sampling more complex oligonucleotide structures during in vitro selection. It has already been observed by Davis and Szostak (2002), for the case of GTP aptamers, that pools based on customized starting sequences may increase the likelihood of finding better aptamer sequences. To further test this hypothesis, we used a DNA sequence designed with our Genetic Filtering method to generate a uniform structure distribution for the starting pool of a SELEX experiment to select for ATP-binding DNA aptamers. We found that after eight rounds of selection cycles, complex structures such as five-way junctions accounted for 20% of the sampled sequences. These experiments confirmed that our methods greatly improved the likelihood of generating sequences of increased structural complexity for SELEX experiments.
RESULTS
Increasing the number of complex structures in SELEX starting pools
Random Filtering method
Our first goal was to increase the percentage of complex structures in starting pools for in vitro selection experiments. We devised a computational method termed Random Filtering, presented in the Materials and Methods section, which selectively increases the number of five-way junctions in DNA pools for SELEX. In very simplified terms, it starts from a random RNA or DNA pool and computes the number of junctions for each sequence in the pool. Each 5J sequence is then mutated at every nonprimer single-stranded site 1 million times so as to calculate the structure distribution of the respective pool design. The pool with the largest percentage of five-way junctions is selected.
Using Vienna RNA to fold 1 million 100-nt random sequences (the choice of Vienna RNA is discussed in the Supplemental Material), we identified 76 5J sequences. These sequences were subjected to Random Filtering to generate a 5J enhanced pool. Figure 1A shows an increase in the structural complexity (using junction order) of a pool generated from Random Filtering (using the 76 5Js as a starting point) as compared with a pool of 1 million 100-nt random sequences. The most frequent structures in the Random Filtering pools are 3Js (48.65%), while the most frequent structures in the random pools are 2Js (64.35%). In the Random Filtering pools, 10.23% of the sequences are 5Js and 15.45% of the sequences are 4Js, compared with 0.01% 5Js and 0.95% 4Js in the random pools.
FIGURE 1.

Comparison of average structural distribution (percent of sequences displaying a given junction order: either one-way, two-way, three-way, four-way, or five-way junction structures) of 1 million sequences for random RNA/DNA pools (dark gray) and RNA/DNA pools generated by our Random Filtering process (light gray). (A) Without primer sites, Random Filtering has greatly increased the percentage of high-complex structures such as 5Js (by 10%) and 4Js (by 15%). (B) With 15-nt primer sites at both ends, “GGAAGAGATGGCGAC” and 15 “AGCTGATCCTGATGG” (light gray). Random Filtering has greatly increased the percentage of high complex structures such as 5Js (by 6%) and 4Js (by 10%).
Since primer sites (short-fixed sequences at the 5′- and 3′-ends required for amplification) are generally used in SELEX experiments, we considered the effect that including them would have on structural diversity. We used a 15-nt 5′ primer site “GGAAGAGATGGCGAC” and a 15-nt 3′ primer site “AGCTGATCCTGATGG,” thereby occupying 30 fixed positions in our starting sequence. This leaves only 70 nt that can be modified for the design of the starting pool, decreasing the sequence length from 100, making it considerably more difficult to find complex structures (Sabeti et al. 1997).
From a 100-nt pool of 20 million random sequences containing these primer sites, we obtained only 35 sequences exhibiting 5Js. These sequences were used to generate a pool using Random Filtering. Compared with 76 5Js found in a 1-million 100-nt random pool without primer sites, 5Js are 40-fold less abundant in a 100-nt pool containing 30-nt fixed primer sites. According to Sabeti et al. (1997), a 5J motif in a 70-nt random pool is about 100- to 200-fold less abundant than a 5J motif in a 100-nt random pool. However, primer sites can participate in the formation of junctions, even though the effective pool length has been shortened. Therefore, the 5Js abundances in a 100-nt pool containing 30-nt primer sites should be higher than a simple 70-nt random pool. More discussion of the relationship between sequence length and structure distribution can be found in the Supplemental Material.
Similar to the case without primer sites, Random Filtering produces a more structurally complex pool (Fig. 1B). The most frequent structures in our Random Filtering pools are 3J sequences (45.76%), while the most frequent structures in random pools are 2J (71.72%). In Random Filtering pools, 6.65% of the sequences are 5Js and 9.58% of the sequences are 4Js, compared with 0.00% 5Js and 0.19% 4Js in random pools. Therefore, Random Filtering increases the percentage of highly complex structures available for selection in the SELEX experiments. The sequence shown in Figure 2 called RFPool A has 19 single-stranded positions (indicated as “N”), excluding primer sites and resulted in the largest number of 5Js (31.13%) (Table 1).
FIGURE 2.

Sample secondary structure of the RFPool A DNA sequence including primer sites “GGAAGAGATGGCGAC” and “AGCTGATCCTGATGG” designed to generate the highest percentage of 5J sequences (31.13%). Image generated by Mfold. The sequence has 19 single-stranded positions, excluding primer sites (noted as “N”). As a starting pool for SELEX experiments, these 19 single-stranded positions are considered as random (N) and the other positions are considered as fixed.
TABLE 1.
In silico and in vitro structural distribution for the RFPool

The RFPool A was synthesized, amplified, cloned, and sequenced, and these sample sequences were folded using ViennaRNA. Of the 17 sequences recovered, 11.76% of them formed five-way junctions (see Table 1; sequences available in Supplemental Table S1).
Designing a diverse SELEX starting pool with uniform structural distribution
Genetic Filtering method
Recent findings show that increased structural diversity of the starting oligonucleotide pool can enhance the possibility of finding novel aptamers with improved activity (Carothers et al. 2004, 2006). However, random pools are not structurally diverse and heavily favor simple structures such as hairpin loops (Gevertz et al. 2005). In the previous section we showed how the Random Filtering method enriches a pool with complex structures such as five-way junctions. In this section, we introduce a new method, termed Genetic Filtering, to design a diverse starting pool for in vitro selection with a uniform structure distribution, i.e., a pool with 20% of each 1J, 2J, 3J, 4J, and 5J structures.
For a given pair of primer sites, Genetic Filtering first creates an initial generation of oligonucleotide pool designs. See Figure 2 for an example of a pool design. Each pool design is assigned a fitness score that indicates how close a pool derived from that design is to the desired uniform structure distribution. New generations of pool designs are created by selecting designs from previous generations with better (i.e., smaller) fitness scores and applying three types of operations: mutation, copy, and crossover. Genetic Filtering creates several generations of pool designs (typically between 500 and 3000) until either a pool design with uniform structure distribution is obtained or the fitness score of the best pool design remains unchanged for a number of generations.
We further modified the fitness score for pool designs in Genetic Filtering such that designs without a desired number of single-stranded (random) positions were penalized. In our case, we chose 24 as the ideal number of single-stranded positions. The reason for this lies in the SELEX experiment. Most SELEX experiments begin with a starting pool consisting of nanomoles of sequences, as this is a reasonable amount of DNA that can be synthesized. This translates to ∼1014–1015 sequences. Given that four possible bases can be inserted at any random sequence position and 1015 ≈ 425, this implies a target number of random positions in the pool design of ∼25 is sufficient for the entire sequence space to be sampled once in a given experiment. Here, we chose to include 24 random positions in our pool design to ensure complete sequence coverage in our starting pool (i.e., allowing four expected copies for every different sequence).
We ran our Genetic Filtering method on a 100-nt pool design with primer sites “ATACCAGCTTATTCAATT” and “AGATAGTAAGTGCAATCT” for 571 generations with a run-time of 2 h, 10 min on a 200 processor cluster. The best fitness score decreases steadily with each generation until it remains steady after ∼500 generations (Fig. 3). The distribution is close to a uniform distribution (Table 2) with a distance from the optimal of 2.95. The best pool design, GFPool1, is also shown in Figure 3. The results of other Genetic Filtering pools with different primer sites or number of variable positions are described in the Supplemental Table S2 and Supplemental Figures S3 and S4.
FIGURE 3.

The best fitness value observed so far at each generation of Genetic Filtering (primer sites “ATACCAGCTTATTCAATT” and “AGATAGTAAGTGCAATCT”) Parameters: NG = 10,000 NoChargeCutoff = 500 Pcrossover = 0.0 Pmutation = 0.9 Pposition = 0.0156 Pcopy = 0.1 (see Materials and Methods). The best fitness score (i.e., smallest value), which indicates its closeness to the desired uniform structure distribution, decreases steadily with each generation until it remains steady after ∼500 generations. (Inset) Sample secondary structure of the DNA pool sequence (GFPool1). This 100-nt in-length pool, containing 24 random nucleotide positions, was synthesized and used in selection experiments against ATP. Image generated by Mfold.
TABLE 2.
In silico structural distribution, distance from the optimal, and number of single-stranded positions present for DNA pools with primer sites, as well as experimental structural distributions prior to starting SELEX and after eight rounds of selection using ATP as the target

Result of SELEX experiment with improved pool design
SELEX experiments were performed using our uniform structure pool, GFPool1, generated by our Genetic Filtering method, in an effort to confirm that our design approach yielded a pool that allowed for an increased range of complex structures to be sampled during the selection process. Selections were performed to find DNA sequences that showed affinity for ATP (Huizenga and Szostak 1995). In the original ATP aptamer SELEX experiments, the random DNA pool used to screen for ATP binders consisted of ∼2 × 1014 different sequences made up of a 72-nt random region flanked by defined primer binding sites. After eight rounds of selection, the random region of the sequence with the best binding affinity was determined to be 5′-GACTGGGCTTGTGCTTGGGGGAGTATTGCGGAGGAAAGCGGCCCTGCTGAAGTGGGATACATGTGGATACCC -3′ (JW Szostak, pers. comm.). This sequence, with its flanking primer sites, is of low complexity, predicted to form a 2J. Given the extensive study of this system, we chose ATP as the target for testing our uniform structure pool, GFPool1, generated by our Genetic Filtering method. A total of eight rounds of SELEX were performed, equivalent to the original procedure by Szostak. The recovery of DNA after each round, corresponding to the amount of DNA binding to the ATP target, was monitored using UV-vis absorbance and fluorescence measurements. With each round, an increased percentage of the pool DNA bound to the target was observed (see Fig. 4). DNA from GFPool1 prior to selections as well from the round eight enriched pool was cloned and sequences were obtained. The secondary structures were analyzed using Vienna RNA. Table 2 shows the distribution of junction structures obtained for both the starting and enriched pools (sequences can be found in Supplemental Tables S3, S4). Of the 19 sequences obtained experimentally from the nonenriched starting pool, 1Js, 2Js, 3Js, 4Js, and 5Js were found. Additionally, 2Js, 3Js, and 5Js were found in the enriched pool, confirming not only the structural diversity of this designed pool, but also that complex structures were retained after several rounds of enrichment.
FIGURE 4.

Percent recovery of DNA eluted from the ATP selection column versus selection round. With each round, an increased percentage of the pool DNA bound to the target was observed (recovery determined using UV-Vis and fluorescence measurements).
Two of the 5J sequences, ATPF4 and ATPF8 (see Supplemental Table S4), were tested for binding affinity to ATP, yielding dissociation constant (Kd) values of 24.4 μM and 3.7 μM, respectively (see Supplemental Fig. S5). By comparison, a Kd of 6 μM is reported for the published ATP aptamer sequence (Huizenga and Szostak 1995). It is important to note that the published ATP aptamer sequence underwent extensive optimization and minimization prior to binding-affinity experiments. The fact that one 5J generated from our SELEX experiments shows improved binding over that optimized system, while the other 5J does not, suggests that complexity alone does not guarantee better target binding, but higher complexity structures do have the potential to yield better aptamers. This clearly underscores the need for structural diversity, not just structural complexity, in the initial starting pool.
DISCUSSION
Analysis of existing aptamers have revealed that the majority of RNA sequences, which are selected from random in vitro selection experiments, have simple structures with low degrees of complexity, and that the highly complex structures are far less abundant (Khoo et al. 2002; Zinnen et al. 2002; Laserson et al. 2004). For example, aptamers that bind to ATP, chloramphenicol, neomycin B, and streptomycin all have linear stem–loop or slightly branched structures (Laserson et al. 2004). In a recent computational analysis of in vitro RNA random pools (Gevertz et al. 2005), it was shown that random pools are not structurally diverse. They heavily favor simple topological structures due to incomplete and insufficient random sequence sampling. The structure distribution for random pools shown in Figure 1, which is consistent with the analytical results in Sabeti et al. (1997), illustrates that 99.80% of structures in a random pool are expected to be 1J, 2J, or 3J structures. This lack of structural diversity in random pools suggests that complex structure motifs with high-order junctions would be rare in selected aptamers.
Recent experimental findings show that enhancing the structural diversity of RNA/DNA pools increases the chance of finding novel aptamers with higher binding affinity (Carothers et al. 2004, 2006). To increase the number of functional RNA/DNA sequences available in the starting pool for in vitro selection, the starting pool should be engineered to have a uniform distribution over all possible structures (Gevertz et al. 2005).
In this study, we present two new systematic computational pool design approaches that increase the covered search space for in vitro selection of complex aptamers: Random Filtering and Genetic Filtering. Compared with close to zero five-way junctions and around 0.2% four-way junctions in a typical random DNA pool with 106 structures of 100 nucleotides each, Random Filtering generated pools of the same size, where >10% of the sequences are five-way junctions and >15% of the sequences are four-way junctions. Very similar results were found for 100-nt DNA pools where 30 nt are fixed primer sites. Our RFPool A (designed via Random Filtering) was synthesized, and a few sequences were determined and folded using ViennaRNA. In a subset of only 17 sequences, 11.76% were found to be five-way and 11.76% were four-way junctions. Compared with a completely random pool, our Random Filtering method can dramatically increase the likelihood of obtaining high-order junctions experimentally.
Our second method, Genetic Filtering, can generate DNA pool designs of 100 nt each, where 30 nt are fixed primer sites, with distributions in silico that are very close to a uniform structure distribution, i.e., highly diverse. Analysis of a small set of the sequences obtained from the GFPool1 (designed via Genetic Filtering) in vitro showed significant structural diversity, with each of the 1J, 2J, 3J, 4J, and 5J structures present at levels of at least 10%. Complex 5J structures accounted for 26% of the sequences prior to enrichment. After GFPool1 was subjected to eight rounds of selection against the target ATP, complex 5Js accounted for 20% of the sampled sequences. The likelihood of observing this concentration of 5J structures in a random pool, where we find only 35 of 20 million sequences resulting in 5J structures, is ∼10−10. These results confirm that our suggested approach not only leads to better access to more complex structures for in vitro selection experiments, but also that SELEX is capable of preserving highly complex structures if they are present in the initial DNA pool. When the binding affinities of two 5J sequences from this enriched pool were tested, one sequence (ATPF8) displayed stronger affinity for ATP than Huizenga and Szostak's low-complexity aptamer, while the other (ATPF4) showed weaker affinity. This suggests that higher complexity sequences have the potential to be better aptamers, but will not always lead to improved binding. Thus, structural diversity, not just structural complexity, is an important characteristic of an ideal starting pool for SELEX. Our Genetic Filtering method is particularly aimed at designing such pools.
It may initially appear that the structural diversity of a pool resulting from our filtering approaches will be overly constrained given the stretches of fixed positions in the stems. However, an examination of a number of structures resulting from the same pool design demonstrates that while the sequence in these regions is constrained, the corresponding structure is not. Examples of different junction structures formed from the pool in Figure 2 are shown in Supplemental Figure S6. These sample structures illustrate that the same fixed sequence region may participate in a stem in one structure while forming a loop in another. For example, in the 5J structure shown in Supplemental Figure S6A, the sequence GCGT starting at base 61 participates in a stem, while the same fixed sequence forms a loop in the corresponding 4J structure illustrated in Supplemental Figure S6B.
While Inverse Folding associated with the ViennaRNA package was developed to find sequences that fold into a predefined structure (Hofacker 2003), it cannot be applied for the design of starting pools for in vitro selection. This is primarily due to the fact that Inverse Folding cannot generate sequences containing primer sites directly, which is required for these in vitro experiments. As shown in Supplemental Figure S7, adding primers to complex structures post-design can have a dramatic effect. From 20 million 5Js of length 70 nt generated by Inverse Folding, <1% of engineered sequences retained their structure upon the addition of two primer sites of length 15 nt. In addition, we compared the Inverse Folding method and the Random Filtering method, and found that Random Filtering generates more complex structures than Inverse Folding, making our method more suitable to generate structurally diverse pools for in vitro experiments (a description of the methods and results are found in Supplemental Fig. S8).
Schlick and coworkers (Kim et al. 2007a,b, 2010) developed a computational approach for designing structured RNA pools by modeling the pool generation process using mixing matrices. We compared our method with the mixing matrices approach. Applying the mixing matrix approach to create a pool with a uniform distribution of 12 tree structures (see Supplemental Material) resulted in error rates of 60% (using one mixing matrix) and 38.9% (using two mixing matrices). In contrast, Genetic Filtering reduced the error to 35.23% (with one single pool, which is equivalent to one mixing matrix). The structural distribution and pool design obtained by this experiment is shown in Supplemental Figure S9. For in vitro experiments, our method requires only one synthesis, whereas the mixing matrices approach needs one synthesis for each mixing matrix. Therefore, our method is also less costly and less time consuming compared with the mixing matrices approach.
Our results complement the mixing matrices approach in several ways. Random Filtering and Genetic Filtering are able to add more complex structures to the pool. For example, the most complex structures created via mixing matrices are 4J structures, whereas Random Filtering and Genetic Filtering can increase and control, respectively, the number of 5J structures in the pool. The mixing matrices approach also does not cover the inclusion of primer sites. However, primer-binding regions are required in essentially all in vitro selection experiments. Since the primer regions are considered in our approach, our designed pools can be directly implemented into in vitro selection experiments. While we showed how Genetic Filtering generates pools with uniform structure distributions, the method can be used to design any given target structure distribution, not only for junction structures, but also for tree structures. The approach can also be applied to design pools, including specific substructures such as common structural components of aptamers.
In conclusion, we have developed two methods for designing improved starting pools for functional nucleic acid selection experiments: Random Filtering and Genetic Filtering. Random Filtering yields a DNA pool (with and without primer-binding regions) with significant enrichment in the number of highly complex structures present. Genetic Filtering allows us to engineer diverse DNA pools with a nearly uniform distribution over all possible structures from 1Js to 5Js. A uniform pool design created with Genetic Filtering was synthesized and subjected to a SELEX experiment for ATP binders. We found that after eight rounds of selection, complex 5J structures still accounted for a sizeable percentage of the pool, confirming that our methods greatly improved generation of high-complexity structures, and that these structures were preserved during the selection process. The disparate binding affinity of two 5J sequences from this enriched pool confirmed that higher complexity sequences have the potential to lead to better aptamers, but certainly do not guarantee improved binding. This suggests that SELEX pool designs should aim to increase structural complexity, while still preserving a diversity of both low and high complexity structures. Future work will examine in more depth whether these designed pools lead more generally to aptamers with improved binding characteristics.
MATERIALS AND METHODS
Computational method: Random Filtering
Random Filtering is a computational method to selectively increase the number of five-way junctions in DNA pools for SELEX. Random Filtering proceeds as follows: We start from a random RNA or DNA pool and use ViennaRNA (see discussion below) to generate their secondary structures. We then compute the number of junctions for each sequence with the CountJunctions algorithm outlined below. Each 5J sequence is then mutated at every single-stranded position by randomly choosing one of four bases (ACGT) to substitute for the original. This process is repeated 1 million times for each 5J sequence so as to calculate the structure distribution of each respective pool design. Sequences are only mutated at nonprimer positions. The pool with the largest percentage of five-way junctions is selected. It was initially unclear whether using a million rounds of mutations was sufficient to determine the distribution with a reasonable degree of accuracy. Supplemental Table S5 shows a typical example for a pool's structure distribution determined with different numbers of mutations. Each experiment was repeated 100 times, and the numbers shown represent the average and variance for each value. The averages are very stable for different numbers of mutations. However, the variance is clearly reduced with additional experiments, reaching very close to zero when the number of mutations reaches 1 million. Hence, 1 million mutations are expected to be sufficient to reliably estimate the structure distribution.
Secondary structure prediction was performed using the ViennaRNA 1.6.5 (Hofacker 2003) software package, in which RNAfold generates a single RNA/DNA secondary structure prediction through energy minimization based on a dynamic programming algorithm originally developed by Zuker and Stiegler (1981). Our choice of ViennaRNA for secondary structure prediction is discussed in the Supplemental Material. ViennaRNA (Hofacker 2003) provides as output a nucleotide pairing list indicating for each nucleotide whether or not it is paired with another nucleotide, and if so, to which. From this information we then determine the number of junctions in the RNA/DNA secondary structure using the CountJunctions algorithm presented in detail in the Supplemental Material.
Computational method: Genetic Filtering
Genetic Filtering is a computational method to design diverse DNA pools for SELEX with uniform structure distribution, i.e., 20% 1J, 2J, 3J, 4J, and 5J structures each. A graphical overview of our method is given in Figure 5 and a more detailed Genetic Filtering pseudo-code is provided in the Supplemental Material. For a given pair of primer sites, Genetic Filtering first generates an initial generation of pool designs. Each pool design is assigned a fitness score that indicates its closeness to the desired uniform structure distribution. New generations of pool designs are then obtained by selecting designs from previous generations with better (i.e., smaller) fitness scores, and then applying three types of operations: mutation, copy, and crossover. The initial generation of pool designs is based on a set of five-way junctions that contain the given primer sites. The reason for selecting a set of five-way junctions as the initial pool designs is that it is possible to obtain low-complexity structures through the mutation of high-complexity structures (see Fig. 1), but the opposite is highly unlikely. This was also reported by a recent investigation (Chiuman and Li 2006), where 5Js were derived from the pre-existing and less-complex structures with 3Js through evolutionary pathways, but it was observed that the possibility is very small. A similar experiment is shown in Supplemental Table S6. Mutating 50 different 3J sequences 1 million times each, for a total of 50 million sequences generated only 162 5J structures.
FIGURE 5.

Graphical overview of the Genetic Filtering algorithm as described in the text.
Given an initial seed population of pool designs based on 5J structures, our genetic algorithm proceeds as shown in Figure 5. Note that, all sequences (i.e., pool designs) always contain the given primer sites. Each sequence (pool design) is assigned a fitness score that indicates its closeness to the desired structure distribution. For a sequence S, let jcti be the percentage of i-way junctions counted after mutating its single-stranded positions 104 times, and let djcti be the desired percentage of i-way junctions (i = 1, …, 5). The fitness score for S is calculated as 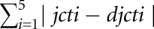 . The desired percentage of i-way junctions is the uniform structure distribution and each djcti is set to 20%. Note that we perform 104 instead of 106 mutations of single-stranded positions for the fitness score calculations. The number of mutations is reduced for fitness score estimation to save computation time. As discussed in the Results section, our Genetic Filtering method takes hours on a 200 processor PC cluster. As shown in Supplemental Table S5, the structure distribution estimated with 104 mutations is close to that estimated with 106 mutations. Furthermore, we confirm our final result by recalculating the fitness score with 106 mutations.
. The desired percentage of i-way junctions is the uniform structure distribution and each djcti is set to 20%. Note that we perform 104 instead of 106 mutations of single-stranded positions for the fitness score calculations. The number of mutations is reduced for fitness score estimation to save computation time. As discussed in the Results section, our Genetic Filtering method takes hours on a 200 processor PC cluster. As shown in Supplemental Table S5, the structure distribution estimated with 104 mutations is close to that estimated with 106 mutations. Furthermore, we confirm our final result by recalculating the fitness score with 106 mutations.
Another consideration for the calculation of the fitness score is that the maximum size of a RNA/DNA pool that can be synthesized and manipulated is typically 1014–1015, which is ∼424–425. We chose our pool design to have 24 single-stranded positions, excluding primer sites, in order to ensure complete sequence space coverage. Hence, when each sequence (pool design) is assigned a fitness score, we penalize sequences whose number of single-stranded positions (num_ss) is not equal to the desired number of single-stranded positions (dnum_ss), and less penalty is given to pool designs with fewer single-stranded positions. For details see Algorithm Fitness_function_with_penalty in the Supplemental Material.
After the fitness scores have been calculated for the current population of pool designs, biased selection (based on fitness scores) is applied to select a new intermediate population of pool designs with better fitness. Here, we use a roulette wheel selection method to select pool designs from the current population, where each pool design is chosen with a probability that corresponds to its relative fitness. By repeatedly spinning the roulette wheel, a new intermediate population is selected and fitter individuals have a greater chance to be selected than weaker ones. Crossover, mutation, and copy are then applied to the intermediate population to create the next generation. For the crossover method, we first choose two sequences S1 and S2 from the intermediate population of pool designs, and then generate a random number called randomPosition between two parameters, Low and High. To create the next generation, we switch the subsequences of S1 and S2 from position randomPosition to position length–randomPosition, where length is the total length of each sequence. The crossover method is designed to potentially switch substructures between two sequences. This is clearly not always the case, but when it happens, new pool designs of interest can be generated. When a sequence in the intermediate population is selected for mutation, we generate a random number between 0 and 1 for each position of the sequence excluding primer sites. If the number is less than a parameter Pposition, we mutate the position by choosing with equal likelihood one of the other three nucleotides. The mutated sequence will then enter the next generation. When a sequence in the intermediate population is selected for a copy operation, the sequence is simply copied unchanged to the next generation. The above crossover, mutation, and copy methods have parameters Pcrossover, Pmutation, and Pcopy, respectively, which represent the likelihood of applying the respective method to a given sequence. Furthermore, for every 10 generations we re-add the best sequence found so far to the new generation of pool designs.
Genetic Filtering generates a number of generations of pool designs (typically between 500 and 3000) until either a pool design with uniform structure distribution is found, or the score of the best pool design remains unchanged for a number of generations, or a maximum number NG of generations is reached.
DNA experiments
Reagents for DNA synthesis were purchased from Glen Research. PCR and acrylamide-gel components were purchased from BioShop. All other chemicals were purchased from Sigma-Aldrich.
Oligonucleotide library and primer synthesis
The oligonucleotide pools were synthesized on a 1-μmol scale using a MerMade 6 Oligonucleotide synthesizer (BioAutomation Corporation). The sequence, GFPool1, was obtained from the Genetic Filtering procedure. This library consisted of ∼1014–1015 single-stranded DNA fragments, comprising two primer binding sites necessary for PCR (shown in bold): 5′-ATACCAGCTTATTCAATTGCNNNNGCAATTNNNGTCNGGACNNNNGTTCNGACNNTCGGCGNNNCGCCGANCTATCTNNNNNAGATAGTAAGTGCAATCT. A small amount of this pool was set aside prior to SELEX for cloning and sequencing experiments.
The following primers used for amplification and cloning of the selected oligonucleotides were synthesized: Primer1: 5′-ATACCAGCTTATTCAATT-3′ and Primer2: 5′-AGATTGCACTTACTATCT-3′. In order to isolate the single-stranded aptamers from the double-stranded PCR product, the primers were synthesized with the following modifications: ModPrimer 1: 5′-fluorescein-ATACCAGCTTATTCAATT and ModPrimer 2: 5′-poly-dA20-HEG- AGATTGCACTTACTATCT -3′. Similarly, the RFPool A and the primers required for PCR amplification were synthesized with the following sequences:
RFPool A: 5′-GGAAGAGATGGCGACGCTACTCCCATCNGGTACCGTNNNNACGGTATTNNGACGCNNNNNGCGTCNNNACACNNNNGTGTGGATCAGCTGATCCTGATGG
RF Primer 1: 5′-GGAAGAGATGGCGACGCT and RFPrimer 2: 5′-CCATCAGGATCAGCTGAT
All synthesized DNA was purified by 12% polyacrylamide gel electrophoresis (PAGE) followed by cleanup using Microcon YM-3 Centrifugal Filter Devices.
SELEX experiments
ATP-agarose and unmodified-agarose columns, 0.25 mL in volume, were washed with ∼10 mL of column buffer (300 mM NaCl, 5 mM MgCl2, 20 mM Tris Hcl at pH 7.6). The oligonucleotide library, suspended in 1 mL of column buffer was heated for 5 min at 75°C, then cooled to room temperature over 20 min. The pool was then immediately loaded into the unmodified agarose column and left to incubate with mild shaking at room temperature for 30 min. The column was washed with 4 mL of column buffer, collecting the DNA that did not bind to the unmodified agarose material to be used for the selection process. After this preselection, eight rounds of positive selection were performed as follows. The DNA library was subjected to 5 min of heating and a 20-min incubation at room temperature, followed by immediate incubation with the ATP-modified agarose column, as described above. After 30 min, sequences that did not have an affinity for ATP were washed away with 5 mL of column buffer. To obtain the sequences bound to the ATP, the column was incubated for 10 min with 0.5-mL elution buffer (40 mM Tris HCl, 3.5 M urea at pH 8) at 80°C and removed using centrifugation. Five elution fractions were collected and the DNA was purified using an ultrafiltration stirred cell (Millipore). This DNA was quantified using UV-visible spectroscopy and fluorescence.
The entire selected oligonucleotide pool was amplified in five to 15 parallel PCR reactions. Each reaction consisted of 0.1 M Tris HCl (pH 9), 50 mM KCl, 1% Triton X-100, 1.9 mM MgCl2, 0.3 mM dNTP mix, 1 μM each primer and 5 U of Taq DNA polymerase. The DNA was initially melted for 10 min at 94.0°C, followed by 25 cycles of 94.0°C (1 min), 47.0°C (1 min), and 72.0°C (1 min). Final extension occurred at 72.0°C for 10 min after the last cycle. PCR products were dried down, heated at 55°C for 5 min in the presence of formamide, and run on a 12% denaturing PAGE to separate the double-stranded product. The fluorescein-labeled DNA strand (the selected sequences) could be identified using an Alpha Imager UV-illuminator. The corresponding DNA bands were cut from the gel and extracted using the freeze/rapid thaw method described by Chen and Ruffner (1996) in 10 mM Tris HCl buffer (pH 7.4). After purifying the DNA on the stirred cell and resuspending it in column buffer, the DNA could be used for the next selection round.
Cloning and sequencing
The selected oligonucleotides from SELEX round 8 and a small amount of the starting library were amplified using the unmodified primers (Primers 1 and 2) and cloned using a StrataClone PCR Cloning Kit (Agilent Technologies). The colonies were grown on LB-ampicillin agar plates for 16 h at 37°C, and colonies of interest were selected via blue-white screening. Each colony was removed carefully from the agar and vortexed in 50 uL of deionized water. These samples were sent for direct sequencing at the University of Calgary University Core DNA Services using the T3 promoter primer and T7 promoter primer. Sequencing results were analyzed and vector sequences were deleted from the total sequence so as to retain only the data that represent the selected random sequences.
Dissociation constant (Kd) experiments
A total of 1 mg of ATP agarose (∼23 nanomoles of ATP) in a microcentrifuge filter tube was washed with column buffer and then exposed to varying concentrations of 5′-fluorescein labeled sequences (from 1 nM to 20 μM) in 100 μL of column buffer. After vortexing, the tubes were centrifuged at 10,000g for 10 min. A total of 100 μL of 90°C column buffer was added and vortexed briefly, followed again by centrifugation at 10,000g for 10 min. The fluorescence of the eluted DNA was recorded and the dissociation constants were evaluated by minimizing the residual values between calculated and observed experimental Δ fluorescence data using the solver feature of Microsoft Excel (Fylstra et al. 1998; Nenov and Fylstra 2003).
SUPPLEMENTAL MATERIAL
Supplemental material can be found at http://www.rnajournal.org.
ACKNOWLEDGMENTS
We thank Yingfu Li (Biochemistry, McMaster University) for initially pointing out the problem. Thanks to Yasmine Miguel for assisting with binding studies. This work was made possible through support from the Natural Sciences and Engineering Council of Canada (NSERC), the Canadian Foundation for Innovation (CFI), the Ontario Research Fund (ORF), and Carleton University.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2102210.
REFERENCES
- Batey RT, Gilbert SD, Montange RK 2004. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature 432: 411–415 [DOI] [PubMed] [Google Scholar]
- Breaker RR 2004. Natural and engineered nucleic acids as tools to explore biology. Nature 432: 838–845 [DOI] [PubMed] [Google Scholar]
- Carothers JM, Oestreich SC, Davis JH, Szostak JW 2004. Informational complexity and functional activity of RNA structures. J Am Chem Soc 126: 5130–5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carothers JM, Davis JH, Chou JJ, Szostak JW 2006. Solution structure of an informationally complex high-affinity RNA aptamer to GTP. RNA 12: 567–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Ruffner DE 1996. Modified crush-and-soak method for recovering oligodeoxynucleotides from polyacrylamide gel. Biotechniques 21: 820–822 [DOI] [PubMed] [Google Scholar]
- Chiuman W, Li Y 2006. Revitalization of six abandoned catalytic DNA species reveals a common three-way junction framework and diverse catalytic cores. J Mol Biol 357: 748–754 [DOI] [PubMed] [Google Scholar]
- Davis JH, Szostak JW 2002. Isolation of high-affinity GTP aptamers from partially structured RNA libraries. Proc Natl Acad Sci 99: 11616–11621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington AD, Szostak JW 1990. In vitro selection of RNA molecules that bind specific ligands. Nature 346: 818–822 [DOI] [PubMed] [Google Scholar]
- Famulok M, Verma S 2002. In vivo-applied functional RNAs as tools in proteomics and genomics research. Trends Biotechnol 20: 462–466 [DOI] [PubMed] [Google Scholar]
- Fylstra D, Lasdon L, Watson J, Waren A 1998. Design and use of the Microsoft Excel Solver. Interfaces 28: 29–55 [Google Scholar]
- Gan HH, Pasquali S, Schlick T 2003. Exploring the repertoire of RNA secondary motifs using graph theory; implications for RNA design. Nucleic Acids Res 31: 2926–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevertz J, Gan HH, Schlick T 2005. In vitro RNA random pools are not structurally diverse: A computational analysis. RNA 11: 853–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann T, Patel DJ 2000. Adaptive recognition by nucleic acid aptamers. Science 287: 820–825 [DOI] [PubMed] [Google Scholar]
- Hodgson DR, Suga H 2004. Mechanistic studies on acyl-transferase ribozymes and beyond. Biopolymers 73: 130–150 [DOI] [PubMed] [Google Scholar]
- Hofacker IL 2003. Vienna RNA secondary structure server. Nucleic Acids Res 31: 3429–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tackerl M, Schuster P 1994. Fast folding and comparison of rna secondary structures Chemical Monthly 125: 167–188 [Google Scholar]
- Huizenga DE, Szostak JW 1995. A DNA aptamer that binds adenosine and ATP. Biochemistry 34: 656–665 [DOI] [PubMed] [Google Scholar]
- Isaacs FJ, Dwyer DJ, Collins JJ 2006. RNA synthetic biology. Nat Biotechnol 24: 545–554 [DOI] [PubMed] [Google Scholar]
- Jaeger L, Wright MC, Joyce GF 1999. A complex ligase ribozyme evolved in vitro from a group I ribozyme domain. Proc Natl Acad Sci 96: 14712–14717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo D, Perez C, Mohr I 2002. Characterization of RNA determinants recognized by the arginine- and proline-rich region of Us11, a herpes simplex virus type 1-encoded double-stranded RNA binding protein that prevents PKR activation. J Virol 76: 11971–11981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft JS, Zhou K, Grech A, Jubin R, Doudna JA 2002. Crystal structure of an RNA tertiary domain essential to HCV IRES-mediated translation initiation. Nat Struct Biol 9: 370–374 [DOI] [PubMed] [Google Scholar]
- Kim N, Gan HH, Schlick T 2007a. A computational proposal for designing structured RNA pools for in vitro selection of RNAs. RNA 13: 478–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Shin JS, Elmetwaly S, Gan HH, Schlick T 2007b. RagPools: RNA-As-Graph-Pools–a web server for assisting the design of structured RNA pools for in vitro selection. Bioinformatics 23: 2959–2960 [DOI] [PubMed] [Google Scholar]
- Kim N, Izzo JA, Elmetwaly S, Gan HH, Schlick T 2010. Computational generation and screening of RNA motifs in large nucleotide sequence pools. Nucleic Acids Res. doi: 10.1093/nar/gkq282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight R, De Sterck H, Markel R, Smit S, Oshmyansky A, Yarus M 2005. Abundance of correctly folded RNA motifs in sequence space, calculated on computational grids. Nucleic Acids Res 33: 5924–5935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laserson U, Gan HH, Schlick T 2004. Searching 2D RNA geometries in bacterial genomes. In Proceedings of the 12th Annual Symposium on Computational geometry. pp. 373–377 [Google Scholar]
- Lau MW, Cadieux KE, Unrau PJ 2004. Isolation of fast purine nucleotide synthase ribozymes. J Am Chem Soc 126: 15686–15693 [DOI] [PubMed] [Google Scholar]
- Lee JF, Hesselberth JR, Meyers LA, Ellington AD 2004. Aptamer database. Nucleic Acids Res 32: D95–D100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legiewicz M, Wichlacz A, Brzezicha B, Ciesiolka J 2006. Antigenomic delta ribozyme variants with mutations in the catalytic core obtained by the in vitro selection method. Nucleic Acids Res 34: 1270–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley DM, Clegg RM, Diekmann S, Seeman NC, Von Kitzing E, Hagerman PJ 1995. A nomenclature of junctions and branchpoints in nucleic acids. Nucleic Acids Res 23: 3363–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenov IP, Fylstra DH 2003. Interval methods for accelerated global search in the Microsoft Excel Solver. Reliable Computing 9: 143–159 [Google Scholar]
- Ohuchi SJ, Ikawa Y, Shiraishi H, Inoue T 2002. Modular engineering of a Group I intron ribozyme. Nucleic Acids Res 30: 3473–3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Unrau PJ, Bartel DP 1997. Accessing rare activities from random RNA sequences: The importance of the length of molecules in the starting pool. Chem Biol 4: 767–774 [DOI] [PubMed] [Google Scholar]
- Schwalbe H, Buck J, Furtig B, Noeske J, Wohnert J 2007. Structures of RNA switches: Insight into molecular recognition and tertiary structure. Angew Chem Int Ed Engl 46: 1212–1219 [DOI] [PubMed] [Google Scholar]
- Scott WG, Murray JB, Arnold JR, Stoddard BL, Klug A 1996. Capturing the structure of a catalytic RNA intermediate: The hammerhead ribozyme. Science 274: 2065–2069 [DOI] [PubMed] [Google Scholar]
- Serganov A, Yuan YR, Pikovskaya O, Polonskaia A, Malinina L, Phan AT, Hobartner C, Micura R, Breaker RR, Patel DJ 2004. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem Biol 11: 1729–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganov A, Huang L, Patel DJ 2008. Structural insights into amino acid binding and gene control by a lysine riboswitch. Nature 455: 1263–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P, Sefah K, Yang CJ, Tan W 2006. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc Natl Acad Sci 103: 11838–11843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup GA, Breaker RR 1999. Nucleic acid molecular switches. Trends Biotechnol 17: 469–476 [DOI] [PubMed] [Google Scholar]
- Thodima V, Pirooznia M, Deng Y 2006. RiboaptDB: A comprehensive database of ribozymes and aptamers. BMC Bioinformatics Suppl 27: S6 doi: 10.1186/1471-2105-7-S2-S6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuerk C, Gold L 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249: 505–510 [DOI] [PubMed] [Google Scholar]
- Wilson DS, Szostak JW 1999. In vitro selection of functional nucleic acids. Annu Rev Biochem 68: 611–647 [DOI] [PubMed] [Google Scholar]
- Wilson TJ, Nahas M, Ha T, Lilley DM 2005. Folding and catalysis of the hairpin ribozyme. Biochem Soc Trans 33: 461–465 [DOI] [PubMed] [Google Scholar]
- Yoshioka W, Ikawa Y, Jaeger L, Shiraishi H, Inoue T 2004. Generation of a catalytic module on a self-folding RNA. RNA 10: 1900–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinnen SP, Domenico K, Wilson M, Dickinson BA, Beaudry A, Mokler V, Daniher AT, Burgin A, Beigelman L 2002. Selection, design, and characterization of a new potentially therapeutic ribozyme. RNA 8: 214–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M, Stiegler P 1981. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res 9: 133–148 [DOI] [PMC free article] [PubMed] [Google Scholar]