

Abstract
In recent years, 4-hydroxy-2-nonenal (4-HNE) has emerged as an important second messenger in cell cycle signaling. Here we demonstrate that 4-HNE induces signaling for apoptosis via both, the Fas mediated extrinsic and the p53 mediated intrinsic pathways in HepG2 cells. 4-HNE induces Fas-mediated DISC independent apoptosis pathway by activating ASK1, JNK and caspase-3. In parallel treatment of 4-HNE to HepG2 cells also induces apoptosis by p53 pathway through activation of Bax, p21, JNK, and caspase-3. Exposure of HepG2 cells to 4-HNE leads to the activation of both Fas and Daxx, promotes the export of Daxx from the nucleus to cytoplasm and facilitates Fas-Daxx binding. Depletion of Daxx by siRNA results in potentiation of apoptosis indicating that Fas-Daxx binding in fact is inhibitory to Fas mediated apoptosis in cells. 4-HNE-induced translocation of Daxx is also accompanied by the activation and nuclear accumulation of HSF1 and up-regulation of heat shock protein Hsp70. All these effects of 4-HNE in cells can be attenuated by ectopic expression of hGSTA4-4, the isozyme of glutathione S-transferase with high activity for 4-HNE. Through immunoprecipitation and liquid chromatography–tandem mass spectrometry, we have demonstrated covalent binding of 4-HNE to Daxx. We also demonstrate that 4-HNE modification induces phosphorylation of Daxx at Ser668 and Ser671 to facilitate its cytoplasmic export. These results indicate that while 4-HNE exhibits toxicity through several mechanisms, in parallel it evokes signaling for defense mechanisms to self regulate its toxicity and can simultaneously affect multiple signaling pathways through its interactions with membrane receptors and transcription factors/ repressors.
Reactive oxygen species (ROS) produced during exposure of cells to UV radiation, heat shock, or xenobiotics and during metabolic processes cause the oxidation of polyunsaturated fatty acids in membrane lipid bilayers that are one of the early targets of ROS. Previous studies have indicated that the exposure of cells to even minimal transient stress causes substantial lipid peroxidation (LPO) (1, 2). These studies show that transient exposure of cells to UV, H2O2, or oxidants leads to a significant (~50%) rise in the levels of 4-hydroxy-2-nonenal (4-HNE) which is a stable end-product of LPO. 4-HNE has been widely implicated in the mechanisms of oxidant toxicity and also in cell cycle signaling (3–9) but most of the associated mechanisms are not completely understood. Liver being the major site for metabolism, and biotransformation of xenobiotics is constantly exposed to ROS. In the event of insufficient levels of defense mechanisms such as free-radical scavengers or antioxidants, increasing levels of lipid hydroperoxides and peroxides can be produced in liver by self-perpetuating chain reactions resulting in the formation of 4-HNE that can exert toxicity through interactions with cellular macromolecules, including proteins, lipids, and nucleic acids. For example, chronic alcohol consumption (10), high-iron diet (11), high-fat diet (12), or exposure to hepatotoxic agents like CCl4 markedly elevate the intracellular concentration of 4-HNE from its basal constitutive levels and damage hepatocytes and the liver (13, 14). 4-HNE is also believed to be involved in the mechanisms of diseases such as atherosclerosis (15, 16), diabetes (17), Alzheimer’s disease (18, 19), Parkinson's disease (20), cataract (21), and cancer (22, 23).
In recent past (1, 2, 6–9), studies in our laboratories have clearly shown that signaling for apoptosis by many oxidants including superoxide anion generated by xanthine-xanthine oxidase system is mediated through 4-HNE and it could be inhibited by accelerating disposal of 4-HNE by forced over expression of GSTA4-4 which is highly specific for 4-HNE but is not involved in the detoxification of ROS such as H2O2, O2•−. A multitude of studies by other investigators (3–5, 24), and in our laboratories indicate a global role of 4-HNE in modulation of cellular processes. Studies in our lab have shown that the signaling for proliferation, transformation, apoptosis, and differentiation is associated with alterations in the intracellular levels of 4-HNE in a wide variety of cells (9, 25–28). These findings pose an intriguing question as to how 4-HNE is able to exert such a multitude of effects on cellular processes.
During the present studies we have addressed this question by investigating the role of 4-HNE mediated signaling in the pathways associated with the regulation of programmed cell death in a liver derived cell line HepG2. Specifically, we have investigated the effect of increased 4-HNE concentrations in cells by direct exposure or by oxidant treatment on different pathways leading to apoptosis. Conversely, we have evaluated the role of 4-HNE in signaling of these pathways by lowering its intracellular concentration through the transfection of cells with hGSTA4. We have also studied the interactions of 4-HNE with some of the key proteins involved in these pathways. Furthermore, to examine the in vivo significance of these findings we have also studied some of these effects of 4-HNE in the liver tissues of Gsta4 null mice where 4-HNE levels are consistently maintained at high levels due to its impaired disposition (29). The results of these studies show that 4-HNE causes toxicity to HepG2 cells via necrosis and apoptosis induced by more than one pathway. These findings integrate the mechanisms for the multifarious effects of 4-HNE on cellular processes suggesting that 4-HNE through direct interactions with membrane receptors, transcription factors, and transcription repressors regulates trafficking, and the signaling functions of key proteins to affect various cellular processes.
MATERIALS AND METHODS
Materials
4-Hydroxynonenal was purchased from Cayman Chemical (Ann Arbor, MI). Bradford reagent, bis-acrylamide, and SDS for SDS-PAGE were obtained from BioRad (Hercules, CA). The apoptosis detection system (CaspACE FITC-VAD-FMK in situ marker) was purchased from Promega Inc. (Madison, WI). The cytoplasmic and nuclear protein extraction kit was acquired from Imgenex Co. (San Diego, CA), protein A/G-agarose from Santa Cruz Biotechnology (Santa Cruz, CA), JNK inhibitor SP6000125 from A–G Scientific (San Deigo, CA), and Western blot stripping buffer from Pierce Co. (Rockford, IL). All other reagents and chemicals were purchased from Sigma-Aldrich (St. Louis, MO). The cell culture medium RPMI-1640, geneticin (G418), Lipofectamine 2000 transfection reagent and fetal bovine serum were from GIBCO (Invitrogen, Carlsbad, CA).
Cell lines and Culture Conditions
The HepG2 human hepatocarcinoma cells purchased from the American Type Culture Collection were cultured in RPMI-1640 supplemented with 10% fetal bovine serum, 1% of a stock solution containing 10,000 IU/mL penicillin and 10 mg/mL streptomycin in an incubator at 37°C under a humidified atmosphere containing 5% CO2.
Preparation of cell extracts and Western blot analysis
Cells were collected, washed with cold PBS and then incubated in 100 µL of RIPA lysis buffer (50 mM Tris-HCl, pH 7.5; 1% NP-40; 150 mM NaCl; 1 mg ml−1 aprotinin; 1 mg ml−1 leupeptin; 1 mM Na3VO4; 1 mM NaF) at 4°C for 30 min. Cell debris was removed by centrifugation at 12,000 g for 10 min at 4°C. Protein concentrations were determined by Bradford assay (30) as described in standard protocol. Cell extracts were separated on SDS polyacrylamide gels (4–20%), and transferred onto nitrocellulose (Bio-Rad). Membranes were blocked with 5% fat-free milk at room temperature for 60 min, and incubated overnight at 4°C with the appropriate primary antibody in 5% milk in Tris-buffered saline (TBS) containing 50 mM NaF and 0.05% Tween 20. After three times washing with T-TBS (Tris-buffered saline containing 0.05% Tween 20), the membrane was incubated with the appropriate secondary antibody at room temperature for 2 h. After washing again with T-TBS, the membrane was treated with Super signal ‘West Pico’ chemiluminescent reagent (Pierce, Rockford, IL) as per manufacturer's instructions, and exposed to Hyperfilm ECL film (Amersham) at room temperature. Isolation of nuclear and cytoplasmic fractions was achieved by Imgenex nuclear extraction kit as per the manufacturer’s instructions (Imgenex, San Deigo, CA).
Stable transfection with pTarget and hGSTA4
HepG2 cells at a density of 5 × 105 cells per 100 mm Petri dish were plated for the transfection. Petri dishes having >50% confluent cells were used for the transfection. The cells were transfected with 24 µg of either empty pTarget-T vector (VT) or the pTarget vector with the open reading frame (ORF) of the hGSTA4 sequence (hGSTA4-Tr), using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) as per the manufacturer’s instructions.
Transfection of Daxx siRNA in HepG2 cells
Small interfering RNA (siRNA) transfection experiments against Daxx were performed using double-stranded RNA synthesized by Dharmacon (ON-TARGET Plus SMARTpool, Dharmacon, Chicago, IL). Briefly, HepG2 cells (2 × 105 cells per well) were plated in a six-well tissue culture plate, in 2 mL normal growth medium supplemented with FBS. Cells were cultured at 37°C until 60–80% confluency. For each transfection, 100 nM double-stranded non-targeting control siRNA (Dharmacon, used as control), or Daxx-specific siRNA were transfected into HepG2 cells using DharmaFECT 4 transfection reagent (Dharmacon) according to the manufacturer’s protocol. Cells were harvested at appropriate time points and the silencing of Daxx was examined by Western blotting.
Immunofluorescence studies
HepG2 cells were grown to 50% confluence on glass cover slips in 12-well plates. The cells were exposed to 20 µM 4-HNE. Untreated cells remained as controls. Treated and untreated cells were incubated for 2 h, washed twice with ice-cold PBS (pH 7.4), fixed with 4% paraformaldehyde for 30 min and then permeabilized with 0.1% Triton X-100 for 30 min. The slides were then washed with PBS, incubated with 5% goat serum in PBS for 2 h, and then incubated with anti-Daxx or anti-HSF1 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:50 in PBS containing 1% goat serum for overnight at 4°C temperature. After washing with ice cold PBS, the cover slips were incubated with FITC-labeled goat anti-rabbit immunoglobulin G (Southern Biotech, USA) diluted 1:200 in PBS containing 1% goat serum for 2 h at room temperature in dark. The cover slips were then washed three times with ice cold PBS and mounted on glass slides with 20 µL of VectaShield medium containing DAPI (1.5 µg/mL) (Vector Laboratories, Inc., USA). The slides were examined using LSM 510 Meta confocal system equipped with an inverted microscope (Axio Observer Z1, Carl Zeiss).
In situ caspase-3 assay for Apoptosis
Cells (2 × 104) were treated with 0–40 µM 4-HNE or with 250 ng/mL Fas agonistic CH11 antibodies, that are known to induce apoptosis, for 2 h at 37°C. Apoptotic cells were detected by staining with in situ marker (10 µM, CaspACE FITC-VAD-FMK, Promega) for 30 min in the dark. The slides were fixed with 4% paraformaldehyde for 30 min, rinsed with PBS twice, mounted in a medium containing 1.5 µg/mL DAPI and observed under Olympus AX70 fluorescence microscope.
Chromatin Immunoprecipitation (ChIP) assay
To determine whether after nuclear translocation HSF1 binds to Hsp70 promoter, ChIP assay was performed using the ChIP-IT kit from Active Motif (Carlsbad, CA) following the manufacturer's instructions. Briefly, 5 × 105 cells were grown and treated with 20 µM 4-HNE for 2 h and fixed with 1% formaldehyde. Cells were washed with PBS followed by the addition of glycine stop solution and washing with PBS. The cells were collected and resuspended in lysis buffer, incubated for 10 min on ice, vortexed for 10s, and centrifuged for 10 min at 4,000 g at 4°C. The DNA pellet was resuspended in shearing buffer, sonicated and the sheared DNA sample was centrifuged. The resultant supernatant was incubated with positive control IgG as well as negative control IgG (provided by Active Motif) and anti-HSF1 IgG (Santa Cruz, CA) overnight at 4°C. Protein G beads were added to the reaction mixtures and incubated for 2 h at 4°C. The beads were pelleted by centrifugation followed by washing, and eluted with 100 µL of ChIP elution buffer. Eluted DNA samples were purified using the DNA purification mini columns and amplified by PCR using the control primers and negative control primers (provided by Active Motif) and hHsp70 primers. PCR products were analyzed by running on 1% agarose gels.
Preparation of 4-HNE-Daxx adduct and identification of 4-HNE binding sites by liquid chromatography–tandem mass spectrometry (LC–MS/MS)
4-HNE adducts of purified bacterially expressed human Daxx (1 mg/mL) were prepared by reacting with 2 mM 4-HNE in 0.1 M phosphate buffer, pH 7.4, at 37°C for 2 h. The enrichment of 4-HNE-modified peptides from the proteolytic digest of Daxx was accomplished by using solid-phase hydrazide (SPH) reagent as described previously (31) and analyzed by LC–MS/MS analyses (32, 33). LC–MS/MS was performed on a hybrid linear ion trap-FTICR (7-Tesla) mass spectrometer (LTQ-FT, Thermo Finnigan, San Jose, CA) equipped with a nanoelectrospray ionization source and operated with the Xcalibur (version 2.2) and Tune Plus (version 2.2) data acquisition software. MS/MS data generated by data dependent acquisition via the LTQ-FT were extracted by BioWorks version 3.3 and searched against a composite IPI human (version 3.47, number of entries is 144164 × 2) protein sequence database containing both forward and randomized sequences using the Mascot v 2.2 (Matrix Science, Boston, MA) search algorithm. (34, 35) MS/MS spectra of 4-HNE modified peptides were visually inspected to verify peptide fragment ions. The MS-Product module of Protein Prospector (http://prospector.ucsf.edu) was used to calculate the m/z values of b- and y-type ions of 4-HNE-modified Daxx tryptic peptides, obtained during CID-MS/MS, where His residues were replaced with 4-HNE-His Michael adducts (M + 156).
Statistical Analysis
The data are expressed as the mean ± SD for each group. The statistical significance was determined by Student's t test and was set at p < 0.05.
RESULTS
4-HNE causes apoptosis and necrosis in HepG2 cells
The cytotoxicity of 4-HNE to HepG2 cells was evaluated by MTT assay and apoptosis measured by flow cytometry, caspase activation, and PARP cleavage. In MTT assay (see supporting information), 4-HNE concentrations ranging from 10 to 100 µM gradually decreased cell viability corresponding to an IC50 value of 53 ± 2.39 µM (n = 8). Based on these results, 4-HNE concentrations of 5–40 µM were used to examine its effect on apoptotic signaling in HepG2 cells. 4-HNE-induced proteolytic cleavage of caspase-3 and PARP was also monitored. In the effector stage of apoptosis, caspase-3 is activated by its proteolytic cleavage into 17 and 12 kDa fragments. Like wise, poly (ADP-ribose) polymerase (PARP), which is normally involved in DNA repair, DNA stability, and other cellular events, is cleaved by members of the caspase family during early apoptosis. Results presented in Figure 1A showed that 4-HNE caused a dose dependent increase in 17 kDa fragment from the procaspase-3 and that of the 89 kDa cleavage product from PARP. 4-HNE-induced apoptosis in HepG2 cells was further analyzed by flow cytometry. Results presented in Figure 1B showed that after treatment with different concentrations of 4-HNE ranging from 0–100 µM for 2 h, the viability of cells decreased from 86.5% to 28.3% with an increase in the percent of late apoptotic cells from 9.7 to 16.1% in a dose dependent manner. A significant increase in necrotic cell population i.e. 31.8% and 55.4%, was observed in cells treated with 80 and 100 µM of 4-HNE respectively (Figure 1C). These results indicated that initial response to sub-lethal doses of 4-HNE (5–40 µM) treatment predominantly caused apoptotic cell death that ultimately proceeded to necrosis of HepG2 cells at lethal 4-HNE concentrations (80–100 µM).
Figure 1. Effect of 4-HNE on HepG2 cells.
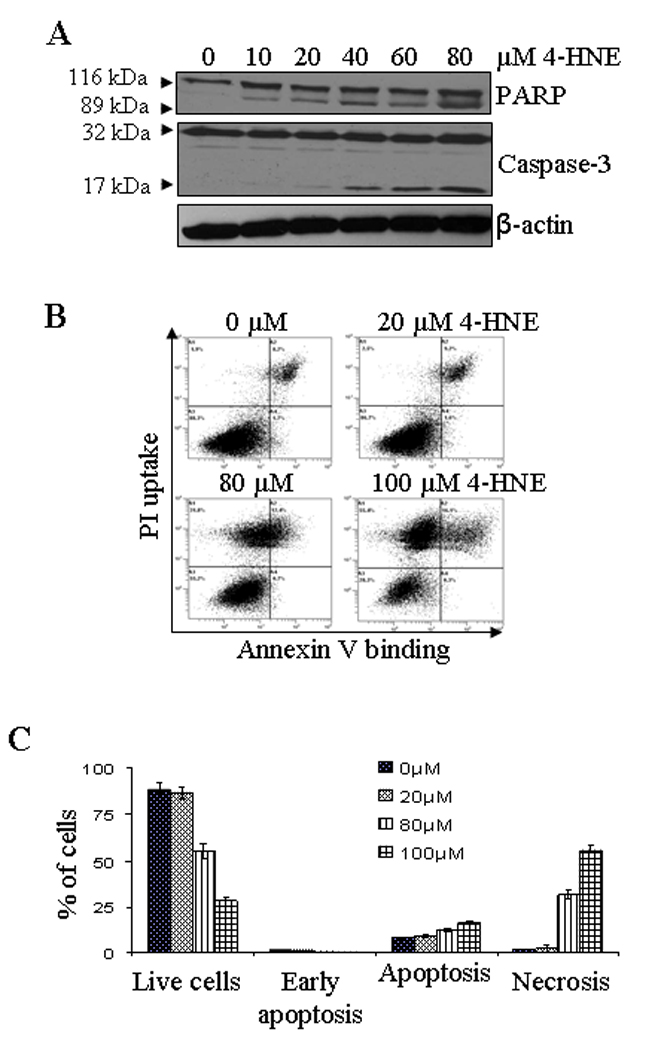
(A) Western blot analysis showing the caspase-3 and PARP cleavage: HepG2 cells were treated with different concentrations of 4-HNE (0–80 µM) for 2 h at 37°C. The cells were scraped, collected and then washed with ice cold PBS, and the cell lysates were prepared as described in Materials and methods section. The cells extracts (50 µg of protein) were subjected to Western blot analyses using anti-caspase-3 and anti-PARP antibodies. Anti-β-actin antibody was used as loading control. (B) Flow cytometry analysis of cells treated with 4-HNE: HepG2 cells (4 × 105) were grown and treated with different concentrations of 4-HNE for 2 h at 37°C in full serum medium. After treatment, the cells were harvested by trypsinization and washed with ice cold PBS and resuspended in 400 µL of cold Annexin binding buffer containing 5 µL of Annexin V-FITC and 5 µL of 0.1 mg/mL propidium iodide. The cells were incubated at room temperature for 10 min. in the dark and were analyzed by flow cytometry using a Beckman Coulter Cytomics FC500 flow cytometer. (C) Bar graph showing the percent of cells present in apoptosis vs necrosis of the panel B.
Over-expression of hGSTA4-4 inhibits 4-HNE induced apoptosis
HepG2 cells were stably transfected with hGSTA4 and over expression of hGSTA4-4 protein was confirmed by the results of Western blot analysis (Figure 2A). GST activity towards 4-HNE was found to be enhanced in hGSTA4 transfected cells along with the expected decrease in the constitutive 4-HNE levels (data not presented). 4-HNE-induced apoptosis in the empty vector and hGSTA4 transfected cells was analyzed by using CaspACE™ FITC-VAD-FMK in situ marker that binds to the cleaved caspase-3. Results of these experiments showed that the hGSTA4 transfected cells acquired significant resistance to 4-HNE-induced apoptosis as compared to the empty vector transfected HepG2 cells (Figure 2B upper panel). hGSTA4 transfected cells also acquired significant resistance to Doxorubicin (DOX)-induced apoptosis (Figure 2B lower panel). Since DOX-induced apoptosis has been attributed to generation of ROS, these results suggest the role of 4-HNE in the mechanism of apoptosis caused by oxidants in general (6, 7).
Figure 2. Effect of hGSTA4 transfection on 4-HNE and doxorubicin-induced apoptosis.
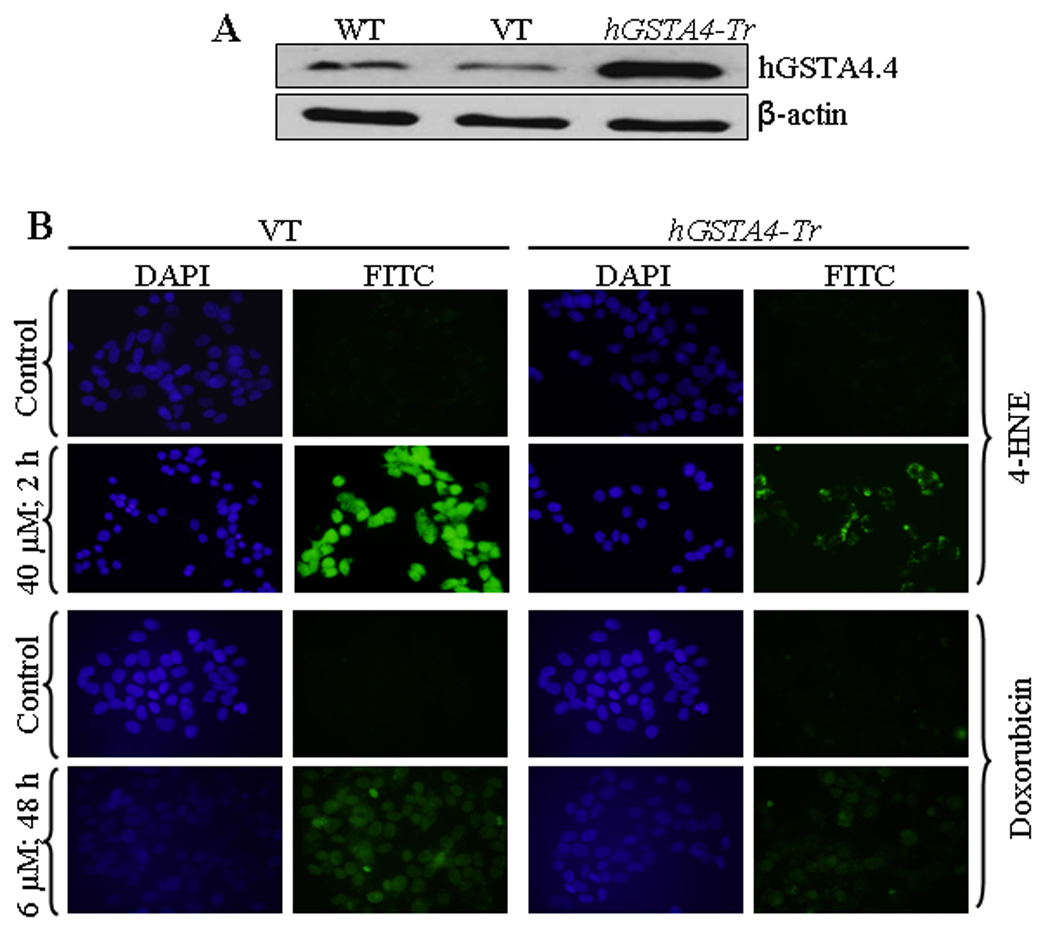
(A) Western blot analysis shows the expression of hGSTA4-4 in wild type, empty vector- (VT) and hGSTA4 transfected HepG2 cells. (B) In situ analysis of activated caspase-3 in VT and hGSTA4 transfected HepG2 cells. The cells (2 × 104) were treated with either 40 µM 4-HNE for 2 h or 6 µM doxorubicin for 48 h. The activation of caspase-3 in these cells was examined by staining with 10 µM CaspACE™ FITC-VAD-FMK in situ marker according to the manufacturer’s instructions. The slides were mounted with Vectashield DAPI mounting medium and observed with a fluorescence microscope (Olympus) using the standard filter sets for DAPI and FITC.
4-HNE and Fas mediated apoptosis
4-HNE activates Fas
To elucidate the mechanism of 4-HNE-induced apoptosis in HepG2 cells, we analyzed first the effect of 4-HNE on the expression of Fas. Results of Western blot analyses (Figure S1A and B in supporting information) indicated that 4-HNE caused a time, and dose dependent induction of Fas in HepG2 cells. These results were consistent with the previously reported induction of Fas by 4-HNE in HLE B-3, Jurkat, and CRL2571 cells (26, 28) indicating that 4-HNE mediated induction of Fas is not limited to a specific cell types.
Interaction of 4-HNE with Fas is required for apoptosis
To address the question, whether or not the interaction of 4-HNE was required to induce apoptosis, HepG2 cells were coated with the antagonistic Fas monoclonal antibodies (B-10) to mask cell surface Fas. The B-10 antibodies bind to cell surface Fas but unlike the agonistic CH11 antibodies that cause apoptosis, these antibodies do not induce apoptosis (36) and this was also confirmed during the present studies by examining their effects on HepG2 cells (data presented later in this section). 4-HNE induced apoptosis was significantly inhibited in B-10 coated cells indicating that Fas-4-HNE interaction was required for apoptosis (Figure 3A). It may be noted that 4-HNE induced apoptosis was not completely inhibited in B-10 coated cells. Although, cells coated with antagonistic Fas antibodies (B-10) were resistant to 4-HNE-induced apoptosis, a small population of cells still underwent apoptosis suggesting the involvement of other signaling pathways.
Figure 3. Effect of 4-HNE on Fas mediated apoptosis in HepG2 cells.
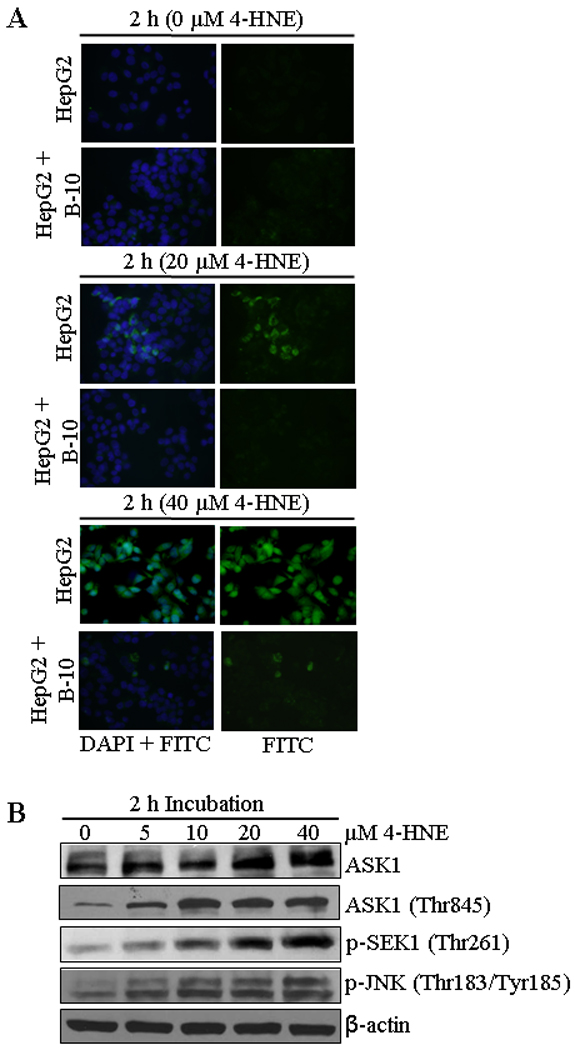
(A) In situ detection of Fas-mediated apoptosis in HepG2 cells. 2 × 104 cells were grown on glass cover slides and after pretreatment with or without antagonistic anti-Fas (B-10) antibodies for 2 h (250 ng/mL) HepG2 cells were treated with 0, 20 and 40 µM 4-HNE for 2 h followed by the addition of in situ caspase marker and fixation. The slides were mounted with Vectashield DAPI mounting medium and observed under a fluorescence microscope (Olympus). The photographs were taken at 400× magnification. (B) The cells were treated with 0, 5, 10, 20 and 40 µM 4-HNE for 2 h at 37°C. Total protein lysates (30 µg) were analyzed by western blotting for ASK1, p-ASK1 (Thr845), p-SEK1 (Thr261), and p-JNK (Thr183/Tyr185). β-actin was used as a loading control.
4-HNE causes the activation of ASK1-SEK1-JNK-Caspase-3
Previous studies have shown that Fas-mediated apoptosis during stress conditions is accompanied by the activation of ASK1, SEK1 and JNK, the kinases involved in the downstream signaling for apoptosis (37–41). It has also been shown that 4-HNE directly interacts with Fas and JNK (28, 42). We therefore, examined the effect of 4-HNE on these down stream apoptotic signaling molecules in HepG2 cells. Results of these studies showed a dose dependent activation of ASK1, p-ASK1 (Thr845), p-SEK1 (Thr261) and p-JNK (Thr183/Tyr185) (Figure 3B). The observed activation of ASK1, SEK1 and JNK by 4-HNE in present studies was rapid and sustained suggesting that a sustained activation and phosphorylation of JNK may be needed for 4-HNE-induced apoptosis. This prediction was supported by the results of experiments showing that pretreatment of HepG2 cells with the JNK inhibitor SP600125 made these cells resistant to apoptosis by 4-HNE (Figure S2, supporting information). We have previously demonstrated that in Jurkat cells, Fas mediated apoptosis by 4-HNE is independent of FADD. Results presented in Figure S3A and B (see supporting information) show the lack of FasL and FADD induction and also the lack of FADD binding to Fas as indicated by the results of immunoprecipitation experiments with 4-HNE treated cells (Figure S3C, supporting information). These results further support our previous observations that 4-HNE affects Fas mediated signaling for apoptosis through activation of ASK1 and JNK and is independent of DISC and this phenomenon is not limited to specific cell types.
Effect of 4-HNE on Daxx expression, phosphorylation, and trafficking
Death domain-associated protein Daxx that was originally cloned as a CD95/Apo-1 (Fas)-interacting protein (37) was shown to bind to the cytoplasmic domain of Fas and suggested to activate signaling for apoptosis during oxidative stress in different types of cells (40, 43). Therefore, we studied the effect of 4-HNE on Daxx in HepG2 cells and the results of these experiments presented in Figure 4 showed that 4-HNE treatment caused a time and concentration dependent induction of Daxx. Since its discovery, the sub-cellular localization of Daxx has been a controversy. Several studies have suggested that Daxx is predominantly a nuclear protein in unstressed cells, which under the conditions of oxidative stress, translocates to cytoplasm and binds to the cytoplasmic domain of Fas (41, 44). The effect of 4-HNE on Daxx trafficking and sub-cellular localization, was therefore analyzed by Western blot and immunofluorescence. Furthermore, the interaction of Daxx with Fas was examined by immunoprecipitation studies. Results of Western blot analysis of 4-HNE treated cells (Figure 5A and B) showed a marked increase of Daxx in the cytoplasmic fraction indicating that 4-HNE facilitated the export of Daxx from nucleus to cytoplasm and these results were further confirmed by immunofluorescence studies (Figure 5C).
Figure 4. 4-HNE induced expression of Daxx and phospho-Daxx in HepG2 cells.
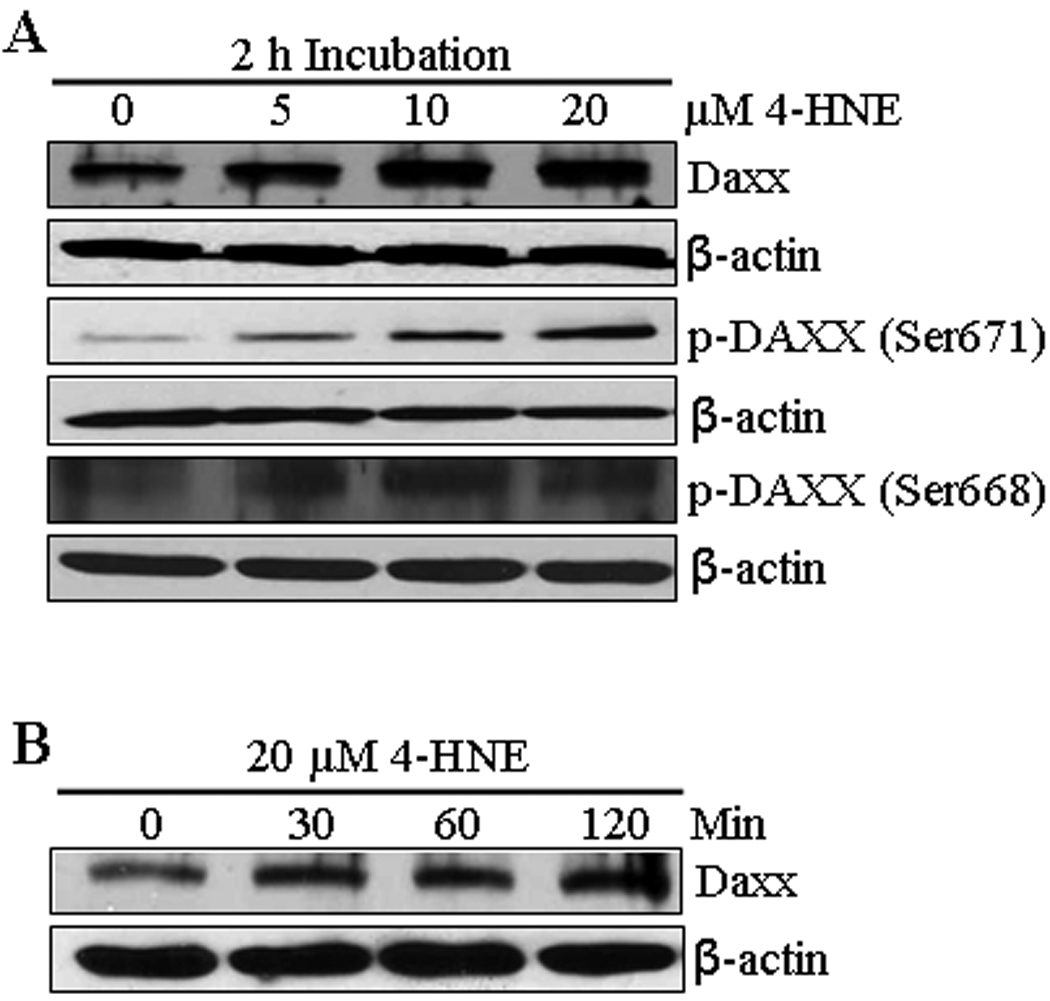
(A) The cells were treated with 0, 5, 10, and 20 µM 4-HNE for 2 h at 37°C. Total protein lysates (30 µg) were analyzed by Western blotting for the expression of Daxx, p-Daxx (Ser671) and p-Daxx (Ser668). β-actin was used as a loading control. (B) The cells were treated with 20 µM 4-HNE for 0, 30, 60, 120 min at 37°C. Total protein lysates were subjected to western blot analyses for Daxx expression. β-actin was used as a loading control.
Figure 5. Effect of 4-HNE induced cytoplasmic translocation of nuclear Daxx in HepG2 cells.
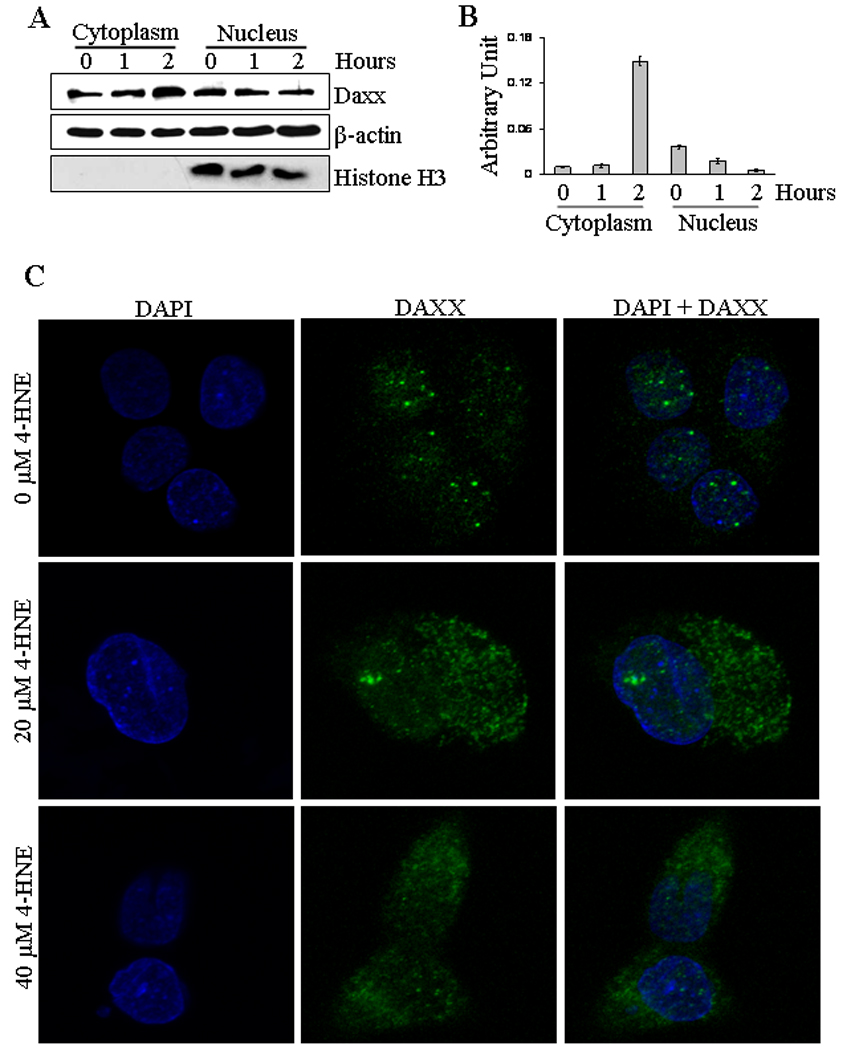
(A) The cells (4 × 105) were grown and treated with 20 µM 4-HNE for 0, 1 and 2 h, separately, at 37°C. After completion of incubation, cells were scraped, collected and washed with ice cold PBS. Cytoplasmic and nuclear extracts of the pelleted cells were prepared using Imgenex kit as per manufacturer’s instructions. The extracts (50 µg of protein) were subjected to western blot analysis for the detection of Daxx, β-actin and Histone H3. Antibodies against β-actin and Histone H3 were used to determine the purity of the cytoplasmic and nuclear fractions respectively. (B) Bar chart showing the densitometric analysis of Daxx bands of immunoblot of panel A. (C) Confocal immunofluorescence analysis of nuclear Daxx translocation. HepG2 cells were grown on glass cover slips and untreated and treated (20 µM 4-HNE for 2 h) cells were fixed, permeabilized and incubated with polyclonal anti-Daxx antibody, followed by fluorescein (FITC) conjugated secondary antibody. DAPI staining shows the nucleus. Slides were analyzed using Zeiss LSM 510META laser scanning fluorescence microscope.
The translocation of Daxx from nucleus to cytoplasm may be mediated through its phosphorylation on Ser and Thr residues (45, 46). Western blot analysis of the extracts of 4-HNE treated HepG2 cells indicated that 4-HNE caused phosphorylation of Daxx at Ser671 and Ser668 residues (Figure 4A). Together, these results show that 4-HNE promotes the translocation of Daxx from nucleus to cytoplasm through the phosphorylation of these residues. These results also suggest, that 4-HNE generated during oxidative stress may be the actual causative factor for the previously reported (41, 44) translocation of Daxx to cytoplasm during oxidative stress.
Interactions of 4-HNE with Fas and Daxx
We have examined the possible interaction of Daxx with 4-HNE and its binding to Fas in order to get insight into the mechanisms of 4-HNE-induced Fas-mediated apoptosis. The extracts from 4-HNE treated cells were immunoprecipitated with Daxx antibodies, and the resulting immunoprecipitates were probed separately with anti-Fas or anti-4-HNE 11-S antibodies (Figure 6A). The results of Western blot analysis showed that Daxx binds with Fas and also to 4-HNE as indicated by the presence of 4-HNE Daxx-adduct in the immunoprecipitate. When the immunoprecipitates obtained using Fas antibodies were probed with anti-Daxx antibody, the results (Figure 6B) further confirmed the binding of Daxx with Fas. Together, the results of these experiments indicated that 4-HNE interacts with both Fas and Daxx and it promotes the binding of Fas to Daxx (Figure 6C).
Figure 6. Co-immunoprecipitation of Fas and Daxx.
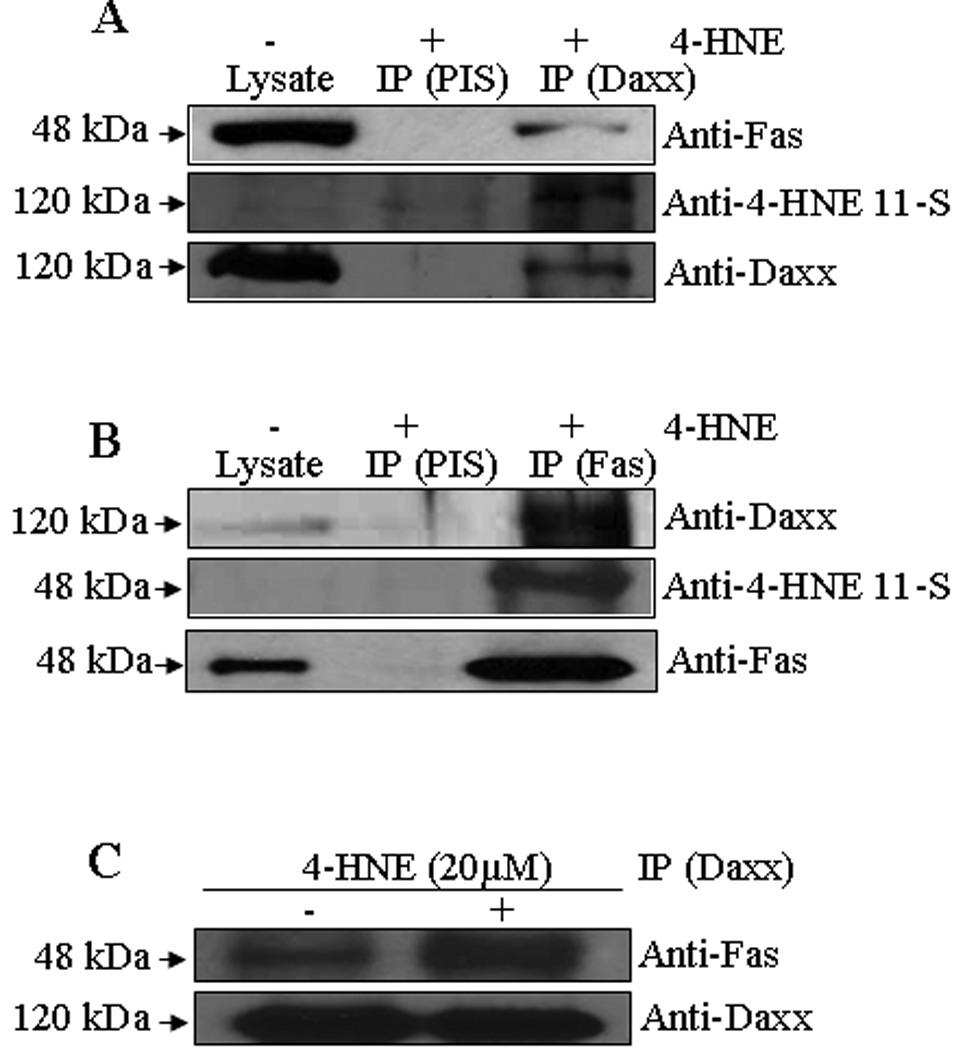
Total protein lysates were collected from control and 4-HNE-treated (20 µM for 2 h) HepG2 cells. The cell lysates were immunoprecipitated with anti-Fas and anti-Daxx antibodies as described in the materials and methods followed by Western blotting to check the expression of different proteins as indicated in the panel. Immunoprecipitation with pre-immune serum (PIS) was used as control. (A) The immunoprecipitate of Daxx (IP Daxx) were probed with antibodies indicated in the panel shows the presence of Fas, 4-HNE-Daxx adduct and Daxx. (B) The immunoprecipitate of Fas (IP Fas) was probed with antibodies indicated in the panel and shows the presence of Daxx and 4-HNE-Fas adduct. (C) The cell lysates were immunoprecipitated with anti-Daxx antibodies from control and 4-HNE-treated (20 µM for 2 h) HepG2 cells followed by Western blotting to check the expression of Fas and Daxx proteins as indicated in the panel.
4-HNE covalently binds to Daxx
To better understand binding of 4-HNE to Daxx, we performed an ex vivo modification of bacterially expressed purified human Daxx (see supporting information) with 4-HNE. After digestion of the modified protein with trypsin and using solid-phase hydrazide enrichment, LC–MS/MS analysis of Daxx identified four tryptic peptides with 4-HNE modification. All of these adducts showed preferential attachment of 4-HNE to His residues via a Michael addition mechanism (+156 Da). A representative CID-MS/MS spectrum of the doubly-charged 4-HNE-carbonylated Daxx tryptic peptide, VDSPSH*GLVTSSLCIPSPAR (where H* indicates 4-HNE Michael adduct on the His), at m/z 1118.58 is shown in Figure S4A (supporting information). The observed 4-HNE neutral loss peak at m/z 1040.8 (loss of 78 m/z from [M+2H]2+ ion) served as a diagnostic for the presence of H* in the peptide. The Mascot ion score was 67.8 (46.9 indicated identity or extensive homology) that surpassed the defined probability-based threshold for identification for this particular peptide derived from Daxx. Based on our results (Figure S4, supporting information), His residues at position 300, 404, 691 and 711 in Daxx were modified preferentially (Table 1).
Table 1.
Binding sites of 4-HNE in the amino acid sequence of Daxx identified by LC-MS/MS.
| Peptide Sequence | Modifications* |
|---|---|
| H*SLGLPR | 4-HNE (+156) |
| LQGTSSH*SADTPEASLDSGEGPSGMASQGCPSASRa | 4-HNE (+156) |
| VDSPSH*GLVTSSLCIPSPARa | 4-HNE (+156) |
| LSQTPH*SQPPRPGTCKa | 4-HNE (+156) |
Methionine (M) oxidation and cysteine (C) carbamidomethylation also occurred and introduced, respectively, during sample preparation for LC-MS/MS analysis, when these residues were present. These modifications were, therefore, considered irrelevant from the study’s point of view and not indicated.
Daxx silencing sensitizes cell to apoptosis
The role of Daxx, in particular its ability to promote or inhibit apoptosis, still remains controversial (41, 44). Some reports suggest that the death domain of Fas can directly activate apoptosis signal-regulating kinase 1 (ASK1) facilitating the translocation of Daxx from nucleus to cytoplasm. The interaction of translocated Daxx with the death domain of Fas has been shown to mediate cellular apoptosis via the Fas-Daxx-ASK1-JNK signaling pathway (47). On the other hand, it has been demonstrated that the binding of Daxx to Fas is not necessarily an apoptotic signal (48–51) and 4-HNE induced Daxx translocation from nucleus to the cytoplasm and it’s binding to Fas, in fact inhibits apoptosis (28). Therefore, we examined the role of Daxx in 4-HNE induced Fas mediated apoptosis in HepG2 cells. For these studies, we silenced the expression of Daxx by siRNA to evaluate the effect of Daxx depletion on 4-HNE-Fas mediated apoptosis. In HepG2 cells transfected with Daxx siRNA, the level of Daxx was only about 11% that of the cells transfected with scrambled siRNA after 48 hours incubation indicating a successful knock down of Daxx expression by siRNA (Figure 7A). Upon depletion of Daxx, cells became remarkably more sensitive to 4-HNE-induced apoptosis as indicated (Figure 7B and C) by the enhanced activation of caspase-3 and JNK in Daxx depleted cells as compared to the controls. Moreover, Daxx depleted cells also became more sensitive to the apoptosis caused by agonistic CH11 Fas antibodies, as indicated by the onset of increased apoptosis in Daxx-deficient cells treated with these antibodies (Figure 7C, lower two panels). Together, these results suggested that the binding of Daxx to Fas inhibits Fas mediated apoptosis and Fas-Daxx binding was not necessary for either activation of JNK and caspase-3 or the onset of apoptosis as reported previously (37).
Figure 7. Silencing of Daxx expression in HepG2 cells by siRNA and potentiation of apoptosis by 4-HNE and CH11 antibody.
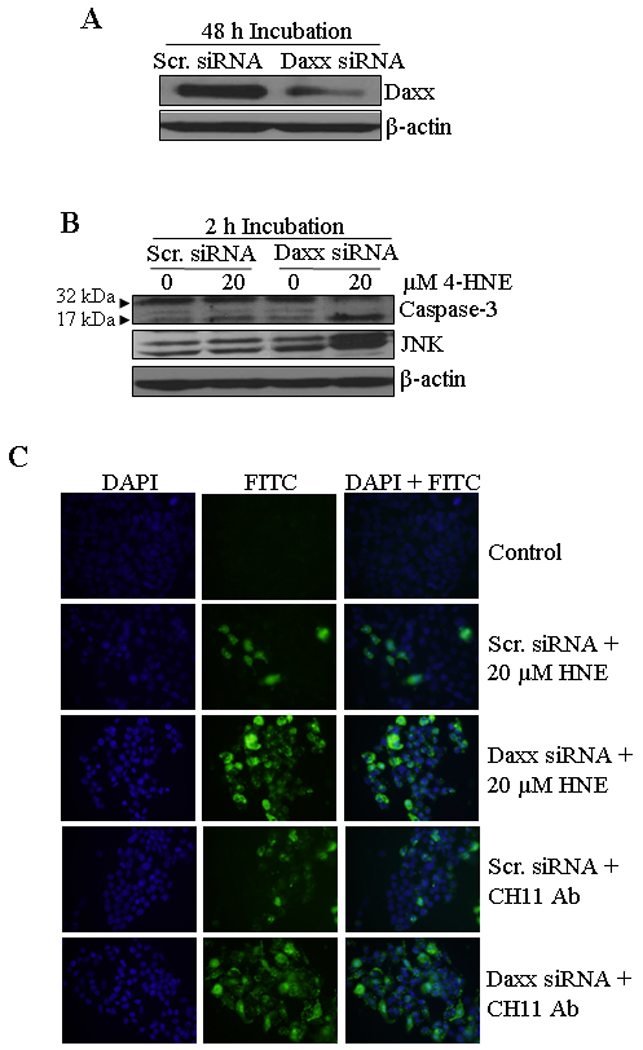
(A) Silencing of Daxx was performed by the ON-TARGET plus SMART pool Daxx siRNA as per the manufacturer’s instructions (Thermo Scientific Dharmacon) and control cells were treated with ON-TARGET plus Non-targeting siRNA in a similar way. After transfection, the cells were harvested after 48 h and the expression level of Daxx was examined by Western blot analysis. (B) Western blot analysis showing the enhanced activation of caspase-3 and JNK in Daxx depleted cells after 4-HNE treatment (20 µM HNE for 2 h). (C) Enhanced activation of apoptosis was also performed by In Situ immunofluorescence study. The cells (2 × 105) were grown on glass cover slips in twelve-well plate and transfected with non targeting siRNA or Daxx siRNA. After 48 h of siRNA transfection, the cells were treated with either 50 µg/well Fas-agonistic antibodies (CH11) or 20 µM 4-HNE for 2 h at 37°C. The activation of caspase-3 in these cells was examined by CaspACE FITC-VAD-FMK in situ marker as per the manufacturer’s instructions and then observed under a fluorescence microscope.
4-HNE causes the activation and translocation of HSF1 to the nucleus and induction of Hsp70
Daxx is known to be involved in transcriptional control of many genes by interacting directly with several transcription factors (41, 44, 52). The results of present studies showed that 4-HNE induced HSF1 in HepG2 cells in a time dependent manner (Figure 8A). 4-HNE also induced HSF1 translocation from cytoplasm to nucleus as indicated by the results of Western blot analysis of cytoplasmic and nuclear fractions (Figure 8B and C) and immunofluorescence analysis (Figure 8E). These results are consistent with 4-HNE mediated translocation of HSF1 from cytoplasm to nucleus in colon cancer RKO cells reported previously (53, 54). Activation and translocation of HSF1 is known to be an indication of the transcriptional up-regulation of stress-responsive heat shock proteins like Hsp70. Consistent with this prediction, 4-HNE was found to induce the expression of Hsp70 in HepG2 cells (Figure 8A). 4-HNE induced a robust accumulation of HSF1 within the nuclear compartment and a dense patchy staining of HSF1 was observed at a higher concentration (Figure 8E, see arrows). To demonstrate that this patchy nuclear staining for HSF1 was due to its binding to Hsp70 promoter sequence, we performed ChIP assay in the nuclear extracts of HepG2 cells treated with 4-HNE using HSF1 antibodies for immunoprecipitation. No signal was seen upon immunoprecipitation with negative control IgG. The positive control in which the antibodies used were against RNA polymerase II gave the expected positive signal validating our assay system. Neither the positive nor the negative controls were affected by 4-HNE treatment. The results presented in Figure 8D clearly indicated that the binding of HSF1 to the Hsp70 promoter was higher in 4-HNE treated HepG2 cells as compared to untreated cells, indicating that 4-HNE induced the activation and nuclear accumulation of HSF1 accompanied with the transcriptional up-regulation of Hsp70.
Figure 8. 4-HNE induced expression of heat shock factor 1 and heat shock protein Hsp70 in HepG2 cells.
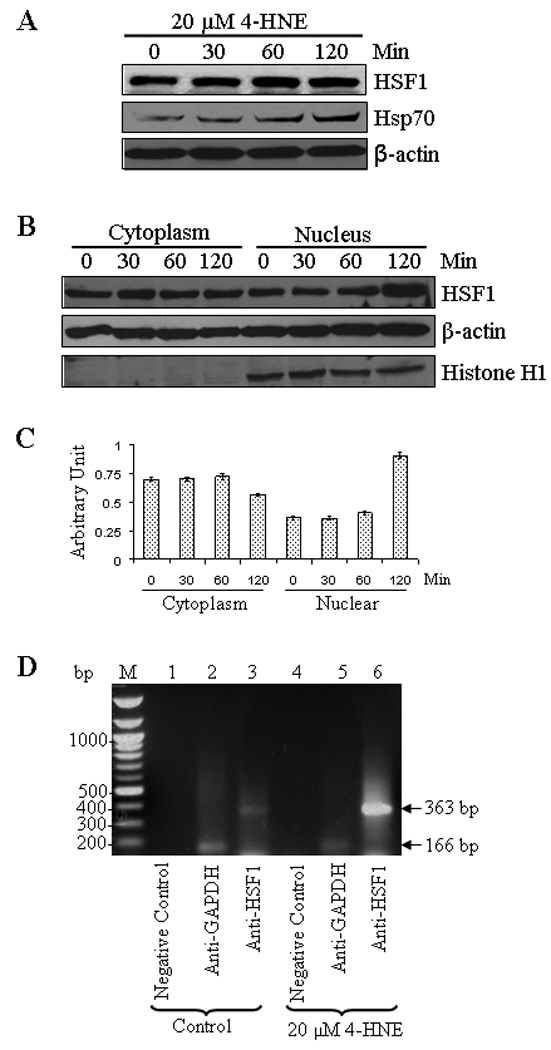
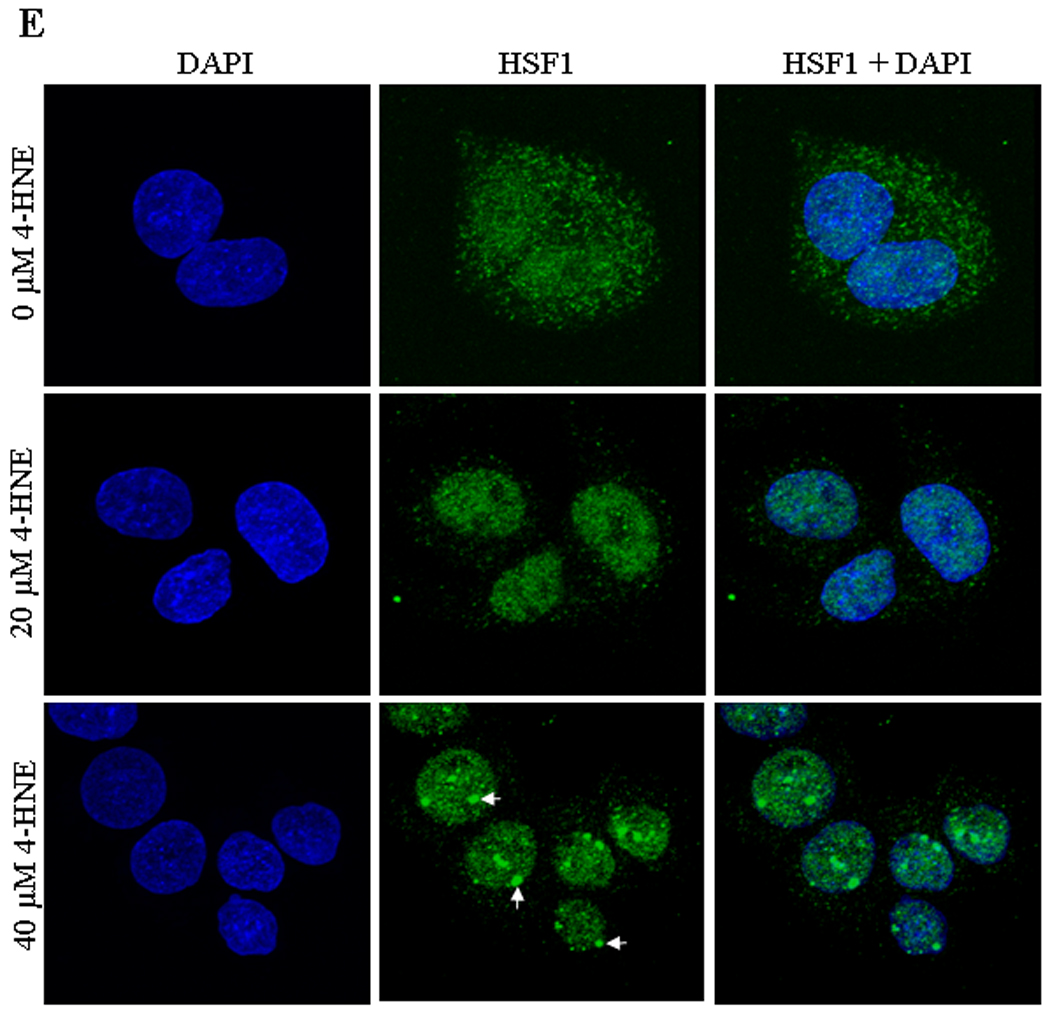
(A) The cells were treated with 20 µM 4-HNE for different time points at 37°C. Total protein lysates were analyzed by western blotting for HSF1, and Hsp70. β-actin was used as a loading control. (B) Effect of 4-HNE induced nuclear translocation of HSF1 in HepG2 cells was analyzed by Western blot analysis. The cells (4 × 105) were grown and treated with 20 µM 4-HNE for 0, 30, 60 and 120 min, separately, at 37°C. After 4-HNE incubation, cells were scraped, collected and washed with ice cold PBS. Cytoplasmic and nuclear extracts of the pelleted cells were prepared according to the Imgenex kit. The extracts (30 µg of protein) were subjected to western blot analysis by using anti-HSF1, anti-β actin and anti-Histone H1 antibodies. Histone H1 antibody was used as the purity control of cytoplasmic and nuclear fractions. (C) Bar chart showing the densitometric analysis of HSF1 bands of immunoblot of panel B. (D) Chromatin binding of HSF1 by ChIP assay. HepG2 cells (4 × 105) were grown and treated with 20 µM 4-HNE for 2 h followed by fixing with 1% formaldehyde for 10 min. Cells were scraped, and the chromatin was sheared by the protocol given under materials and methods. The chromatin was immunoprecipitated using the negative control IgG (provided by Active Motif), positive control IgG (provided by Active Motif), and HSF1 IgG (Santa Cruz, CA) followed by binding with protein G beads. The chromatin was eluted from the protein G beads and was amplified by PCR using the control primers as well as negative control primers (provided by Active Motif) and hHsp70 primers. PCR products were analyzed by running on 1% agarose gel. (E) Confocal immunofluorescence analysis of HSF1 translocation. HepG2 cells were grown on glass cover slips and untreated and treated (20 or 40 µM 4-HNE for 2 h) cells were fixed, permeabilized and incubated with polyclonal anti-HSF1 antibody, followed by fluorescein (FITC) conjugated secondary antibody. DAPI staining shows the nucleus. Slides were analyzed using Zeiss LSM 510META laser scanning fluorescence microscope.
4-HNE induces p53, phospho-p53, and activates p21, JNK, and Bax
To examine a possible role of 4-HNE in the p53 mediated apoptotic pathway, we determined the effect of 4-HNE on the expression of p53 and its phosphorylation in HepG2 cells. The results of these studies showed that at 20 µM concentration, 4-HNE caused a time dependent induction of p53 and phosphorylated p53 at Ser15 (Figure 9). Since activation and phosphorylation of p53 in response to oxidative stress has been associated with apoptosis through the induction of its target genes including p21 and Bax, we examined the effect of 4-HNE on the expression of its down stream targets. During apoptosis, Bax has been shown to undergo a conformational shift, and inserts into the outer mitochondrial membrane where it is believed to induce the opening of the mitochondrial membrane resulting in the release of cytochrome c and other pro-apoptotic factors from the mitochondria, leading to activation of caspases (55, 56). Consistent with one of the mechanisms of p53 mediated apoptosis, treatment of HepG2 cells with 4-HNE resulted in the activation of pro-apoptotic proteins Bax and p21. In parallel, the expression of anti-apoptotic Bcl-xL was found to be down regulated in 4-HNE treated cells. To determine the effect of 4- HNE on caspase-3 independent apoptosis in these cells, we analyzed the expression of AIF in 4-HNE treated cells. No significant change in the expression of AIF in control and 4-HNE treated cells (data not shown) suggests that 4-HNE does not affect AIF mediated caspase-independent apoptosis of these cells. The activation and phosphorylation of p53 along with p53 client pro-apoptotic proteins indicate that in addition to Fas mediated apoptosis 4-HNE also activates p53 mediated intrinsic apoptotic pathway. However, our results showing the resistance of Fas (B-10) antibodies coated HepG2 cells to 4-HNE induced apoptosis demonstrate that, as compared to Fas-mediated apoptosis, the relative contribution of p53-mediated apoptosis was only minimal (Figure 3A). No significant effect on 4-HNE induced activation of p53, JNK and Bax in B-10 coated cells (Figure S5, supporting information) suggest that the activation of p53 pathway is not dependent on Fas-4-HNE interaction. Further studies are needed to evaluate the physiological significance and relative contributions of Fas and p53 mediated pathways in 4-HNE induced apoptosis.
Figure 9. Effect of 4-HNE on p53 mediated intrinsic apoptotic pathway.
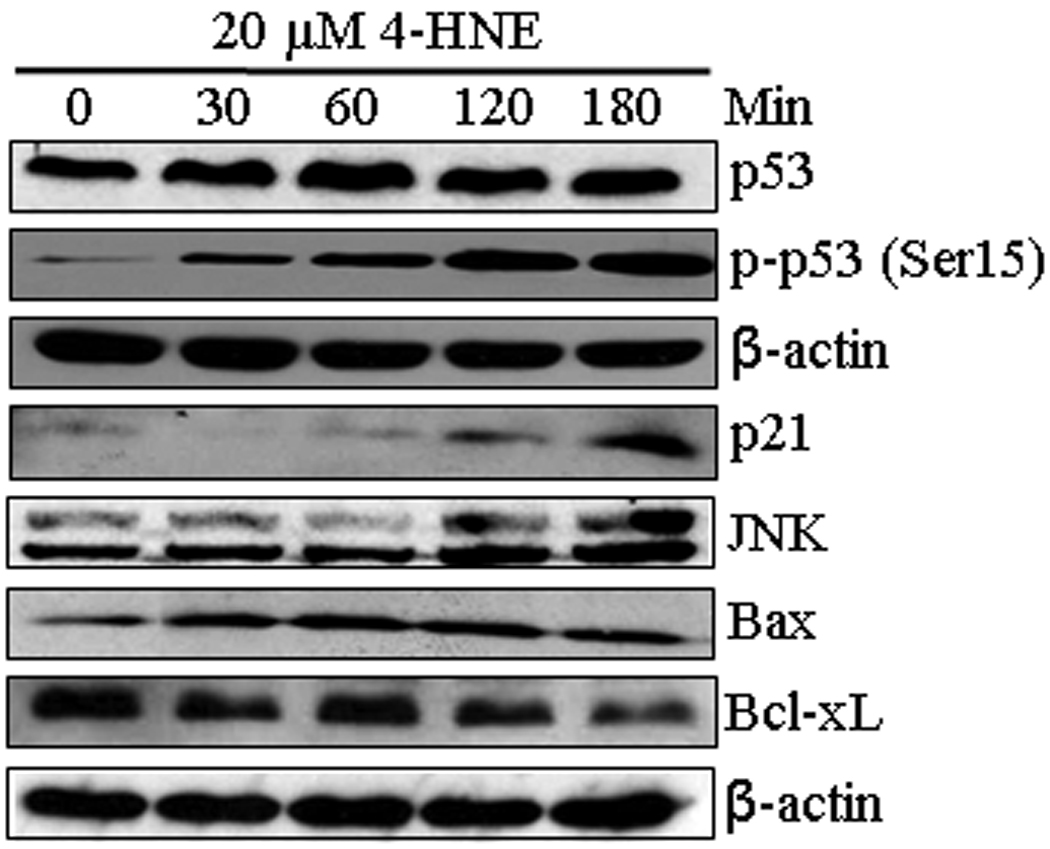
HepG2 cells were treated with 20 µM 4-HNE for the indicated time points at 37°C. Total protein lysates were collected as described under Materials and methods section. The lysates were analyzed by western blotting for p53, p-p53 (Ser15), p21, JNK, Bax and Bcl-xL. β-actin was used as a loading control.
Over-expression of hGSTA4-4 inhibits the activation of Fas and p53 mediated apoptotic pathways
To investigate any possible role of hGSTA4-4 in the regulation of Fas and p53 mediated apoptosis, we studied the effect of hGSTA4-transfection on the components of these pathways. Results of these experiments presented in Figure 10A showed that the activation of the component of both these pathways (Fas, Daxx, ASK1, p53, Bax and JNK) was significantly suppressed in hGSTA4 transfected cells as compared to that with vector-transfected cells. These results demonstrate an important regulatory role of hGSTA4-4 during apoptosis induced by oxidative stress via the Fas and p53 pathways and are consistent with reported rapid induction of this isozyme in stress conditions that lead to 4-HNE generation (26, 57).
Figure 10. Effect of 4-HNE on the expression of various proteins in hGSTA4 overexpressing HepG2 cells and Gsta4 null mice.
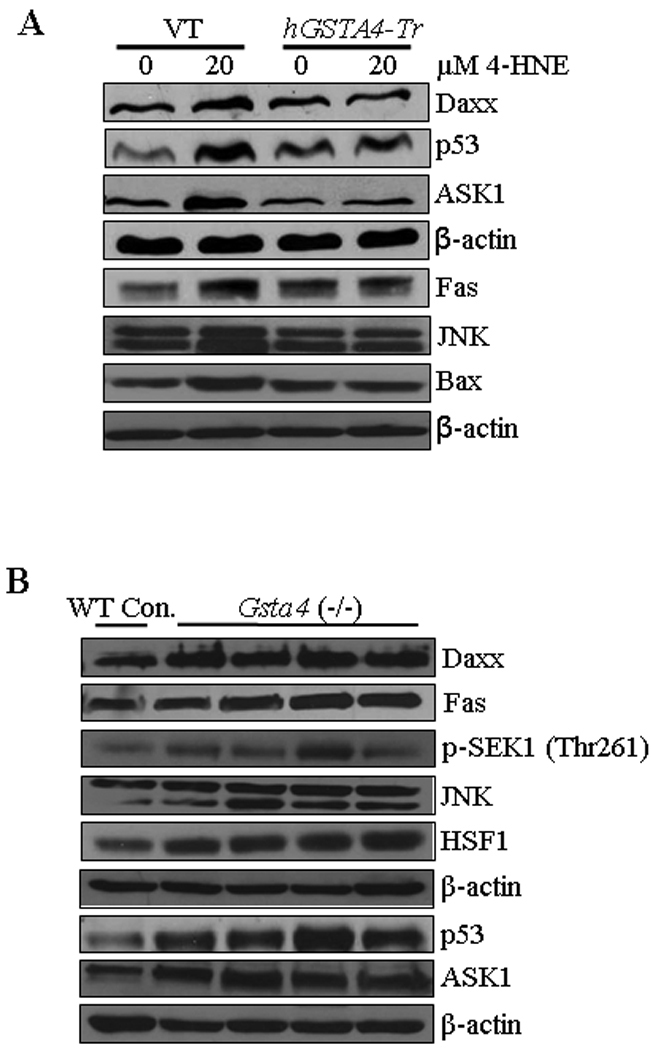
(A) Effect of 4-HNE (20 µM, 2 h) on the expression of Daxx, p53, ASK1, Fas, JNK and Bax in empty vector- and hGSTA4-transfected HepG2 cells (B) Expression analysis of apoptotic proteins in liver samples of normal and mGSTA4 (−/−) mice. Mice liver samples were homogenized in ice-cold RIPA buffer and centrifuged at 12,000 g and the supernatants were subjected to western blot analysis for the expression of Daxx, Fas, p-SEK1 (Thr261), JNK, HSF1, p53, and ASK1. β-actin was used as the loading control.
Increased 4-HNE levels in liver of mGsta4 null mice activate Fas and p53 mediated apoptotic pathways in vivo
The in vitro cell culture studies presented above were extended to in vivo conditions using the liver tissues from mGsta4 null mice which have been shown to contain approximately 3-fold higher concentration of 4-HNE in liver (29). To examine whether the effects of 4-HNE in cell cultures were also reflected in vivo, we compared the expression of Fas, Daxx, p53, HSF1, ASK1, p-SEK1, and JNK in the tissues of mGsta4 (−/−) and wild-type (+/+) mice. Consistent with our prediction, results of Western blot analysis presented in Figure 10B showed that the expression of these proteins was remarkably up regulated in liver tissues of mGsta4 (–/–) mice examined in this study. These results demonstrated a role of 4-HNE in signaling, in vivo and are consistent with our earlier suggestion (25, 58) that LPO and 4-HNE in particular, play an important role in oxidative stress induced signaling.
DISCUSSION
4-HNE, which was initially thought to be merely a toxic end-product of LPO, has emerged as an important second messenger signaling molecule in recent years. A multitude of studies (See reviews: 3–9, 24) over the past 10 years attest to its role in the regulation of gene expression, and modulation of various cellular processes including proliferation, transformation, differentiation, cell cycle regulation, and apoptosis. The mechanisms through which 4-HNE exerts such a wide variety of effects are largely unknown. For example, it remains to be determined as to how 4-HNE exerts seemingly opposite effects such as proliferation and programmed cell death in a concentration dependent manner. Present studies validate the important signaling role of 4-HNE and in addition, provide significant insight into the possible mechanisms through which 4-HNE exerts its multifarious effects in cells.
Here we demonstrate that 4-HNE causes toxicity to HepG2 cells through apoptosis and necrosis in a dose dependent manner. At sub lethal concentrations of 4-HNE, these cells undergo apoptosis via two separate pathways. Results of these studies clearly demonstrate that 4-HNE binds to Fas, and affects the downstream signaling mediated by this death receptor and that this 4-HNE-induced apoptosis is independent of the classical Fas-mediated apoptosis through FADD and DISC (59) because 4-HNE neither activates FADD nor promotes the binding of Fas to FADD. However, 4-HNE induced, Fas-mediated apoptosis involves the activation of ASK1, JNK and caspase-3 but does not induce FasL or the constituents of DISC. As indicated by the lack of apoptosis by 4-HNE in cells coated with antagonistic Fas antibodies, our results clearly demonstrate that binding of 4-HNE with Fas is essential for the execution of apoptosis. This is further indicated by abrogation of 4-HNE-induced activation of ASK1, JNK, and caspase-3 in cells when Fas is masked by coating with antibodies. Our results showing inhibition of apoptosis and lack of the activation of ASK1 and JNK in cells over expressing hGSTA4-4 pinpoint these effects to 4-HNE because this enzyme is highly specific for the conjugation of 4-HNE to GSH. 4-HNE induced activation of p53 and down stream targets including p21 and Bax in HepG2 cells also suggest the involvement of p53 mediated pathway in the apoptotic mechanisms of these cells but the contribution of this pathway seems to be minimal. All these effects of 4-HNE can also be abrogated by GSTA4-4 over expression. These finding are consistent with previous studies suggesting important role of GST isozymes in the regulation of stress-induced signaling, by attenuating 4-HNE concentrations (8) as well as by inhibiting JNK (60–62).
These signaling effects of 4-HNE observed in in vitro cell lines appear to mirror in the liver tissues of mice which have higher basal levels of 4-HNE due to the disruption of Gsta4 gene. This is important because it may enhance our understanding of the mechanism involved in manifestation of the toxicity of ROS and oxidants, and the role of relevant defense mechanisms in vivo. Even though Gsta4 (−/−) mice are more sensitive to oxidant toxicity, they seem to have no apparent toxic manifestations in stress free conditions and in fact may have an increased life span (63). Thus the question may arise as to why these mice do not show any apparent toxicity despite activation of Fas and p53 mediated apoptotic pathways. The answer to this question may lie in our results showing that in parallel to its apoptotic signaling, 4-HNE invokes signaling for the defense against its own toxicity. First, by inhibiting its apoptotic effect by facilitating cytoplasmic export of Daxx and its binding with Fas and second, by activating HSF1 initiated stress responsive transcription of protective heat shock proteins such as Hsp70. The involvement of 4-HNE in promoting the cytoplasmic translocation of Daxx is further confirmed by our results demonstrating a direct binding of 4-HNE with Daxx, and phosphorylation of Daxx at Ser668 and Ser671 that is reported (45, 46) to be required for the translocation of Daxx from nucleus to cytoplasm. Once in cytoplasm Daxx binds to Fas and inhibits its own apoptotic effect that may be an important defense mechanism to check apoptosis. Our results clearly demonstrate that the binding of Fas to Daxx does indeed inhibit apoptosis (28, 48–51) rather than causing a pro-apoptotic effect as suggested in some earlier studies (37, 43, 47, 64, 65). The potentiation of the apoptotic effect of the agonistic CH11 anti-Fas antibodies in Daxx depleted cells further substantiates the anti-apoptotic role of Daxx and indicates that its inhibitory role is not limited to oxidative stress/ 4-HNE mediated apoptosis.
In addition to Daxx mediated inhibition of apoptosis, in parallel 4-HNE induces trafficking of HSF1 from cytoplasm to nucleus to promote transcription of stress responsive genes to reinforce the defense response. In RKO cells 4-HNE mediated translocation of HSF1 has been shown and a role of HSF1 in anti-apoptotic mechanisms has been demonstrated (53, 54). Present studies through Western blot and immunofluorescence analyses provide clear evidence for the trafficking of HSF1 from cytoplasm to nucleus in HepG2 cells where it promotes the expression of Hsp70 indicating that this effect of 4-HNE is general and not limited to a specific cell type. Since the effect of 4-HNE in the liver of Gsta4 null mice are similar to those observed in in vitro cell culture studies, the defense mechanisms evoked due to the elevated levels of 4-HNE in these mice may also explain the observed increased life span of Gsta4 null mice (63) without any apparent toxicity. The effects of 4-HNE on Daxx, HSF1, and p53 reported here may perhaps include other transcription repressors/factors. If so 4-HNE should have a profound effect on the expression of client genes. This prediction finds strong support from the results of several studies by us (26–28, 66) and other investigators (5, 67–69) showing profound changes in the expression of a multitude of genes when 4-HNE concentrations are altered in cells. It may be argued that the steady state 4-HNE concentrations in cells are reported (24) to be much lower than those used in present studies. However, it should be pointed out that during oxidative stress the intracellular concentration could be even higher than used in these studies (70).
Since our results indicated the binding of 4-HNE to Fas, HSF1, p53 and Daxx, proteomics studies were carried out with Daxx to demonstrate 4-HNE binding to its specific residues. Through these studies, we have identified several binding sites, although possible functional consequences of the binding are yet to be determined. It is possible that observed concentration dependent effects of 4-HNE may be determined by the differential reactivity of selective target residue(s) of key proteins such as Daxx for 4-HNE binding that may evoke differential signaling. An interesting novel paradigm seems to emerge from the results of present studies. It is possible that through interactions with membrane receptors such as Fas, EGF, VEGF, transcription factor such as p53, HSF1, and transcription repressors such as Daxx, 4-HNE could have a global effect on the expression of genes regulating a variety of cellular processes. The cellular concentrations of 4-HNE and its relative affinity to the residues of target proteins may be determinants of these interactions and consequent effects. Our previous studies have shown that suppression of 4-HNE levels in cells of at least two different types leads to profound alteration in the expression of genes in these cells and results in their transformation (27, 66). Thus, it seems likely that 4-HNE, through direct interactions with some key proteins such as those investigated in the present studies, can have profound effects on gene expression and consequently on various cellular processes. This would also imply that the Alpha class GSTs including GSTA4-4 that regulate LPO and 4-HNE levels in cells (25, 57, 71), play an important role in stress-induced signaling. Ongoing studies in our lab suggest that 4-HNE does indeed affect the signaling pathways mediated by VEGF, and VEGFR, in a concentration dependent manner but numerous conclusive studies will have to be conducted to substantiate this implication.
Supplementary Material
ACKNOWLEDGEMENT
Funding: This work was supported in part by NIH Grants ES012171, EY04396 (YCA), AG025384 (LP) and CA77495 (SA), and by the Welch Foundation (LP, endowment number BK-0031).
We greatly appreciate the gift of human Daxx cDNA from Dr. Alnawaz Rehemtulla, (Department of Radiation Oncology and Radiology, University of Michigan, Ann Arbor, MI 48109-2200, USA). We thank Xiangle Sun (Core Facility at the University of North Texas Health Science Center) for helping with flow Cytometry.
Abbreviations
- 4-HNE
4-hydroxy-2-nonenal
- DOX
doxorubicin (Adriamycin)
- GST
glutathione S-transferase
- siRNA
small interfering RNA
- Daxx
death domain-associated protein
- ASK1
apoptosis signal-regulating kinase 1
- SEK1
stress-activated protein kinase/extracellular-signal regulated kinase kinase
- JNK
c-Jun NH2-terminal kinase
- HSF1
heat shock factor 1
- WT
wild type
- IP
immunoprecipitation
- ChIP
chromatin immunoprecipitation
- DAPI
4',6-diamidino-2-phenylindole
- PARP
Poly (ADP-ribose) polymerase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- Hsps
Heat shock proteins
- AIF
apoptosis inducing factor
- FADD
Fas-associated death domain
- DISC
death-inducing signaling complex
Footnotes
SUPPORTING INFORMATION AVAILABLE
Additional Materials and Methods, and Figures S1–S5 as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Cheng JZ, Sharma R, Yang Y, Singhal SS, Sharma A, Saini MK, Singh SV, Zimniak P, Awasthi S, Awasthi YC. Accelerated metabolism and exclusion of 4-hydroxynonenal through induction of RLIP76 and hGST5.8 is an early adaptive response of cells to heat and oxidative stress. J. Biol. Chem. 2001;276:41213–41223. doi: 10.1074/jbc.M106838200. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, Sharma A, Sharma R, Patrick B, Singhal SS, Zimniak P, Awasthi S, Awasthi YC. Cells preconditioned with mild, transient UVA irradiation acquire resistance to oxidative stress and UVA-induced apoptosis: role of 4-hydroxynonenal in UVA-mediated signaling for apoptosis. J. Biol. Chem. 2003;278:41380–41388. doi: 10.1074/jbc.M305766200. [DOI] [PubMed] [Google Scholar]
- 3.Dianzani MU. 4-hydroxynonenal from pathology to physiology. Mol. Aspects Med. 2003;24:263–272. doi: 10.1016/s0098-2997(03)00021-9. [DOI] [PubMed] [Google Scholar]
- 4.Nakashima I, Liu W, Akhand AA, Takeda K, Kawamoto Y, Kato M, Suzuki H. 4-Hydroxynonenal triggers multistep signal transduction cascades for suppression of cellular functions. Mol. Aspects Med. 2003;24:231–238. doi: 10.1016/s0098-2997(03)00018-9. [DOI] [PubMed] [Google Scholar]
- 5.Barrera G, Pizzimenti S, Dianzani MU. 4-Hydroxynonenal and regulation of cell cycle: effects on the pRb/E2F pathway. Free Radic. Biol. Med. 2004;37:597–606. doi: 10.1016/j.freeradbiomed.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 6.Awasthi YC, Sharma R, Cheng JZ, Yang Y, Sharma A, Singhal SS, Awasthi S. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Mol. Aspects Med. 2003;24:219–230. doi: 10.1016/s0098-2997(03)00017-7. [DOI] [PubMed] [Google Scholar]
- 7.Awasthi YC, Yang Y, Tiwari NK, Patrick B, Sharma A, Li J, Awasthi S. Regulation of 4-hydroxynonenal-mediated signaling by glutathione S-transferases. Free Radic. Biol. Med. 2004;37:607–619. doi: 10.1016/j.freeradbiomed.2004.05.033. [DOI] [PubMed] [Google Scholar]
- 8.Awasthi YC, Ansari GA, Awasthi S. Regulation of 4-hydroxynonenal mediated signaling by glutathione S-transferases. Methods Enzymol. 2005;401:379–407. doi: 10.1016/S0076-6879(05)01024-4. [DOI] [PubMed] [Google Scholar]
- 9.Awasthi YC, Sharma R, Sharma A, Yadav S, Singhal SS, Chaudhary P, Awasthi S. Self-regulatory role of 4-hydroxynonenal in signaling for stress-induced programmed cell death. Free Radic. Biol. Med. 2008;45:111–118. doi: 10.1016/j.freeradbiomed.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sampey BP, Stewart BJ, Petersen DR. Ethanol-induced modulation of hepatocellular extracellular signal-regulated kinase-1/2 activity via 4-hydroxynonenal. J. Biol. Chem. 2007;282:1925–1937. doi: 10.1074/jbc.M610602200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsukamoto H, Horne W, Kamimura S, Niemel O, Parkkila S, Yl-Herttuala S, Brittenham GM. Experimental liver cirrhosis induced by alcohol and iron. J. Clin. Invest. 1995;96:620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nanji AA, Zhao S, Sadrzadeh SMH, Dannenberg AJ, Tahan SR, Waxman DJ. Markedly enhanced cytochrome P450 2E1 induction and lipid peroxidation is associated with severe liver injury in fish oil-ethanol-fed rats. Alcohol Clin. Exp. Res. 1994;18:1280–1285. doi: 10.1111/j.1530-0277.1994.tb00119.x. [DOI] [PubMed] [Google Scholar]
- 13.Dwivedi S, Sharma R, Sharma A, Zimniak P, Ceci JD, Awasthi YC, Boor PJ. The course of CCl4 induced hepatotoxicity is altered in mGSTA4-4 null (−/−) mice. Toxicology. 2006;218:58–66. doi: 10.1016/j.tox.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Kamimura S, Gaal K, Britton RS, Bacon BR, Triadafilopoulos G, Tsukamoto H. Increased 4-hydroxynonenal levels in experimental alcoholic liver disease: Association of lipid peroxidation with liver fibrogenesis. Hepatology. 1992;16:1014–1021. doi: 10.1002/hep.1840160225. [DOI] [PubMed] [Google Scholar]
- 15.Uchida K, Toyokuni S, Nishikawa K, Kawakishi S, Oda H, Hiai H, Stadtman ER. Michael addition-type 4-hydroxy-2-nonenal adducts in modified low-density lipoproteins: markers for atherosclerosis. Biochemistry. 1994;33:12487–12494. doi: 10.1021/bi00207a016. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y, Yang Y, Trent MB, He N, Lick SD, Zimniak P, Awasthi YC, Boor PJ. Glutathione-S-transferase A4-4 modulates oxidative stress in endothelium: possible role in human atherosclerosis. Atherosclerosis. 2004;173:211–221. doi: 10.1016/j.atherosclerosis.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 17.Awasthi S, Singhal SS, Yadav S, Singhal J, Vatsyayan R, Zajac E, Luchowski R, Borvak J, Gryczynski K, Awasthi YC. A Central Role of RLIP76 in Regulation of Glycemic Control. Diabetes. 2010;59:714–725. doi: 10.2337/db09-0911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J. Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 19.Montine KS, Olson SJ, Amarnath V, Whetsell WO, Graham DG, Jr, Montine TJ. Immunohistochemical detection of 4-hydroxy-2-nonenal adducts in Alzheimer's disease is associated with inheritance of APOE4. Am. J. Pathol. 1997;150:437–443. [PMC free article] [PubMed] [Google Scholar]
- 20.Selley ML. (E)-4-hydroxy-2-nonenal may be involved in the pathogenesis of Parkinson's disease. Free Radic. Biol. Med. 1998;25:169–174. doi: 10.1016/s0891-5849(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 21.Awasthi S, Srivatava SK, Piper JT, Singhal SS, Chaubey M, Awasthi YC. Curcumin protects against 4-hydroxy-2-trans-nonenal-induced cataract formation in rat lenses. Am. J. Clin. Nutr. 1996;64:761–766. doi: 10.1093/ajcn/64.5.761. [DOI] [PubMed] [Google Scholar]
- 22.Zarkovic N, Tillian MH, Schaur J, Waeg G, Jurin M, Esterbauer H. Inhibition of melanoma B16-F10 growth by lipid peroxidation product 4-hydroxynonenal. Cancer Biother. 1995;10:153–156. doi: 10.1089/cbr.1995.10.153. [DOI] [PubMed] [Google Scholar]
- 23.Hammer A, Ferro M, Tillian HM, Tatzber F, Zollner H, Schauenstein E, Schaur RJ. Effect of oxidative stress by iron on 4-hydroxynonenal formation and proliferative activity in hepatomas of different degrees of differentiation. Free Radic. Biol. Med. 1997;23:26–33. doi: 10.1016/s0891-5849(96)00630-2. [DOI] [PubMed] [Google Scholar]
- 24.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 25.Cheng JZ, Singhal SS, Saini M, Singhal J, Piper JT, Van Kuijk FJ, Zimniak P, Awasthi YC, Awasthi S. Effects of mGST A4 transfection on 4-hydroxynonenal-mediated apoptosis and differentiation of K562 human erythroleukemia cells. Arch. Biochem. Biophys. 1999;372:29–36. doi: 10.1006/abbi.1999.1479. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Sharma R, Patrick B, Sharma A, Jeyabal VP, Reddy PM, Saini MK, Dwivedi S, Dhanani S, Ansari NH, Zimniak P, Awasthi S, Awasthi YC. Regulation of CD95 (Fas) expression and Fas-mediated apoptotic signaling in HLE B-3 cells by 4-hydroxynonenal. Biochemistry. 2006;45:12253–12264. doi: 10.1021/bi060780+. [DOI] [PubMed] [Google Scholar]
- 27.Sharma R, Brown D, Awasthi S, Yang Y, Sharma A, Patrick B, Saini MK, Singh SP, Zimniak P, Singh SV, Awasthi YC. Transfection with 4-hydroxynonenal-metabolizing glutathione S-transferase isozymes leads to phenotypic transformation and immortalization of adherent cells. Eur. J. Biochem. 2004;271:1690–1701. doi: 10.1111/j.1432-1033.2004.04067.x. [DOI] [PubMed] [Google Scholar]
- 28.Sharma R, Sharma A, Dwivedi S, Zimniak P, Awasthi S, Awasthi YC. 4-Hydroxynonenal self-limits fas-mediated DISC-independent apoptosis by promoting export of Daxx from the nucleus to the cytosol and its binding to Fas. Biochemistry. 2008;47:143–156. doi: 10.1021/bi701559f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engle MR, Singh SP, Czernik PJ, Gaddy D, Montague DC, Ceci JD, Yang Y, Awasthi S, Awasthi YC, Zimniak P. Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: generation and analysis of mGsta4 null mouse. Toxicol. Appl. Pharmacol. 2004;194:296–308. doi: 10.1016/j.taap.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 30.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 31.Roe MR, Xie H, Bandhakavi S, Griffin TJ. Proteomic Mapping of 4-Hydroxynonenal Protein Modification Sites by Solid-Phase Hydrazide Chemistry and Mass Spectrometry. Anal. Chem. 2007;79:3747–3756. doi: 10.1021/ac0617971. [DOI] [PubMed] [Google Scholar]
- 32.Rauniyar N, Stevens SM, Jr, Prokai-Tatrai K, Prokai L. Characterization of 4-hydroxy-2-nonenal-modified peptides by liquid chromatography-tandem mass spectrometry using data-dependent acquisition: neutral loss-driven MS3 versus neutral loss-driven electron capture dissociation. Anal. Chem. 2009;81:782–789. doi: 10.1021/ac802015m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rauniyar N, Prokai-Tatrai K, Prokai L. Identification of carbonylation sites in apomyoglobin after exposure to 4-hydroxy-2-nonenal by solid-phase enrichment and liquid chromatography-electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 2010 doi: 10.1002/jms.1725. (In press) DOI 10.1002/jms.1725. [DOI] [PubMed] [Google Scholar]
- 34.Stevens SM, Jr, Rauniyar N, Prokai L. Rapid characterization of covalent modifications to rat brain mitochondrial proteins after ex vivo exposure to 4-hydroxy-2-nonenal by liquid chromatography-tandem mass spectrometry using data-dependent and neutral loss-driven MS3 acquisition. J. Mass Spectrom. 2007;42:1599–1605. doi: 10.1002/jms.1349. [DOI] [PubMed] [Google Scholar]
- 35.Rauniyar N, Prokai L. Detection and identification of 4-hydroxy-2-nonenal Schiff-base adducts along with products of Michael addition using data-dependent neutral loss-driven MS3 acquisition: method evaluation through an in vitro study on cytochrome c oxidase modifications. Proteomics. 2009;9:5188–5193. doi: 10.1002/pmic.200900116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fadeel B, Thorpe CJ, Yonehara S, Chiodi F. Anti-Fas IgG1 antibodies recognizing the same epitope of Fas/APO-1 mediate different biological effects in vitro. Int. Immunol. 1997;9:201–209. doi: 10.1093/intimm/9.2.201. [DOI] [PubMed] [Google Scholar]
- 37.Yang X, Khosravi-Far R, Chang HY, Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell. 1997;89:1067–1076. doi: 10.1016/s0092-8674(00)80294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 39.Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science. 1998;281:1860–1863. doi: 10.1126/science.281.5384.1860. [DOI] [PubMed] [Google Scholar]
- 40.Soh Y, Jeong KS, Lee IJ, Bae MA, Kim YC, Song BJ. Selective activation of the c-Jun N-terminal protein kinase pathway during 4-hydroxynonenal-induced apoptosis of PC12 cells. Mol. Pharmacol. 2000;58:535–541. doi: 10.1124/mol.58.3.535. [DOI] [PubMed] [Google Scholar]
- 41.Salomoni P, Khelifi AF. Daxx: death or survival protein? Trends Cell Biol. 2006;16:97–104. doi: 10.1016/j.tcb.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 42.Parola M, Robino G, Marra F, Pinzani M, Bellomo G, Leonarduzzi G, Chiarugi P, Camandola S, Poli G, Waeg G, Gentilini P, Dianzani MU. HNE interacts directly with JNK isoforms in human hepatic stellate cells. J. Clin. Invest. 1998;102:1942–1950. doi: 10.1172/JCI1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khelifi AF, D'Alcontres MS, Salomoni P. Daxx is required for stress-induced cell death and JNK activation. Cell Death Differ. 2005;12:724–733. doi: 10.1038/sj.cdd.4401559. [DOI] [PubMed] [Google Scholar]
- 44.Lindsay CR, Morozov VM, Ishov AM. PML NBs (ND10) and Daxx: from nuclear structure to protein function. Front Biosci. 2008;13:7132–7142. doi: 10.2741/3216. [DOI] [PubMed] [Google Scholar]
- 45.Song JJ, Lee YJ. Tryptophan 621 and serine 667 residues of Daxx regulate its nuclear export during glucose deprivation. J. Biol. Chem. 2004;279:30573–30578. doi: 10.1074/jbc.M404512200. [DOI] [PubMed] [Google Scholar]
- 46.Ecsedy JA, Michaelson JS, Leder P. Homeodomain-interacting protein kinase 1 modulates Daxx localization, phosphorylation, and transcriptional activity. Mol. Cell Biol. 2003;23:950–960. doi: 10.1128/MCB.23.3.950-960.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ko YG, Kang YS, Park H, Seol W, Kim J, Kim T, Park HS, Choi EJ, Kim S. Apoptosis signal-regulating kinase 1 controls the proapoptotic function of death-associated protein (Daxx) in the cytoplasm. J. Biol. Chem. 2001;276:39103–39106. doi: 10.1074/jbc.M105928200. [DOI] [PubMed] [Google Scholar]
- 48.Michaelson JS, Bader D, Kuo F, Kozak C, Leder P. Loss of Daxx, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes Dev. 1999;13:1918–1923. doi: 10.1101/gad.13.15.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michaelson JS, Leder P. RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J. Cell Sci. 2003;116:345–352. doi: 10.1242/jcs.00234. [DOI] [PubMed] [Google Scholar]
- 50.Chen LY, Chen JD. Daxx silencing sensitizes cells to multiple apoptotic pathways. Mol. Cell. Biol. 2003;23:7108–7121. doi: 10.1128/MCB.23.20.7108-7121.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zobalova R, Swettenham E, Chladova J, Dong LF, Neuzil J. Daxx inhibits stress-induced apoptosis in cardiac myocytes. Redox Rep. 2008;13:263–270. doi: 10.1179/135100008X308975. [DOI] [PubMed] [Google Scholar]
- 52.Boellmann F, Guettouche T, Guo Y, Fenna M, Mnayer L, Voellmy R. DAXX interacts with heat shock factor 1 during stress activation and enhances its transcriptional activity. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4100–4105. doi: 10.1073/pnas.0304768101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jacobs AT, Marnett LJ. Heat shock factor 1 attenuates 4-Hydroxynonenal-mediated apoptosis: critical role for heat shock protein 70 induction and stabilization of Bcl-XL. J. Biol. Chem. 2007;282:33412–33420. doi: 10.1074/jbc.M706799200. [DOI] [PubMed] [Google Scholar]
- 54.Jacobs AT, Marnett LJ. HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins. J. Biol. Chem. 2009;284:9176–9183. doi: 10.1074/jbc.M808656200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat. Rev. Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 56.Wang XW. Role of p53 and apoptosis in carcinogenesis. Anticancer Res. 1999;19:4759–4771. [PubMed] [Google Scholar]
- 57.Sharma A, Sharma R, Chaudhary P, Vatsyayan R, Pearce V, Jeyabal PV, Zimniak P, Awasthi S, Awasthi YC. 4-Hydroxynonenal induces p53-mediated apoptosis in retinal pigment epithelial cells. Arch. Biochem. Biophys. 2008;480:85–94. doi: 10.1016/j.abb.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Y, Sharma R, Zimniak P, Awasthi YC. Role of alpha class glutathione S-transferases as antioxidant enzymes in rodent tissues. Toxicol. Appl. Pharmacol. 2002;182:105–115. doi: 10.1006/taap.2002.9450. [DOI] [PubMed] [Google Scholar]
- 59.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 60.Gilot D, Loyer P, Corlu A, Glaise D, Lagadic-Gossmann D, Atfi A, Morel F, Ichijo H, Guguen-Guillouzo C. Liver protection from apoptosis requires both blockage of initiator caspase activities and inhibition of ASK1/JNK pathway via glutathione S-transferase regulation. J. Biol. Chem. 2002;277:49220–49229. doi: 10.1074/jbc.M207325200. [DOI] [PubMed] [Google Scholar]
- 61.Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z. Regulation of JNK signaling by GSTp. EMBO J. 1999;18:1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Romero L, Andrews K, Ng L, O’Rourke K, Maslen A, Kirby G. Human GSTA1-1 reduces c-Jun N-terminal kinase signalling and apoptosis in Caco-2 cells. Biochem J. 2006;400:135–141. doi: 10.1042/BJ20060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singh SP, Niemczyk M, Saini D, Sadovov V, Zimniak L, Zimniak P. Disruption of the mGsta4 gene increases life span of C57BL mice. J. Gerontol. A Biol. Sci. Med. Sci. 2010;65:14–23. doi: 10.1093/gerona/glp165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim KS, Hwang HA, Chae SK, Ha H, Kwon KS. Upregulation of Daxx mediates apoptosis in response to oxidative stress. J. Cell Biochem. 2005;96:330–338. doi: 10.1002/jcb.20545. [DOI] [PubMed] [Google Scholar]
- 65.Boehrer S, Nowak D, Hochmuth S, Kim SZ, Trepohl B, Afkir A, Hoelzer D, Mitrou PS, Weidmann E, Chow KU. Daxx overexpression in T-lymphoblastic Jurkat cells enhances caspase-dependent death receptor- and drug-induced apoptosis in distinct ways. Cell Signalling. 2005;17:581–595. doi: 10.1016/j.cellsig.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 66.Patrick B, Li J, Jeyabal PV, Reddy PM, Yang Y, Sharma R, Sinha M, Luxon B, Zimniak P, Awasthi S, Awasthi YC. Depletion of 4-hydroxynonenal in hGSTA4-transfected HLE B-3 cells results in profound changes in gene expression. Biochem. Biophys. Res. Commun. 2005;334:425–432. doi: 10.1016/j.bbrc.2005.06.099. [DOI] [PubMed] [Google Scholar]
- 67.Fazio VM, Barrera G, Martinotti S, Farace MG, Giglioni B, Frati L, Manzari V, Dianzani MU. 4-Hydroxynonenal, a product of cellular lipid peroxidation, which modulates c-myc and globin gene expression in K562 erythroleukemic cells. Cancer Res. 1992;52:4866–4871. [PubMed] [Google Scholar]
- 68.Ruef J, Rao GN, Li F, Bode C, Patterson C, Bhatnagar A, Runge MS. Induction of rat aortic smooth muscle cell growth by the lipid peroxidation product 4-hydroxy-2-nonenal. Circulation. 1998;97:1071–1078. doi: 10.1161/01.cir.97.11.1071. [DOI] [PubMed] [Google Scholar]
- 69.Chiarpotto E, Domenicotti C, Parola D, Vitali A, Nitti M, Pronzato MA, Biasi F, Cottalasso D, Marinari UM, Dragonetti A, Cesaro P, Isidoro C, Poli G. Regulation of rat hepatocyte protein kinase C beta isoenzymes by the lipid peroxidation product 4-hydroxy-2,3-nonenal: a signaling pathway to modulate vesicular transport of glycoproteins. Hepatology. 1999;29:1565–1572. doi: 10.1002/hep.510290510. [DOI] [PubMed] [Google Scholar]
- 70.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 71.Zhao T, Singhal SS, Piper JT, Cheng J, Pandya U, Clark-Wronski J, Awasthi S, Awasthi YC. The role of human glutathione S-transferases hGSTA1-1 and hGSTA2-2 in protection against oxidative stress. Arch. Biochem. Biophys. 1999;367:216–224. doi: 10.1006/abbi.1999.1277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.