

Abstract

Stimulated by the substantial challenge of synthesizing the complex and sensitive stereogenic allene-containing core of (−)-peridinin, the first stereocontrolled coupling of haloallenes with boronic acids has been achieved. This new method and the principles that emerged during its development stand to enable the more efficient and flexible preparation of a wide range of natural products, pharmaceuticals, and intermediates that possess a stereogenic allene motif. This new reaction was harnessed to achieve the first completely stereocontrolled total synthesis of (−)-peridinin. This synthesis was accomplished using only one reaction iteratively to assemble four fully functionalized building blocks with complete stereoretention at each initially halide or boron-bearing carbon. This synthesis elevates the capacity of the iterative cross-coupling strategy to an unprecedented benchmark. Moreover, the efficient and highly modular nature of this synthesis promises to enable systematic dissection of the heretofore enigmatic structure/function relationships that underlie the protein-like antilipoperoxidant activities of this remarkable small molecule natural product.
Deficiencies of antilipoperoxidant proteins have been associated with atherosclerosis, rheumatoid arthritis, and cancer, and may contribute to an accelerated aging process.1 Small molecules with the capacity to replicate the functions of these proteins could therefore have a major positive impact on human health. In contrast to most carotenoids,2 the C37-norcarotenoid peridinin (1)3 may exert antilipoperoxidant activities primarily through self-preserving mechanisms, including catalytic quenching of 1O2 and decreasing membrane permeability to other reactive oxygen species.4 Isolation from natural sources is very inefficient,3b however, and the sensitive and stereogenic allene moiety central to the structure of 1 has made its stereoselective synthesis very challenging.5,6 As a result, the underpinnings of these activities have been only minimally explored.4,7 To facilitate systematic studies of such small molecules with protein-like functions, we recently introduced a simple, efficient, and flexible synthesis strategy that involves the iterative cross-coupling (ICC) of bifunctional haloboronic acids masked as the corresponding N-methyliminodiacetic acid (MIDA) boronates.8 Enabled by this ICC approach and the development of new methodology for the highly stereoretentive Suzuki-Miyaura (SM) cross-coupling of haloallenes, we herein report the first fully stereocontrolled total synthesis of (−)-1.
Guided by the ICC strategy,8 we applied only SM transforms to retrosynthesize 1 into four building blocks, BB1–BB4, having all of the required functionality pre-installed in the correct oxidation states and with the desired stereochemical relationships (Scheme 1). We recognized, however, that the disconnection between C8’/C9’ corresponded to a stereoretentive SM coupling with a haloallene. Albeit potentially very useful in the preparation of many natural products,9 this was an unprecedented transformation10 that first required development.
Scheme 1.

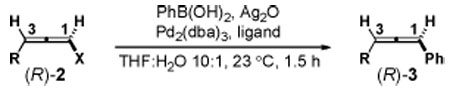
In related Negishi couplings,11 chloro- and bromoallenes yield products representing net stereochemical inversion, whereas the corresponding iodoallenes tend to favor stereoretention. These results were attributed to two competing mechanisms involving direct oxidative addition (OA) at C–X or indirect SN2’-like OA12 followed by a suprafacial 1,3 shift of Pd.13 Attempts to form the C8’/C9’ bond in 1 via a Stille coupling have been complicated by similar issues.6 Consistent with these precedents, using conditions similar to those reported to promote the SM coupling of achiral haloallenes,10a the reaction of PhB(OH)2 with enantioenriched chloro- and bromoallenes (R)-2a,b demonstrated net stereoinversion to provide (S)-3a as the major product (Table 1, entries 1–2). In contrast, coupling of iodoallene (R)-2c yielded (R)-3a with a moderate 72% stereoretention (entry 3).
Table 1.
Development of the first stereocontrolled SM coupling of chiral haloallenes.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | 2 | R | X | ligand | 3 | % stereoretention a,b |
| 1 | (R)-2a | t-Bu | CI | PPh3 | (S)-3a | −78 |
| 2 | (R)-2b | t-Bu | Br | PPh3 | (S)-3a | −78 |
| 3 | (R)-2c | t-Bu | I | PPh3 | (R)-3a | 72 |
| 4 | (R)-2d | 3-pentyl | I | PPh3 | (R)-3b | 58 |
| 5 | (R)-2e | n-pentyl | I | PPh3 | (R)-3c | 25 |
| 6 | (R)-2c | t-Bu | I | PFur3 | (R)-3a | 80 |
| 7 | (R)-2c | t-Bu | I | PCy3 | (R)-3a | 50 |
| 8 | (R)-2c | t-Bu | I | Pt-Bu2Me | (R)-3a | 71 |
| 9 | (R)-2c | t-Bu | I | Po-Tol3 | (R)-3a | 91 |
| 10 | (R)-2c | t-Bu | I | Pt-Bu3 | (R)-3a | 93 |
| 11 | (R)-2c | t-Bu | I | XPhos | (R)-3a | 91 |
| 12c | (R)-2c | t-Bu | I | XPhos | (R)-3a | >99d |
| 13c | (R)-2d | 3-pentyl | I | XPhos | (R)-3b | >99 |
| 14c | (R)-2e | n-pentyl | I | XPhos | (R)-3c | 85 |
% stereoretention = ee product/ee starting material (chiral GC, average of 2 runs); negative values reflect net stereoinversion.
Unoptimized GC yields ranged from 10–83%.
Hexane:THF:H2O 9:1:1 was used as solvent.
Isolated yield = 61%.
Building on this starting point, we tested the previously unconfirmed hypothesis11 that maximized steric bulk at C3 of a haloallene could promote stereoretention, presumably by disfavoring SN2’-like OA. Specifically, we evaluated a series of substrates 2c–e having progressively smaller R groups at C3 and indeed observed decreased stereoretention (entries 3–5). Guided by reciprocal logic,14 we hypothesized that bulky phosphine ligands15 would also promote stereoretentive direct OA at the less sterically hindered C1. In fact, in contrast to smaller ligands (entries 3, 6–8), >90% stereoretention was observed for phosphine ligands having cone angles >180° (entries 9–11).16 With air-stable XPhos,15a optimization of solvent led to >99% stereoretention and 61% isolated yield (entry 12). Substantial improvements in stereoretention were also observed for couplings with 2d and 2e under these optimized conditions (entries 13 and 14).
Collectively, these findings revealed that maximized stereoretention in the SM coupling of chiral haloallenes can best be achieved using allenyl iodide substrates having steric bulk at C3 in combination with sterically bulky phosphine ligands. Guided by these general principles, we designed BB4 6b for peridinin (Scheme 1) to include both an allenyl iodide and a sterically bulky, yet still easily removable, TMS ether at C5’.
Preparation of the remaining building blocks was enabled by some highly favorable features of the MIDA boronate platform.8 Specifically, following a hydroboration of alkyne 45b to give pinacol ester 5, a direct transesterification to form the air-stable MIDA boronate BB1 was achieved (Scheme 2).17 This direct transesterification from a boronic ester to a MIDA boronate avoids the intermediacy of unstable boronic acids. The unique compatibilities of the MIDA boronate functional group with a wide range of reaction conditions8c and chromatography8 were utilized to prepare the final two building blocks. Specifically, propynyl MIDA boronate 6 (SI), underwent highly regio- and stereocontrolled molybdenum-catalyzed hydrostannylation18 to yield bis-metalated olefin 7 (Scheme 3). A subsequent metal and halide selective coupling between 7 and lactone 819 provided BB2 as a single stereoisomer.20 The final building block BB3 was prepared via initial bromination of commercially-available 9 followed by regio- and stereoselective elimination to generate the novel tri-substituted bromoalkenyl MIDA boronate 10 as a single stereoisomer20 (Scheme 4). Two cycles of our recently developed methodology8e for stereospecific metal-selective cross-coupling with bis-metalated olefin 1121 followed by stereoretentive iododegermylation22 provided BB3.
Scheme 2.

Scheme 3.

Scheme 4.

The synthesis of 1 was completed using only iterative SM couplings to assemble these four building blocks, thereby completely avoiding mixtures of olefin isomers5,6 (Scheme 5). Hydrolysis of BB1 followed by B-selective cross-coupling with BB2 provided tetraenyl MIDA boronate 13. The corresponding boronic acid proved to be unstable, thus a new tactic was developed to promote the next cycle of B-activation and coupling. Specifically, in a novel transformation, 13 was directly converted into the corresponding pinacol ester, which was an effective intermediate for B-selective coupling with BB3. The resulting highly complex heptaenyl MIDA boronate 14 was stable to chromatography and storage. Conversely, attempts to isolate the heptaenyl boronic acid derived from 14 or to utilize the corresponding pinacol ester were not fruitful. To solve this challenging problem, we hybridized the principles established above for allenyl halide coupling (Table 1) with in situ release of the unstable boronic acid8d,b derived from MIDA boronate 14 to promote the final union with BB4 in good yield and with complete stereoretention. Global desilylation concluded, to the best of our knowledge, the first completely stereocontrolled total synthesis of (−)-peridinin.
Scheme 5.

This efficient and highly modular pathway to 1 stands to facilitate systematic dissection of the structure/function relationships that underlie the self-preserving antilipoperoxidant activities of this small molecule natural product.
Acknowledgement
We thank the NIH (GM090153) for funding and DSM for a gift of (−)-actinol. MDB is an HHMI Early Career Scientist, Beckman Young Investigator, and Sloan Fellow. EMW is an NSF and Eli Lilly Graduate Fellow.
Footnotes
Supporting Information Available: Procedures and spectral and crystallographic data (.cif). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Lewis P, et al. Circulation. 2007;115:2178–2187. doi: 10.1161/CIRCULATIONAHA.106.664250. [DOI] [PubMed] [Google Scholar]; (b) Rister M, Bauermeister K, Gravert U, Gladtke E. Lancet. 1978;311:1094. doi: 10.1016/s0140-6736(78)90933-9. [DOI] [PubMed] [Google Scholar]; (c) Saadat M. Cancer Sci. 2006;97:505–509. doi: 10.1111/j.1349-7006.2006.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hulbert AJ, Pamplona R, Buffenstein R, Buttemer WA. Physiol. Rev. 2007;87:1175–1213. doi: 10.1152/physrev.00047.2006. [DOI] [PubMed] [Google Scholar]
- 2.β-carotene, astaxanthin, and lutein operate through stoichiometric quenching of reactive oxygen species which is self-destructive and produces potentially toxic breakdown products. Garavelli M, Bernardi F, Olivucci M, Robb MA. J. Am. Chem. Soc. 1998;120:10210–10222. Gorman AA, Hamblett I, Lambert C, Spencer B, Standen MC. J. Am. Chem. Soc. 1988;110:8053–8059.
- 3.(a) Schütt F. Ber. Deut. Bot. Ges. 1890;8:9. [Google Scholar]; (b) Haugan JA, Aakermann T, Liaaen-Jensen S. Meth. Enzym. 1992;213:231–245. [Google Scholar]
- 4.(a) Barros MP, Pinto E, Colepicolo P, Pedersen M. Biochem. Biophys. Res. Comm. 2001;288:225. doi: 10.1006/bbrc.2001.5765. [DOI] [PubMed] [Google Scholar]; (b) Pinto E, Catalani LH, Lopes NP, Di Mascio P, Colepicolo P. Biochem. Biophys. Res. Comm. 2000;268:496. doi: 10.1006/bbrc.2000.2142. [DOI] [PubMed] [Google Scholar]
- 5.(a) Yamano Y, Ito M. J. Chem. Soc., Perkin Trans. 1. 1993:1599–1610. [Google Scholar]; (b) Furuichi N, Hara H, Osaki T, Nakano M, Mori H, Katsumura S. J. Org. Chem. 2004;69:7949–7959. doi: 10.1021/jo048852v. [DOI] [PubMed] [Google Scholar]; (c) Olpp T, Brückner R. Angew. Chem. Int. Ed. 2006;45:4023–4027. doi: 10.1002/anie.200600502. [DOI] [PubMed] [Google Scholar]; (d) Vaz B, Domínguez M, Alvarez R, de Lera AR. Chem. Eur. J. 2007;13:1273–1290. doi: 10.1002/chem.200600959. [DOI] [PubMed] [Google Scholar]
- 6. Vaz B, Alvarez R, Brückner R, de Lera AR. Org. Lett. 2005;7:545–548. doi: 10.1021/ol0478281. For studies of Stille couplings with BB4 and alternative conclusions regarding underpinnings of stereocontrol, see: Vaz B, Pereira R, Pérez M, Alvarez R, de Lera AR. J. Org. Chem. 2008;73:6534. doi: 10.1021/jo800756b.
- 7.Studies of peridinin as a harvester of light: Kajikawa T, Hasegawa S, Iwashita T, Kusumoto T, Hashimoto H, Niedzwiedzki DM, Frank HA, Katsumura S. Org. Lett. 2009;11:5006–5009. doi: 10.1021/ol901940g.
- 8.(a) Gillis EP, Burke MD. J. Am. Chem. Soc. 2007;129:6716–6717. doi: 10.1021/ja0716204. [DOI] [PubMed] [Google Scholar]; (b) Lee SJ, Gray KC, Paek JS, Burke MD. J. Am. Chem. Soc. 2008;130:466–468. doi: 10.1021/ja078129x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gillis EP, Burke MD. J. Am. Chem. Soc. 2008;130:14084–14085. doi: 10.1021/ja8063759. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Knapp DM, Gillis EP, Burke MD. J. Am. Chem. Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lee SJ, Burke MD. manuscript in preparation. [Google Scholar]
- 9.Hoffman-Röder A, Krause N. Angew. Chem. Int. Ed. 2004;43:1196. doi: 10.1002/anie.200300628. [DOI] [PubMed] [Google Scholar]
- 10.For prior reports of SM coupling of achiral haloallenes, see: Gillmann T, Weeber T. Synlett. 1994:649–650. Saalfrank RW, Haubner M, Deutscher C, Bauer W, Clark T. J. Org. Chem. 1999;64:6166–6168.
- 11.Elsevier CJ, Vermeer P. J. Org. Chem. 1985;50:3042–3045. [Google Scholar]
- 12.Corey EJ, Boaz NW. Tetrahedron Lett. 1984;25:3059–3062. [Google Scholar]
- 13.The enhanced stereoretention observed with C–I has been attributed to its increased propensity for direct oxidative addition (ref 11 and 6).
- 14.The mechanism of OA with allenyl iodides is unknown. Based on leading studies with aryl iodides (Barrios-Landeros, F.; Carrow, B.P.; Hartwig, J.F. J. Am. Chem. Soc. 2009, 131, 8141–8154), OA may proceed through L2Pd intermediates where sterically bulky phosphines promote reactivity at the less sterically-hindered C1. Alternatively, bulky phosphines may promote an L1Pd pathway in which case the enhanced capacity of L1Pd for direct OA15 would presumably be advantageous.
- 15.(a) Martin R, Buchwald SL. Acc. Chem. Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fu GC. Acc. Chem. Res. 2008;41:1555–1564. doi: 10.1021/ar800148f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hartwig JF, Frederic P. J. Am. Chem. Soc. 1995;117:5373–5374. [Google Scholar]
- 16.(a) Tolman CA. Chem. Rev. 1977;77:313–348. [Google Scholar]; (b) Bachechi F, Burini A, Galassi R, Pietroni BR. J. Mol. Struct. 2005;740:119–123. [Google Scholar]; (c) Rafter E, Gilheany DG, Reek JNH, van Leeuwen PWNM. ChemCatChem. 2010;2:387–391. [Google Scholar]
- 17.The transesterification of aryl pinacol esters to MIDA boronates was also described by M.R. Smith, 237th Am. Chem. Soc. Conference, March, 2009.
- 18.Zhang HX, Guibe F, Balavoine G. J. Org. Chem. 1990;55:1857–1867. [Google Scholar]
- 19.Sorg A, Blank F, Brückner R. Synlett. 2005;8:1286–1290. [Google Scholar]
- 20.Structure was confirmed via single crystal X-ray analysis (SI).
- 21.David-Quillot F, Thibonnet J, Marsacq D, Abarbri M, Duchêne A. Tetrahedron Lett. 2000;41:9981–9984. [Google Scholar]
- 22.Oda H, Morizawa Y, Oshima K, Nozaki H. Tetrahedron Lett. 1984;25:3221–3224. [Google Scholar]