

Abstract
Inactivators of plasminogen activator inhibitor-1 (PAI-1) have been identified as possible treatments for a range of conditions, including atherosclerosis, venous thrombosis, and obesity. We describe the synthesis and inhibitory activity of a novel series of compounds based on bis-arylsulfonamide and aryl sulfonimide motifs that show potent and specific activity towards PAI-1. Inhibitors containing short linking units between the sulfonyl moieties and a 3,4-dihydroxy aryl substitution pattern showed the most potent inhibitory activity, and retained high specificity for PAI-1 over the structurally-related serpin anti-thrombin III (ATIII).
Keywords: PAI-1 inhibitors, bis-arylsulfonamide, aryl sulfonimide
Plasminogen activator inhibitor-1 (PAI-1) is member of the serpin superfamily of protease inhibitors and is the primary inhibitor of urokinase type plasminogen activator (uPA) and tissue type plasminogen activator (tPA).1 At normal physiologic levels PAI-1 plays a significant role in multiple processes, including fibrinolysis, angiogenesis, cell migration, and wound healing.2-6 Pathologic levels of PAI-1 have been connected with obesity and metabolic syndrome, tumor metastasis, and vascular diseases such as venous thrombosis and atherosclerosis, making it an attractive pharmacological target.7-13 However, PAI-1 has proven to be a challenging target for drug design as it is present in multiple conformational forms and has a flexible reactive center loop. Despite the challenges inherent in the development of small-molecule PAI-1 inactivators, several have been reported recently, although each has drawbacks that have impeded further progress in their development.14-22 The most studied of these inhibitors, tiplaxtinin,3,19,23-26 has a reported IC50 in the low micromolar range although PAI-1 inhibitors with IC50 values as low as 0.2 μM have been reported.14,20
Here we report the development of a novel class of PAI-1 inhibitors based on an aryl sulfonamide or aryl sulfonimide motif that shows up to 30-fold more potent inhibitory activity toward PAI-1 than that of tiplaxtinin. Based on insights gained from a screen for anti-PAI-1 activity of a library of structurally diverse compounds,27 we developed a set of compounds designed to probe structural requirements for PAI-1 inactivation. Our general design strategy was to synthesize compounds containing two polyphenolic moieties separated by linking units of various length and composition. Sulfonamides were chosen as a basis for the linking unit due to the moiety's widespread use in pharmaceutical design and the flexible synthetic access to structural diversity it would provide. As we were also interested in the level of specificity for the inhibition of PAI-1, we assayed the set of compounds for activity against anti-thrombin III (ATIII), a structurally similar mammalian serpin.
Bis-arylsulfonamides 3a-d were prepared as shown in Scheme 1. Diamines (1a-d) were treated with 3,4-dimethoxybenzenesulfonyl chloride in the presence of triethylamine to form the bis-arylsulfonamides 2a-d. Exposure of the bis-sulfonamides to boron tribromide in methylene chloride achieved deprotection of the aryl methoxy groups, providing the fully deprotected tetraphenols 3a-d. Bis-sulfonamide 4 was similarly produced by this route, using piperazine as the initial diamine. Variously substituted bis-arylsulfonamides 5-7 were synthesized by analogous routes using the appropriate sulfonyl chlorides. Unsubstituted bis-benzenesulfonamide 8 was synthesized directly by the reaction of benzenesulfonyl chloride and N,N′-diethylethylenediamine in the presence of triethylamine.
Scheme 1.

Preparation of bis-arylsulfonamides: (a) 3,4-dimethoxybenzenesulfonyl chloride, triethylamine, ethyl acetate, 35-100%; (b) 1M BBr3 in CH2Cl2, CH2Cl2, 0 °C to rt, 36-86%.
The synthesis of bis-3,5-difluoro-4-hydroxybenzenesulfonamide 13 proceeded as shown in Scheme 2. A solution of 2,6-difluorophenol in dimethylformamide was treated with iodomethane and anhydrous potassium carbonate to provide 2,6-difluoroanisole (10),28 which was then exposed to chlorosulfonic acid29 to provide the aryl sulfonyl chloride 11. N,N′-Diethylethylenediamine was added dropwise to a solution of 11 in pyridine to give the bis-sulfonamide 12, which was demethylated using BBr3 to provide fully deprotected bis-phenol 13.
Scheme 2.

Preparation of bis-3,5-difluoro-4-hydroxybenzenesulfonamide 13: (a) K2CO3, CH3I, DMF, 50 °C, 65%; (b) chlorosulfonic acid, CH2Cl2, reflux, 2 hr, 85%; (c) N,N′-diethylethylenediamine, Et3N, EtOAc, 40%; (d) 1M BBr3 in CH2Cl2, CH2Cl2, 0 °C to rt, 67%.
Difficulties were encountered in forming aryl sulfonimides 17a-d directly from primary amines 14a-d; instead we installed each sulfonyl group in a stepwise fashion (Scheme 3). 3,4-Dimethoxybenzenesulfonyl chloride was exposed to a solution of the appropriate primary amine 14 and triethylamine in ethyl acetate to provide the respective sulfonamide 15. Sulfonamide 15 was treated with sodium hydride and 3,4-dimethoxybenzenesulfonyl chloride to provide the aryl sulfonimide 16, which was then deprotected with BBr3 as before to provide the PAI-1 inhibitors 17a-d.
Scheme 3.

Preparation of aryl sulfonimides: (a) 3,4-dimethoxybenzenesulfonyl chloride, Et3N, EtOAc, 63-100%; (b) NaH (60% dispersion in oil), 3,4-dimethoxybenzenesulfonyl chloride, DMF, 25-64%; (c) 1M BBr3 in CH2Cl2, CH2Cl2, 0 °C to rt, 48-65%.
Compounds 2b, 3a-d, 4-8, 13, and 17-d were assayed in vitro against PAI-1 and anti-thrombin III (ATIII), a related mammalian serpin (Table 1).30 Initially we varied the length of the linking unit in order to determine the distance between our sulfonamide units that would lead to maximal potency against PAI-1. We found that bis-sulfonamides containing longer linker chains (3c and 3d) were less active than 3a, an otherwise equivalent compound containing a single ethylene linking unit (IC50 = 518 μM, Table 1).31 Replacement of the acidic hydrogens within the sulfonamide units of 3a with ethyl groups resulted in an increase in inhibitor potency of about 55-fold (3b, IC50 = 9.32 μM) as compared to the non-alkylated version. Significantly, the anti-PAI-1 activity of 3b equaled that of tiplaxtinin (IC50 = 9.7 μM).19 Inclusion of the linking unit within a more conformationally-constrained 1,4-piperazinyl ring, as in 4, resulted in a compound that lacked significant inhibitory activity towards PAI-1, indicating that the relative orientation of the aromatic rings that leads to the greatest degree of bioactivity are not available to this molecule.
Table 1.
Inhibitory activity of bis-arylsulfonamides and aryl sulfonimides against PAI-1 and ATIII.
| Cmpd | Structure | PAI-1 Inhibition IC50, μMa | ATIII Inhibition IC50, μMa |
|---|---|---|---|
| 2b | >300 | >300 | |
| 3a | 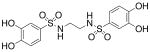 |
518 (±64) |
>3000b |
| 3b | 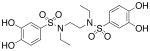 |
9.32 (±0.36) |
>1000b |
| 3c | >3000 | >3000 | |
| 3d | 1384 (±112) |
>3000 | |
| 4 | 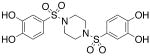 |
>300c | >300 |
| 5 | 343 (±14) |
>1000 | |
| 6 | 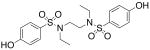 |
>300 | >300 |
| 7 | 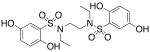 |
>300c | >300 |
| 8 | 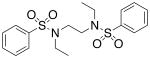 |
>300 | >300 |
| 13 | 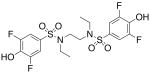 |
128 (±18) |
>3000 |
| 17a | 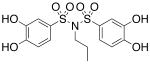 |
2.67 (±0.27) |
>1000 |
| 17b | 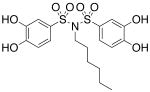 |
0.284 (±0.016) |
>300b |
| 17c | 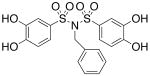 |
4.82 (±0.32) |
>300 |
| 17d | 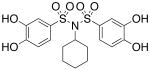 |
0.594 (±0.086) |
>300 |
Each value is the average of at least 3 determinations; standard error is given in parentheses.
<30% of ATIII was inhibited at the highest compound concentration used.
<30% of PAI-1 was inhibited at the highest compound concentration used.
We then investigated the importance of the substitution pattern of the aromatic ring. Given the high potency conferred by the N,N′-ethylethylenediamine linking unit of 3b, we maintained that structural motif while varying the substitution pattern of the aromatic rings. Hydroxyl groups at both the 3- and 4- positions are critical for maximal activity as the compound containing this substitution, 3b, was far more inactivating towards PAI-1 (IC50 = 9.32 μM) than those containing a single hydroxyl group at either the 3-position (5, IC50 = 343 μM) or 4-position (6, IC50 > 300 μM). Flanking the 4-OH moiety with fluorine atoms at the 3- and 5- positions, as in 13, restored some of the activity lost with the absence of the 3-OH group (IC50 = 128 μM), but still resulted in an inhibitor roughly 15-fold less potent than the 3,4-dihydroxy version. Sulfonamide 7, containing a hydroxyl group at each of the 2- and 5- positions, lacked significant potency towards PAI-1, supporting the importance of the 3,4-substitution pattern. Additionally, compounds that lacked hydroxyl groups entirely, such as 2b and 8, displayed no inhibitory activity against PAI-1 at the tested range of concentrations (>300 μM).
In order to confirm that two aryl sulfonyl groups are indeed required for anti-PAI-1 activity, three compounds containing a single such group (18-20, Figure 1) were synthesized by routes analogous to those described in Scheme 1. These compounds showed a complete lack of activity towards PAI-1 at the range of concentrations tested (>3000 μM), confirming that both arylsulfonamide groups are required for inhibitory activity.
Figure 1.

Tested compounds containing a single sulfonamide unit.
Interested in exploring the inhibitory activity of bisarylsulfonyl compounds with even shorter linking units, we synthesized a set of N-substituted aryl sulfonimides, 17a-d. The sulfonimides showed improved potency towards PAI-1, performing 2-fold to 30-fold better in vitro compared to the best of the bis-sulfonamides (cf. Table 1). Inhibitors containing n-hexyl (17b, IC50 = 0.284 μM) and cyclohexyl (17d, IC50 = 0.594 μM) substituents displayed somewhat better activity against PAI-1 compared to n-propyl (17a, IC50 = 2.67 μM) and benzyl (17c, IC50 = 4.82 μM) moieties. In addition, the most potent of the sulfonimides (17b and 17d) proved to be 15- to 34-fold more potent than tiplaxtinin (IC50 = 9.7 μM).
Specificity for the target protein over structurally-related proteins is an important consideration in the drug development process, so to investigate the specificity of these new classes of PAI-1 inhibitors we tested 2b, 3a-d, 4-8, 13, 17a-d, and 18-20 against anti-thrombin III (ATIII), a mammalian serpin that is closely related to PAI-1. None of the compounds displayed significant activity against ATIII, although three compounds (3a, 3b, and 17b) showed <30% inhibition of ATIII at the highest concentration of inhibitor. This lack of inhibitory activity towards ATIII indicates that an eventual drug candidate from these classes of inhibitors may display a high level of specificity for PAI-1 over related mammalian serpins.
In conclusion, we have described the design of a series of novel bis-arylsulfonamides and aryl sulfonimides that display highly potent and selective inhibitory activity towards PAI-1. We have determined the minimal structural requirements for inhibition of PAI-1 by these classes of compounds, as shorter linking units and 3,4-dihydroxy substitution of flanking aromatic rings are essential for maximal inhibitor potency. We will report further work based on this molecular scaffold in the near future.
Supplementary Material
Acknowledgments
The authors wish to thank Robert Cody for his assistance with analysis by Direct Analysis in Real Time (DART) mass spectroscopy. This work was supported by NIH Grants HL55374, HL54710, and HL089407 to DAL.
Footnotes
Supplementary material containing synthetic details, spectroscopic characterization of novel compounds, and assay conditions for this paper is available to all readers.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Yepes M, Loskutoff DJ, Lawrence DA. In: Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Coleman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ, editors. Lippincott Williams & Wilkins; Baltimore: 2006. pp. 365–380. [Google Scholar]
- 2.McMahon GA, Petitclerc E, Stefansson S, Smith E, Wong MK, Westrick RJ, Ginsburg D, Brooks PC, Lawrence DA. J Biol Chem. 2001;276:33964. doi: 10.1074/jbc.M105980200. [DOI] [PubMed] [Google Scholar]
- 3.Leik CE, Su EJ, Nambi P, Crandall DL, Lawrence DA. J Thromb Haemost. 2006;4:2710. doi: 10.1111/j.1538-7836.2006.02244.x. [DOI] [PubMed] [Google Scholar]
- 4.Maquerlot F, Galiacy S, Malo M, Guignabert C, Lawrence DA, d'Ortho MP, Barlovatz-Meimon G. Am J Pathol. 2006;169:1624. doi: 10.2353/ajpath.2006.051053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stefansson S, Lawrence DA. Nature. 1996;383:441. doi: 10.1038/383441a0. [DOI] [PubMed] [Google Scholar]
- 6.Cao C, Lawrence DA, Li Y, Von Arnim CA, Herz J, Su EJ, Makarova A, Hyman BT, Strickland DK, Zhang L. EMBO J. 2006;25:1860. doi: 10.1038/sj.emboj.7601082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andreasen PA. Curr Drug Targets. 2007;8:1030. doi: 10.2174/138945007781662346. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y, Border WA, Yu L, Zhang J, Lawrence DA, Noble NA. J Am Soc Nephrol. 2008;19:329. doi: 10.1681/ASN.2007040510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinsky DJ, Liao H, Lawson CA, Yan SF, Chen J, Carmeliet P, Loskutoff DJ, Stern DM. J Clin Invest. 1998;102:919. doi: 10.1172/JCI307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schafer K, Fujisawa K, Konstantinides S, Loskutoff DJ. FASEB J. 2001;15:1840. doi: 10.1096/fj.00-0750fje. [DOI] [PubMed] [Google Scholar]
- 11.Shah C, Yang G, Lee I, Bielawski J, Hannun YA, Samad F. J Biol Chem. 2008;283:13538. doi: 10.1074/jbc.M709950200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lijnen HR. Thromb Res. 2009;123 4:S46. doi: 10.1016/S0049-3848(09)70143-4. [DOI] [PubMed] [Google Scholar]
- 13.Sobel BE, Taatjes DJ, Schneider DJ. Arterioscler Thromb Vasc Biol. 2003;23:1979. doi: 10.1161/01.ATV.0000091250.53231.4D. [DOI] [PubMed] [Google Scholar]
- 14.Folkes A, Roe MB, Sohal S, Golec J, Faint R, Brooks T, Charlton P. Bioorg Med Chem Lett. 2001;11:2589. doi: 10.1016/s0960-894x(01)00508-x. [DOI] [PubMed] [Google Scholar]
- 15.Gils A, Stassen JM, Nar H, Kley JT, Wienen W, Ries UJ, Declerck PJ. Thromb Haemost. 2002;88:137. [PubMed] [Google Scholar]
- 16.Einholm AP, Pedersen KE, Wind T, Kulig P, Overgaard MT, Jensen JK, Bodker JS, Christensen A, Charlton P, Andreasen PA. Biochem J. 2003;373:723. doi: 10.1042/BJ20021880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crandall DL, Elokdah H, Di L, Hennan JK, Gorlatova NV, Lawrence DA. J Thromb Haemost. 2004;2:1422. doi: 10.1111/j.1538-7836.2004.00829.x. [DOI] [PubMed] [Google Scholar]
- 18.Liang A, Wu F, Tran K, Jones SW, Deng G, Ye B, Zhao Z, Snider RM, Dole WP, Morser J, Wu Q. Thromb Res. 2005;115:341. doi: 10.1016/j.thromres.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 19.Gorlatova NV, Cale JM, Elokdah H, Li D, Fan K, Warnock M, Crandall DL, Lawrence DA. J Biol Chem. 2007;282:9288. doi: 10.1074/jbc.M611642200. [DOI] [PubMed] [Google Scholar]
- 20.Gardell SJ, Krueger JA, Antrilli TA, Elokdah H, Mayer S, Orcutt SJ, Crandall DL, Vlasuk GP. Mol Pharmacol. 2007;72:897. doi: 10.1124/mol.107.037010. [DOI] [PubMed] [Google Scholar]
- 21.Rupin A, Gaertner R, Mennecier P, Richard I, Benoist A, De Nanteuil G, Verbeuren TJ. Thromb Res. 2008;122:265. doi: 10.1016/j.thromres.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Izuhara Y, Takahashi S, Nangaku M, Takizawa S, Ishida H, Kurokawa K, van Ypersele de Strihou C, Hirayama N, Miyata T. Arterioscler Thromb Vasc Biol. 2008;28:672. doi: 10.1161/ATVBAHA.107.157479. [DOI] [PubMed] [Google Scholar]
- 23.Elokdah H, Abou-Gharbia M, Hennan JK, McFarlane G, Mugford CP, Krishnamurthy G, Crandall DL. J Med Chem. 2004;47:3491. doi: 10.1021/jm049766q. [DOI] [PubMed] [Google Scholar]
- 24.Hennan JK, Elokdah H, Leal M, Ji A, Friedrichs GS, Morgan GA, Swillo RE, Antrilli TM, Hreha A, Crandall DL. J Pharmacol Exp Ther. 2005;314:710. doi: 10.1124/jpet.105.084129. [DOI] [PubMed] [Google Scholar]
- 25.Crandall DL, Quinet EM, El Ayachi S, Hreha AL, Leik CE, Savio DA, Juhan-Vague I, Alessi MC. Arterioscler Thromb Vasc Biol. 2006;26:2209. doi: 10.1161/01.ATV.0000235605.51400.9d. [DOI] [PubMed] [Google Scholar]
- 26.Baxi S, Crandall DL, Meier TR, Wrobleski S, Hawley A, Farris D, Elokdah H, Sigler R, Schaub RG, Wakefield T, Myers D. Thromb Haemost. 2008;99:749. doi: 10.1160/TH07-11-0669. [DOI] [PubMed] [Google Scholar]
- 27.Cale JM, Li SH, Warnock M, Su EJ, North PR, Sanders KL, Puscau MM, Emal CD, Lawrence DL. J Biol Chem. doi: 10.1074/jbc.M109.067967. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Zheng J, Hu J. J Org Chem. 2006;71:9845. doi: 10.1021/jo061799l. [DOI] [PubMed] [Google Scholar]
- 29.Adapted from the procedure of: Sato M, Kawashima Y, Goto J, Yamane Y, Chiba Y, Jinno S, Satake M, Iwata C. Eur J Med Chem. 1995;30:403–414.
- 30.See supplementary information for detailed assay conditions and the synthesis and spectroscopic characterization of tested compounds.
- 31.Attempts to synthesize analogous compounds containing a hydrazine-based linking unit were unsuccessful, possibly due to unintended side-reactions similar to those described by Shyam K, Cosby LA, Sartorelli AC. J Med Chem. 1985;28:525. doi: 10.1021/jm00382a027.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.