

Abstract
MicroRNAs (miRNA) are small RNAs that attenuate protein expression by complementary binding to the 3′UTR of a target mRNA. Currently, very little is known about miRNA after cerebral ischemia. In particular, miR-21 is a strong antiapoptotic factor in some biological systems. We investigated the role of miR-21 after stroke in rat. We employed in situ hybridization (ISH) and laser capture microdissection (LCM) in combination with real-time RT-PCR to investigate the expression of miR-21 after stroke. ISH revealed that miR-21 expression was upregulated in neurons of the ischemic boundary zone (IBZ), and quantitative real-time RT-PCR analysis revealed that stroke increased mature miR-21 levels by ~3 fold in neurons isolated from the IBZ by LCM compared to homologous contralateral neurons 2 (n=4; P<0.05) and 7(n=3; P<0.05) days after stroke. In vitro, overexpression of miR-21 in cultured cortical neurons substantially suppressed oxygen and glucose deprivation (OGD)-induced apoptotic cell death, whereas attenuation of endogenous miR-21 by antisense inhibition exacerbated cell death after OGD. Moreover, overexpression of miR-21 in neurons significantly reduced Faslg protein levels and introduction of a miR-21 mimic into 293-HEK cells substantially reduced luciferase activity in a reporter system containing the 3′ untranslated region of Faslg. Our data indicate that overexpression of miR-21 protects against ischemic neuronal death and that downregulation of Faslg, a TNFα family member and an important cell death inducing ligand, targeted by miR-21 likely mediates the neuroprotective effect. These novel findings suggest that miR-21 may be an attractive therapeutic molecule for treament of stroke.
Introduction
Focal cerebral ischemia by middle cerebral artery occlusion (MCAo) induces apoptotic neural cell death that is primarily localized to the area immediately adjacent to the infarcted tissue, known as the ischemic boundary zone (IBZ) [1]. Up to 95% of apoptotic cells are neurons [2]. Neurons in the IBZ may be salvageable, and the viability of these cells has been correlated to positive functional outcomes [3]. Currently, the underlying molecular mechanisms that determine which neurons live or die are not entirely understood.
MicroRNAs (miRNAs) are integral to many biological processes. miRNAs inhibit translation of mRNAs by guiding an inhibitory protein complex, the RNA induced silencing complex (RISC) to the mRNA via complementarity to its 3′ untranslated region (3′UTR), thereby limiting a gene’s functionality, even though transcription of the gene may not be stopped [4]. Apoptosis is an important, highly regulated process, in which induction of miRNAs has been implicated [5–8]. In particular, miRNA-21 (miR-21) has been shown to be a strong antiapoptotic factor [5, 9–11]. It is upregulated in most solid tumors, including breast, colo-rectal and glioma [5, 10, 12]. miR-21 functions–at least in part–by targeting a host of pro-apoptotic genes [13, 14]. Its targets, however, do not all operate in the same direct pathway. Thus, at least in cancer, the antiapoptotic efficacy of miR-21 is increased by inhibiting expression of several genes that all render a common effect.
Despite its relevance in brain tumor, the role of miR-21 in other neurologic diseases has not been investigated. Specifically, its role in stroke has not been reported. In the present study, we measured miR-21 expression in the ischemic brain. Mechanistically, we examined an as yet undescribed miR-21 target, the tumor necrosis factor-α (TNF-α) family member, Fas ligand (Faslg), a transcript predicted to be a miR-21 target by TargetScan, that is involved in ischemia induced apoptosis [15]. FASLG induces cell death by binding to its receptor, FAS, leading to caspase-dependant apoptosis [16, 17]. Our results show that miR-21 is upregulated in neurons of the IBZ, and that when overexpressed in vitro, miR-21 reduces neurons’ sensitivity to apoptosis by targeting Faslg.
Results
miR-21 is expressed in neurons in the IBZ
To examine whether stroke affects miR-21 expression, we performed in situ hybridization (ISH) on brain coronal sections. miR-21 was observed in non-ischemic cells with neuronal morphology, but stronger miR-21 signals were detected in the IBZ of the cortex 2 and 7 days after stroke (Fig. 1A–F). Adjacent sections were also stained for TUNEL, and revealed that areas of high TUNEL reactivity were localized to areas with little miR-21 signal (Fig. 1G, H). Immunostaining revealed that many cells with positive miR-21 signal were also MAP2 positive, a marker of neurons (Fig. 1J), but none was positive for GFAP, a marker of astrocytes, or O4, a marker of oligodendrocyte progenitor cells (data not shown). These data suggest that miR-21 is selectively upregulated in neurons localized to the IBZ.
Fig. 1.

ISH images show abundance of miR-21 in specific areas of cortex. (A) A representative coronal brain section showing distribution of miR-21 in the non-ischemic cortex. (B), (C) Cells in the ipsilateral cortex show a strong miR-21 signal adjacent to the ischemic lesion at 2 and 7 days after stroke. White dashed lines indicate the demarcation between the ischemic core (IC) and the ischemic boundary zone (IBZ), the region directly adjacent to the lesion. (D), (E) and (F) are details marked by the boxes in (A), (B) and (C), respectively. (G), (H) An adjacent section of (C) shows TUNEL reactive cells at low and high magnification, respectively, in the IBZ. The white box in (G) indicates the detail shown in (H). (I) Quantitative comparison of miR-21, -9 and -335 levels at 2d and 7d after stroke in neurons from the ischemic boundary collected by LCM (asterisk indicates P<0.05). (J) miR-21 colocalizes with MAP2 in cortex, confirming that it is expressed in neurons.
ISH is not ideal for absolute quantification of gene expression. Therefore, to quantify dynamic changes in miR-21 in response to stroke, we used laser capture microdissection (LCM) to isolate neurons from the IBZ of the cortex. At 2 days post-MCAo, we found that there is a notable upregulation of miR-21 in ipsilateral neurons compared to those in the homologous region of the contralateral hemisphere, with an average induction of 3.5 ± 1.8 fold (n=4; P<0.05), when normalized to U6 snRNA. At 7d after stroke miR-21 expression remained robust, with an average induction of 3.2 ± 0.5 times contralateral expression (n=3; P<0.05; Fig. 1I). To determine if miR-21 was specifically upregulated, we also quantified two other miRNAs, miR-9 and miR-335. Both have had specific roles described in brain physiology [18, 19]. Neither miR-9 nor miR-335 showed significant changes at either timepoint, and neither were consistently up or downregulated after MCAo at either timepoint (n=3; P>0.2 for all genes/timepoints). Therefore, these data show that stroke specifically induces upregulation of miR-21 in neurons.
miR-21 protects cultured cortical neurons in vitro
To investigate the dynamics of miR-21 in vitro, we employed a model of ischemia in which primary cortical neurons from rat embryos were subjected to oxygen and glucose deprivation (OGD). Surprisingly, we did not observe a significant change in miR-21 expression at either 2 d (0.63 ± 0.24 compared to naïve neurons; n=3; P>0.1), or 7 d (0.78 ± 0.47; n=3; P>0.5; data not shown) after OGD. miR-21 was easily detectable in all samples, suggesting that it is highly expressed in embryonic neurons, but it is unaltered by OGD. This may be due to the fact that the neurons which we employ are immature, and miR-21 is expressed in developing brain tissue [18], or possibly that other factors that do not exist in our in vitro system, such as those secreted by glia, are required for miR-21 induction. Therefore, to test the biologic function of miR-21 in ischemic neurons, we artificially overexpressed miR-21 via a mimic, and knocked it down by antisense inhibition, a common strategy for investigating miRNA function in cell lines as well as primary cultures (e.g. [20, 21]). We then used a live/dead cell assay to determine the proportion of cells that were killed by OGD (Fig. 2A, C). 24h after OGD, cells transfected with miR-67, an inert, C. elegans-derived miRNA not expressed in mammal, were 31.7 ± 4.9% (average ± SD; n=5) dead. In contrast, antisense inhibition of endogenous miR-21 had a significantly higher average proportion of dead cells, 42.2 ± 6.8% (n=5), while fewer cells died upon overexpression of miR-21 (24.6 ± 4.7%; n=5; P<0.05, ANOVA).
Fig. 2.
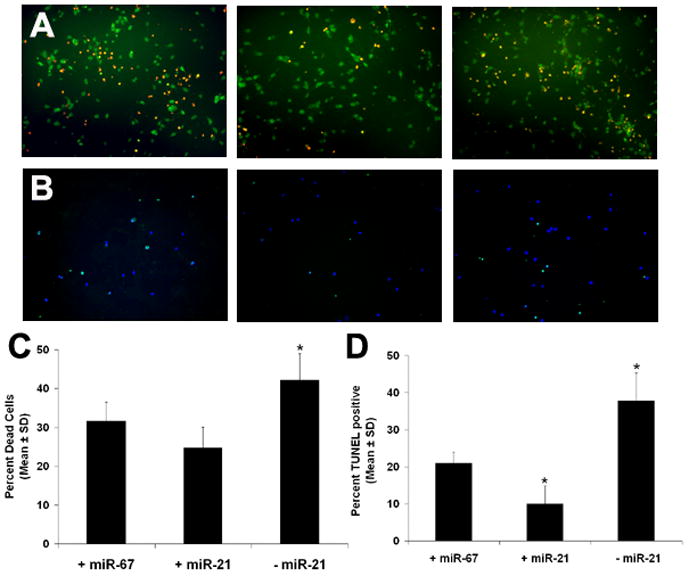
Cultured neurons that were either overexpressed with miR-21, or had endogenous miR-21 knocked down by antisense inhibition were subjected to OGD. Cel-miR-67 was used a negative control miRNA. (A) Green and red color show live and dead neurons, respectively. (B) Green color shows TUNEL reactive neurons; blue is DAPI. (C), (D) Quantitative comparison of the proportion of dead cells and TUNEL reactive cells, respectively, after overexpression and knockdown of miR-21. Asterisks indicate P<0.05 for ad hoc two group comparison t-tests.
Because of the alternate mechanisms of programmed cell death after OGD, namely, necrosis and apoptosis, we further investigated the specific way miR-21 modulates neuronal death. We used the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay to measure the proportion of cells undergoing apoptosis. Inhibition of miR-21 in neurons significantly increased TUNEL positive cells after OGD compared to control cells (37.8 ± 15.8%; n=10), whereas overexpression of miR-21 in neurons suppressed OGD induced TUNEL reactivity (9.9 ± 7.7%; n=10; Fig. 2B, D; P<0.05, ANOVA). Moreover, after overexpression of miR-21 the TUNEL reactivity was not statistically different from naïve neurons that were mock transfected, but had not been subjected to OGD (P>0.4; data not shown). Together, these data demonstrate that miR-21 plays an appreciable role in neuronal death after OGD. Specifically, it modulates apoptotic activity. The statistically insignificant data we gathered from our live/dead assay is likely due to a background level of necrosis, since our TUNEL data all have a nominally lower proportion of reactive cells. When we control for this background, the effect of miR-21 is quite strong.
miR-21 attenuates expression of FASLG protein by binding to Faslg transcript
TargetScan 5.1 and PicTar each predict binding sites for miR-21 on several proapoptotic genes. Among them is Faslg. We focused on this gene because it, and its receptor Fas, are known to induce cell death after stroke [22]. To confirm that FASLG protein is upregulated in cultured cortical neurons after OGD, and can thus contribute to apoptotic death, we performed western blot analysis. Indeed, after OGD, FASLG was induced by 1.37 ± 0.23 fold compared to naïve neurons (n=3, P<0.05; Fig. 3A). Next, we investigated via Western blot analysis whether FASLG protein could be modulated in response to overexpression of miR-21 after OGD. Compared with the control, overexpression of miR-21 knocked down FASLG protein by 32 ± 11% (n=4; P<0.05; Fig. 3B), which completely recoved it to levels we observed in naïve neurons. Furthermore, to determine if overexpression of miR-21 had any impact on Faslg gene expression, we measured the Faslg mRNA level by real-time RT-PCR under the same conditions. Overexpression of miR-21 had no significant effect on Faslg mRNA after OGD compared to miR-67 negative control (1.17 ± 0.34; n=3; P>0.4; Fig. 3B). These data suggest that FASLG expression is modulated post-transcriptionally.
Fig. 3.

(A) Western blot analysis of FASLG protein in cultured cortical neurons before and after OGD. (B) Western blot analysis of FASLG protein expression in cultured cortical neurons overexpressed with miR-21 or miR-67 (negative control) after OGD. (C) Activity of a luciferase plasmid that contains the Faslg miR-21 putative binding site after transfection with miR-67, -21 or -21 and anti-miR-21. Asterisks indicate P<0.05 for post-hoc two tailed t-tests.
Active binding of miR-21 to Faslg has not been shown. To determine if miR-21 actively binds to Faslg, we used a luciferase reporter assay containing a 60bp segment of the Faslg 3′UTR encompassing the putative miR-21 binding site (Faslg-luc; position 418–426 of 3′UTR; see methods for sequence). When Faslg-luc was cotransfected with miR-21 mimic into 293T-HEK cells, the luciferase activity was attenuated by 43 ± 13% compared to cells cotransfected with miR-67 mimic (n=4; P<0.02; Fig. 3C). However, when miR-21 and a miR-21 antisense inhibitor were simultaneously cotransfected with Faslg-luc plasmid, the attenuation completely recovered, showing only a 2 ± 4% difference from the control (n=4; P<0.8; Fig. 3C). To confirm the putative miR-21 binding site, we repeated the above experiment with a mutated Faslg 3′UTR. The sequence differed from the Faslg-luc in that the 9-mer putative binding site was scrambled (Mut-luc; see methods for sequence). After transfection with Mut-luc, there was no statistically significant difference in any experimental group (Fig. 3C), indicating that the 9-mer portion of Faslg that is complementary to miR-21 is necessary for translational inhibition. These data therefore indicate that miR-21 binds to the Faslg transcript via complementarity to its seed sequence.
Discussion
We have found, for the first time, that stroke induces upregulation of miR-21 in IBZ neurons in vivo. Furthermore, overexpression of miR-21 in cultured embryonic neurons abolished OGD-induced apoptotic cell death, which was concurrent with downregulation of protein expression of the predicted miR-21 target, Faslg. Moreover, we demonstrate that miR-21 targeted the Faslg 3′UTR. Thus, the present study indicates that miR-21 plays a critical role in reduction of ischemic cell death by targeting an important cell death inducing ligand. Faslg is a member of the TNFα family of ligands. TNFα signaling is well known to contribute to ischemic injury of neurons [23]. Cultured neurons derived from mice with mutant FASLG that cannot interact with FAS, have been shown to be more resistant to cell death from OGD [24]. Moreover, ischemia induced enhancement of FASLG expression recruits imflammatory cells to the infarcted area [22]. FASLG has therefore been proposed as a therapeutic target for acute treatment of stroke [25].
Our data raise the interesting prospect of using miR-21 for RNAi based stroke therapy. RNAi has been employed preclinically and clinically in treatment of several types of disease including viruses and cancer [26, 27], but has not been employed in stroke therapy. Specifically, Hamar et al. targeted Faslg by synthetic siRNA in ischemic kidney and showed that knockdown of Faslg protects renal cells from apoptosis, and, in fact, whole animals from death [28]. We speculate that overexpression of miR-21 may represent a way to use an endogenous gene to replicate these results in the ischemic brain.
There is strong evidence that miR-21 is protective in other organs and with different types of injury. Cheng et al. have shown that miR-21 is upregulated in response to H2O2 injury in cardiac myocytes, and that it functions to attenuate apoptosis in that cell population [29]. Additionally, cardiac fibroblasts have been shown to increase miR-21 expression specifically in the injured region after myocardial infarction, and that this increase serves a protective role [30]. Moreover, although they did not investigate the function of miR-21, Lei et al. have shown that miR-21 is globally upregulated in response to traumatic brain injury (TBI) [31].
Attenuation of apoptosis in ischemic brain injury is critically important. Currently, only one drug, tissue plasminogen activator (tPA) is available to stroke victims, and it has a limited window of about 4.5 hours from the onset of symptoms in which it can be used for treatment. Furthermore, it is not safe for all patients, leading to only about 5% of stroke sufferers actually receiving tPA therapy [32]. For these reasons, new drugs for the treatment of stroke would be very valuable to those affected. Although preliminary in nature, our data add to the growing evidence that protection of cells after injury via miR-21 induced disruption of apoptosis might be a general phenomenon. Apoptosis reaches its peak level 1 to 2 days after stroke [2]. Therefore, the therapeutic window of compounds that specifically protect against apoptosis may be longer that what is currently available. Further investigation of the effects of miR-21 modulation on neuronal cell death in vivo is warranted.
Materials and Methods
Ethics Statement
The use and care of animals employed in our stroke model, as well as our neuronal culture system, were approved by the Henry Ford Health System Institutional Animal Care and Use Committee, in accordance with all relevant laws of the United States.
Animal model
Male Wistar rats weighing 350 to 450 g were subjected to embolic MCAo, as previously described [33]. Briefly, the animals were anesthetized with 4% isoflurane during induction and thenmaintained with 2% isoflurane in a mixture of 30% O2 and 70% N2O. Body temperature was monitored and maintained at 37°C using a feedback-regulated water heating system. Under the operating microscope (Carl Zeiss, Oberkochen, Germany), the right common carotid artery (CCA), the right external carotid artery (ECA), and the internal carotid artery (ICA) were isolated via a midline incision. A modified PE-50 catheter with a 0.3 mm outer diameter was gently advanced from the ECA into the lumen of the ICA until the tip of thecatheter reached the origin of the MCA (~15 to 16 mm). A single clot (~0.8 μL) along with 2 to 3 μL of 0.9% saline was then gently injected. The catheter was withdrawn immediately after injection, and the right ECAwas ligated.
Tissue processing, ISH and immunofluorescence staining
Using 30% LNA-70% DNA 3′-DIG-labeled miRCURY probes (Exiqon, Vedbaek, Denmark), hybridization was performed according to a published protocol [34]. In brief, rats were perfused with PBS followed by 4% paraformaldehyde. Brains were removed and incubated for 24h in 4% paraformaldehyde, after which they were incubated in 0.5 M sucrose for 24h, then frozen on dry ice. Coronal sections were mounted on charged slides for ISH analysis. Sections were air dried for 1h, then digested in proteinase K (20 μg/ml) for 20min, rinsed in TBS, then fixed in 4% PFA for 10min. Sections were then washed in 0.2% glycine, rinsed and incubated for 30 min in acetylation solution (0.5% acetic anhydride and 0.1M triethanolamine in DEPC-H2O) to inactivate enzymes, then rinsed again. Tissue was blocked at room temperature for 2h in hybridization buffer (50% formamide, 25% 5× SSC, 10% 5× Denhardt’s and 15% DEPC-H2O containing 200 μg/ml yeast RNA, 500 μg/ml salmon sperm DNA and 20 mg/ml Roche blocking reagent), then hybridized overnight at 52°C with LNA probe diluted 1:600 in hybridization buffer. Hybridized miRNAs were visualized with AP. For immunostaining of ISH tissue, coronal sections were blocked in PBS containing 0.5% BSA and 0.05% Tween 20. Incubation in 1° antibody was followed by incubation in a fluorescent 2° antibody. Finally, cells were stained with DAPI diluted 1:7500 in PBS for 10 min to visualize nuclei.
LCM
Brain coronal cryosections (10μm) obtained from rat subjected to 2, 7 and 14 days of MCAo were mounted on LCM membrane slides (Leica Microsystems, Wetzlar, Germany). To identify neurons, coronal sections were immunostained with an antibody against NeuN (1:250; Chemicon, Billerica, MA) using a fast staining protocol [35]. Approximately 2,000 NeuN positive cells in the IBZ were collected by an LMD 6000 system (Leica Microsystems). The IBZ was identified by proximity to ischemic core. The same number of NeuN positive cells collected from the homologous tissue in the contralateral hemisphere was used as control. Cells were lysed in Qiazol reagent (Qiagen, Valencia, CA); total RNA was isolated immediately after cells were collected.
Isolation of total RNA and real-time RT-PCR
For miRNA analysis, cells were lysed in Qiazol reagent and total RNA was isolated using the miRNeasy Mini kit (Qiagen). miRNA was reverse transcribed with the miRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and amplified with TaqMan miRNA assay (Applied Biosystems), which is specific for mature miRNA sequences. For analysis of mRNA, RNA was reverse transcribed with M-MLV reverse transcriptase (Invitrogen), and amplified by SYBR Green reporter (Applied Biosystems) using custom primers (Invitrogen). Analysis of gene expression was carried out by the 2−ΔΔCt method [36].
Primary cortical neuron culture
Pregnant Wistar rats purchased from Charles River were sacrificed at embryonic day 17 (E17) under deep pentobarbital anesthesia. Embryos were removed and the cerebral cortex was dissected out, washed in Hank’s balanced salt solution (HBSS; Gibco, Grand Island, NY), dissociated in 0.125% trypsin, washed again, then plated onto poly-D-lysine coated culture dishes at a density of 105 cells/cm2 in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco) containing 5% fetal bovine serum (FBS) and antibiotics overnight. After 24h, cells were transferred to serum-free Neurobasal medium (Gibco) supplemented with 2% B-27 (Gibco), 500 μM L-glutamine and antibiotics. After 3d, media were changed and further supplemented with 10 nM uridine and 10 nM 5-fluoro-deoxyuridine to kill astrocytes. Cells were incubated for an additional 48h, after which they were subjected to oxygen and glucose deprivation (OGD). To induce OGD, culture medium was replaced with HBSS and cells were placed in an anaerobic chamber continuously perfused with 85%/5%/10% NO2/CO2/H2 for 3h. Cells were then returned to normal culture conditions for 24h prior to analysis.
Primary cortical neuron transfection
Prior to plating, approximately 10x106 neurons were transfected with 50pmol of a miRIDIAN miR-21 mimic, miR-21 inhibitor, or a negative control miRNA mimic (based on cel-miR-67 sequence, which lacks homology in mammal; Dharmacon, Lafayette, CO) via electroporation using the Rat Neuron Nucleofector kit (Lonza, Basel, Switzerland) according to the manufacturer’s protocol.
Western blotting
Cells were lysed in RIPA buffer containing protease inhibitors, and lysate was sonicated then centrifuged for 10 min at >2 × 104g to remove cell debris. Protein concentrations were determined via BCA assay (Thermo Scientific, Waltham, MA). Equal amounts of protein were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were probed with 1° antibodies against Faslg (1:500; Abcam, Cambridge, MA), and β-Actin (1:10000 Abcam) as a control, followed by 2° antibodies conjugated to horseradish peroxidase. Proteins were visualized by enhanced chemiluminescence. Relative intensities were determined by use of Photoshop 6.0 software (Adobe, San Jose, CA).
Live/dead assay and TUNEL staining
Live/dead assay (Invitrogen) was performed on neurons grown on 3.5 cm culture dishes according to manufacturer’s protocol. TUNEL staining was performed on cultured cortical neurons grown on 3.5 cm culture dishes using fluorescein conjugated ApopTag kit (Chemicon) according to the manufacturer’s instructions. 4 experimental groups were used for analysis. Naïve neurons were used to determine baseline TUNEL reactivity. The remaining groups were cells transfected with miRIDIAN miR-21 mimic or a miR-21 inhibitor; neurons transfected with miR-67 were used as control.
Target prediction
Several freewares available on the internet are commonly used to search for potential miRNA-mRNA binding. We searched three such softwares, PicTar (pictar.mdc-berlin.de), TargetScan 5.1 (www.targetscan.org), and miRanda (www.microrna.org). Two, TargetScan and PicTar identified Faslg as having a relatively likely putative binding site for miR-21.
Luciferase activity assay
A pMiR luciferase reporter assay with Faslg 3′UTR (position 392-451; sequence [putative binding site underlined]: GGUGAGAAAGGAUGCUAGGUUUCAUGGAUAAGCUAGAGACUGAAAAAAGCCAGUGUCCCA; mutant sequence: GGUGAGAAAGGAUGCUAGGUUUCAUGUAGAGAUACGAGACUGAAAAAAGC CAGUGUCCCA) was used for analysis (Signosis, Sunnyvale, CA). 293-HEK cells were transfected via electroporation with luciferase reporter and β-galactosidase control vector. HEK cells were cotransfected with miR-67 negative control miRNA, miR-21 mimic, or both the miR-21 mimic and inhibitor. 3 × 104 cells were plated per well on a 96 well plate for 24h, and then analyzed on a Fusion plate reader (PerkinElmer, Weltham, MA) with Luciferase Reporter Assay (Promega, Madison, WI) according to the manufacturer’s protocol.
Statistics
The data are presented as mean ± s.d. One way ANOVA was used for multiple group experiments. Ad hoc two sample t-test was used for two-group comparisons. A value of P<0.05 was taken as significant.
Acknowledgments
This work was supported by National Institute of Neurologic Diseases and Stroke grants PO1 NS42345 and RO1 HL64766 and American Heart Association fellowship 10PRE2730004.
References
- 1.Mattson MP, Culmsee C, Yu ZF. Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res. 2000;301:173–187. doi: 10.1007/s004419900154. [DOI] [PubMed] [Google Scholar]
- 2.Chopp M, Li Y. Apoptosis in focal cerebral ischemia. Acta Neurochir Suppl. 1996;66:21–26. doi: 10.1007/978-3-7091-9465-2_4. [DOI] [PubMed] [Google Scholar]
- 3.Lim SH, Lee JS, Lee JI, Im S, Ko YJ, Kim HW. The quantitative assessment of functional impairment and its correlation to infarct volume in rats with transient middle cerebral artery occlusion. Brain Res. 2008;1230:303–309. doi: 10.1016/j.brainres.2008.07.002. S0006-8993(08)01655-7 [pii] [DOI] [PubMed] [Google Scholar]
- 4.Pillai RS. MicroRNA function: multiple mechanisms for a tiny RNA? RNA. 2005;11:1753–1761. doi: 10.1261/rna.2248605. 11/12/1753 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. 65/14/6029 [pii] [DOI] [PubMed] [Google Scholar]
- 6.Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM. MicroRNA expression and function in cancer. Trends Mol Med. 2006;12:580–587. doi: 10.1016/j.molmed.2006.10.006. S1471-4914(06)00242-5 [pii] [DOI] [PubMed] [Google Scholar]
- 7.Jovanovic M, Hengartner MO. miRNAs and apoptosis: RNAs to die for. Oncogene. 2006;25:6176–6187. doi: 10.1038/sj.onc.1209912. 1209912 [pii] [DOI] [PubMed] [Google Scholar]
- 8.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–222. doi: 10.1016/j.cell.2008.04.001. S0092-8674(08)00449-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Liu W, Chao T, Zhang Y, Yan X, Gong Y, Qiang B, Yuan J, Sun M, Peng X. MicroRNA-21 down-regulates the expression of tumor suppressor PDCD4 in human glioblastoma cell T98G. Cancer Lett. 2008;272:197–205. doi: 10.1016/j.canlet.2008.06.034. S0304-3835(08)00526-0 [pii] [DOI] [PubMed] [Google Scholar]
- 10.Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 2008;283:1026–1033. doi: 10.1074/jbc.M707224200. M707224200 [pii] [DOI] [PubMed] [Google Scholar]
- 11.Papagiannakopoulos T, Shapiro A, Kosik KS. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008;68:8164–8172. doi: 10.1158/0008-5472.CAN-08-1305. 68/19/8164 [pii] [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z, Li Z, Gao C, Chen P, Chen J, Liu W, Xiao S, Lu H. miR-21 plays a pivotal role in gastric cancer pathogenesis and progression. Lab Invest. 2008;88:1358–1366. doi: 10.1038/labinvest.2008.94. labinvest200894 [pii] [DOI] [PubMed] [Google Scholar]
- 13.Kim YJ, Hwang SJ, Bae YC, Jung JS. MiR-21 regulates adipogenic differentiation through the modulation of TGF-beta signaling in mesenchymal stem cells derived from human adipose tissue. Stem Cells. 2009;27:3093–3102. doi: 10.1002/stem.235. [DOI] [PubMed] [Google Scholar]
- 14.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. S0016-5085(07)01002-5 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sairanen T, Karjalainen-Lindsberg ML, Paetau A, Ijas P, Lindsberg PJ. Apoptosis dominant in the periinfarct area of human ischaemic stroke--a possible target of antiapoptotic treatments. Brain. 2006;129:189–199. doi: 10.1093/brain/awh645. awh645 [pii] [DOI] [PubMed] [Google Scholar]
- 16.Becher B, Barker PA, Owens T, Antel JP. CD95-CD95L: can the brain learn from the immune system? Trends Neurosci. 1998;21:114–117. doi: 10.1016/s0166-2236(97)01180-6. S0166-2236(97)01180-6 [pii] [DOI] [PubMed] [Google Scholar]
- 17.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 18.Sathyan P, Golden HB, Miranda RC. Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J Neurosci. 2007;27:8546–8557. doi: 10.1523/JNEUROSCI.1269-07.2007. 27/32/8546 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coolen M, Bally-Cuif L. MicroRNAs in brain development and physiology. Curr Opin Neurobiol. 2009;19:461–470. doi: 10.1016/j.conb.2009.09.006. S0959-4388(09)00128-7 [pii] [DOI] [PubMed] [Google Scholar]
- 20.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439:283–289. doi: 10.1038/nature04367. nature04367 [pii] [DOI] [PubMed] [Google Scholar]
- 22.Rosenbaum DM, Gupta G, D’Amore J, Singh M, Weidenheim K, Zhang H, Kessler JA. Fas (CD95/APO-1) plays a role in the pathophysiology of focal cerebral ischemia. J Neurosci Res. 2000;61:686–692. doi: 10.1002/1097-4547(20000915). 61:6<686::AID-JNR12>3.0.CO;2-7 [pii] [DOI] [PubMed] [Google Scholar]
- 23.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 24.Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J, Krammer PH. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 2001;8:679–686. doi: 10.1038/sj.cdd.4400882. [DOI] [PubMed] [Google Scholar]
- 25.French LE, Tschopp J. Protein-based therapeutic approaches targeting death receptors. Cell Death Differ. 2003;10:117–123. doi: 10.1038/sj.cdd.4401185. 4401185 [pii] [DOI] [PubMed] [Google Scholar]
- 26.Huang DD. The potential of RNA interference-based therapies for viral infections. Curr HIV/AIDS Rep. 2008;5:33–39. doi: 10.1007/s11904-008-0006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pirollo KF, Chang EH. Targeted delivery of small interfering RNA: approaching effective cancer therapies. Cancer Res. 2008;68:1247–1250. doi: 10.1158/0008-5472.CAN-07-5810. 68/5/1247 [pii] [DOI] [PubMed] [Google Scholar]
- 28.Hamar P, Song E, Kokeny G, Chen A, Ouyang N, Lieberman J. Small interfering RNA targeting Fas protects mice against renal ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2004;101:14883–14888. doi: 10.1073/pnas.0406421101. 0406421101 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C. MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol. 2009;47:5–14. doi: 10.1016/j.yjmcc.2009.01.008. S0022-2828(09)00017-0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong S, Cheng Y, Yang J, Li J, Liu X, Wang X, Wang D, Krall TJ, Delphin ES, Zhang C. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem. 2009;284:29514–29525. doi: 10.1074/jbc.M109.027896. M109.027896 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lei P, Li Y, Chen X, Yang S, Zhang J. Microarray based analysis of microRNA expression in rat cerebral cortex after traumatic brain injury. Brain Res. 2009;1284:191–201. doi: 10.1016/j.brainres.2009.05.074. S0006-8993(09)01094-4 [pii] [DOI] [PubMed] [Google Scholar]
- 32.Fields JD, Lindsay K, Liu KC, Nesbit GM, Lutsep HL. Mechanical thrombectomy for the treatment of acute ischemic stroke. Expert Rev Cardiovasc Ther. 2010;8:581–592. doi: 10.1586/erc.10.8. [DOI] [PubMed] [Google Scholar]
- 33.Zhang RL, Chopp M, Zhang ZG, Jiang Q, Ewing JR. A rat model of focal embolic cerebral ischemia. Brain Res. 1997;766:83–92. doi: 10.1016/s0006-8993(97)00580-5. S0006-8993(97)00580-5 [pii] [DOI] [PubMed] [Google Scholar]
- 34.Pena JT, Sohn-Lee C, Rouhanifard SH, Ludwig J, Hafner M, Mihailovic A, Lim C, Holoch D, Berninger P, Zavolan M, et al. miRNA in situ hybridization in formaldehyde and EDC-fixed tissues. Nat Methods. 2009;6:139–141. doi: 10.1038/nmeth.1294. nmeth.1294 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu XS, Chopp M, Zhang RL, Hozeska-Solgot A, Gregg SC, Buller B, Lu M, Zhang ZG. Angiopoietin 2 mediates the differentiation and migration of neural progenitor cells in the subventricular zone after stroke. J Biol Chem. 2009;284:22680–22689. doi: 10.1074/jbc.M109.006551. M109.006551 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. S1046-2023(01)91262-9 [pii] [DOI] [PubMed] [Google Scholar]