

Abstract
Background
Lower levels of myocardial Akt activity in males are associated with a higher incidence of heart failure and worsened cardiac function after ischemia/reperfusion (I/R). While Akt activation by estrogen provides cardioprotection in females, no information exists regarding the effect of testosterone on the myocardial Akt pathway following I/R. We hypothesized that following I/R: 1) endogenous testosterone will decrease myocardial Akt activation in male hearts; 2) endogenous testosterone will mediate downstream signals of Akt, including Bad, Bcl-2 and FOXO3a; 3) administration of exogenous testosterone will recapitulate negative effects on the Akt pathway in castrated male hearts.
Methods and Results
Rat hearts from age-matched adult males, females, castrated males, males with androgen receptor blocker-flutamide, castrated males with chronic 5α-dihydrotestosterone (DHT) implantation or acute testosterone infusion (ATI) (n=9/group) were subjected to I/R (Langendorff). Castration or flutamide treatment significantly upregulated myocardial Akt activation, increased downstream apoptosis-regulatory molecules: p-Bad, Bcl-2, p-FOXO3a, but reduced Fas-L, consistent with decreased myocardial injury in male hearts following I/R. ATI administration, but not chronic DHT, reversed these effects on Akt signaling associated with further exacerbated cardiac dysfunction in castrated males. Notably, lower levels of MnSOD were observed in male hearts, and castration or flutamide treatment restored myocardial MnSOD expression to the levels of females in male hearts after I/R.
Conclusion
Our study represents the initial evidence of testosterone-induced downregulation of the Akt pathway in male hearts following I/R, thereby mediating cardiac injury through decreased p-Bad, reduced ratio of Bcl-2/Bax in the cytoplasm, and increased FOXO3a in the nucleus.
Keywords: sex, myocardium, ischemia, signal transduction, apoptosis
INTRODUCTION
Ischemic heart disease is a leading killer of both men and women. Notably, sex differences in myocardial ischemia/reperfusion (I/R) injury have been well established (1, 2). Males are much more susceptible to I/R-induced cardiac injury with worsened myocardial function compared to females following I/R (3, 4). The increased I/R injury in males has been attributed primarily to a lack of estrogen-mediated protection. However, testosterone may play a negative role in the myocardial response to acute I/R in males. Indeed, our previous studies have clearly indicated that testosterone exacerbates I/R-induced cardiac dysfunction, and enhances myocardial inflammation and apoptotic signaling in male hearts (4). However, the detailed molecular mechanisms responsible for this deleterious effect of testosterone on myocardium subjected to acute I/R remain unclear.
Protein kinase B (Akt) signaling is a mediator of an important intracellular prosurvival pathway that has been shown to protect cardiomyocytes against I/R-induced injury (5, 6). Notably, sex-specific differences exist in myocardial Akt activation with higher levels of nuclear-localized phosphor-Akt (p-Akt) in young women compared to age matched men (7). These differences may be due to the effects of the sex hormones estrogen and/or testosterone. Surprisingly, a majority of studies have been focused on the role of estrogen in myocardial Akt signaling following I/R (8, 9), but little information exists regarding the role of testosterone. In addition, Akt acts as a survival kinase by modulating proapoptotic substrates (e.g., Bad, Forkhead transcription factors [FOXO]), and anti-apoptotic molecules (e.g., Bcl-2) in the heart (6, 10, 11). However, it is unknown what role testosterone plays in modulating myocardial Akt signaling and by which downstream targets testosterone mediates myocardial injury following acute I/R.
Therefore, given the detrimental effects of testosterone on myocardial function and apoptotic signaling following I/R, we hypothesized that: 1) endogenous testosterone will decrease myocardial Akt activation in male heart subjected to I/R; 2) endogenous testosterone will modulate downstream signals, including Bad, Bcl-2, FOXO3a, and FOXO3a-regulated death genes following I/R; 3) administration of exogenous testosterone will reverse the prosurvival effects in the Akt pathway observed in castrated male hearts during I/R.
MATERIALS AND METHODS
All animal studies conformed to the “Guide for the Care and Use of Laboratory Animals” (NIH publication No. 85-23, revised 1996). The protocols were reviewed and approved by the Indiana Animal Care and Use Committee of Indiana University.
Isolated heart perfusion (Langendorff)
Isolated rat heart was performed as previously described (2, 4). Briefly, rat hearts were rapidly excised and the aorta was cannulated. The heart was perfused (70 mm Hg) with oxygenated (95% O2 / 5% CO2) Krebs-Henseleit solution (37° C) and paced at 350 bpm/min except during ischemia. All isolated rat hearts were subjected to the same I/R protocol: 15-minute equilibration, 25-minute global ischemia (37°C), and 40-minute reperfusion.
Experimental Groups
A total of 54 Sprague-Dawley rats (9-11 weeks) (Harlan, Indianapolis, IN) were divided into six groups (n=9/group): female, male, castrated male (Castr M), male treated with flutamide (M+Flut), castrated male with chronic 5α-dihydrotestosterone (DHT) administration (Castr M + DHT), or with acute testosterone infusion (ATI) (Castr M + ATI). Twenty-seven male rats weighing 100-125g (4-5 weeks) underwent bilateral castration followed by a 4-week recovery. Subcutaneous implantation of 21-day release pellets containing 10 mg of flutamide (androgen receptor blocker) or 100 mg of DHT was performed in male (7 weeks) or castrated male rats (8 weeks), respectively. Those animals were utilized after 21 days. ATI (17beta-hydroxy-4-androstenone, 10 ng/ml/min × 5 min) was administered prior to ischemia. All doses of drugs chosen were based on the previous publications and our preliminary results (4, 12).
Western blotting
Heart tissue was homogenized in cold RIPA buffer (Sigma, Saint Louis, MO). The protein extracts (25 μg/lane) were subjected to electrophoresis on a 4-12% Bis-Tris protein gel (Invitrogen, Carlsbad, CA). The membranes were incubated with the following primary antibodies: total-Akt (T-Akt), p-Akt (Thr308), p-Bad (Ser112), T-FOXO3a, p-FOXO3a (Ser253), Bim, Bax, Fas-L (Cell Signaling Technology, Beverly, MA), Bcl-2 (ab-4) (Oncogene Research Products, San Diego, CA), manganese superoxide dismutase (MnSOD) (Sigma) and GAPDH (Biodesign International, Saco, Maine), and then incubated with horseradish peroxidase-conjugated secondary antibodies.
Lactate dehydrogenase assay
Heart tissue from male and female was homogenized as described above and lactate dehydrogenase (LDH) activity was assessed using a Cytotoxicity Detection Kit (Roche Diagnostics Corporation, Indianapolis, IN).
Immunofluorescence for Akt and Apoptosis
The rat hearts (female and male) rapidly frozen and embedded in OCT compound after I/R were sliced into 20-μM sections and stained with an anti-Akt antibody using a standard immunohistochemistry protocol followed by incubation with Texas red-conjugated anti-rabbit antibody (Vector Laboratory, Burlingame, CA). The heart slices were also assessed for apoptosis using a DeadEnd Fluorimetric TUNEL System (Promega Corporation, Madison, WI).
Real-time RT PCR analysis of mRNA levels
Total RNA was extracted from heart tissue using RNA STAT-60 (TEL-TEST, Friendswood, TX). 0.1 μg of total RNA was reverse-transcribed to cDNA using a Cloned AMV First-Strand cDNA Synthesis kit (Invitrogen Life Technologies). SYBR Green Real-time PCR was performed on the 7300 Real-Time PCR system (Applied Biosystems, Foster City, CA) for quantification of Bcl-2, Bax, Bim, Fas-L and MnSOD mRNA levels. The nucleotide sequences of the PCR primers used are listed in table 1.
Table 1.
PCR primers
| Genes | Forward 5’-3’ | Reverse 5’-3’ |
|---|---|---|
| Bcl-2 | AGGATAACGGAGGCTGGGATGCCTTTG | CGTCTTCAGAGACAGCCAGGAGAAATC |
| Bax | TAGCAAACTGGTGCTCAAGGCCCTGTG | AGTCCAGTGTCCAGCCCATGATGGTTC |
| Bim | CACCCATGAGTTGTGACAAGTCAACAC | CTGCCTTATGGAAGCCATTGCACTGAG |
| Fas-L | GAACTGGCAGAACTCCGTGAGTTCACC | AGAGGGTGTGCTGGGGTTGGCTATTTG |
| MnSOD | GGTGGTGGAGAACCCAAAGGAGAGTTG | CCACAGACACAGCTGTCAGTTTCTCC |
| Beta-actin | AACCGTGAAAAGATGACCCAGATCATG | ACAACACAGCCTGGATGGCTACGTAC |
Presentation of data and statistical analysis
All reported values are mean ± SEM. Data were compared using the Student’s t test. P < 0.05 was considered statistically significant.
RESULTS
Sex-specific differences in the myocardial response to I/R
Sex differences were evident in myocardial function following I/R, with markedly worsened recovery of +/- dP/dt in male hearts compared to female hearts (Figure 1A). To examine cardiomyocyte injury, we measured LDH level in cardiac tissue. Greater myocardial damage from I/R leads to higher levels of LDH in the coronary effluent and lower remaining LDH in cardiac tissue. Lower levels of LDH were observed in the male myocardium after I/R (Figure 1B), indicating more LDH released into coronary effluent and thereby, demonstrating more severe myocardial damage in males. A TUNEL assay indicated no significant apoptosis in either female or male hearts after I/R (Figure 1C).
Figure 1.
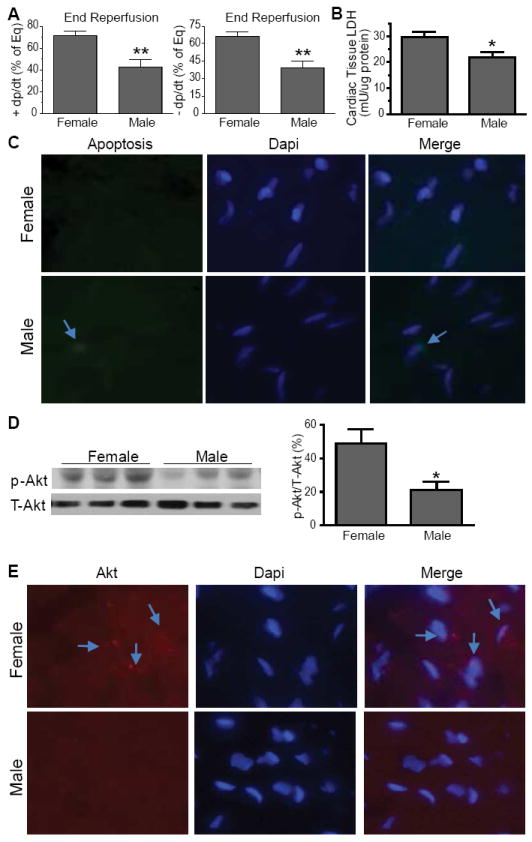
Sex differences in myocardial response to I/R. A. Worsened post-ischemic function +/- dP/dt in male hearts compared to females (N=9 hearts/group). B. Lower levels of LDH remained in male cardiac tissue after I/R (N=4/group). C. Apoptosis in female and male heart after I/R, staining with DAPI (blue) for nuclei and TUNEL assay staining with Fluorescein-12-dUTP (green) for apoptotic cells. D. Decreased Akt activation in male myocardium (N=6/group). Representative immunoblots are shown as 3 lanes/group for Akt. E. Immunofluorescence of heart tissue from females and males, stained with Akt (red) and Dapi (blue). Photomicrographs are shown as 400x magnification. Results are Mean ± SEM, *p<0.05, **p<0.01 vs. female.
In associated with exacerbated myocardial injury, p-Akt levels were significantly lower in male heart subjected to I/R (Figure 1D). Similarly, immunofluorescence indicated higher levels of Akt in the nucleus of female cardiomyocytes compared to males (Figure 1E), suggesting more pronounced Akt activity in female hearts.
Negative effects of endogenous testosterone on myocardial function and PI3K/Akt pathway following I/R
To determine the role of endogenous testosterone in myocardial function and Akt signaling in male hearts following I/R, we employed castration to deplete endogenous testosterone or flutamide treatment to block the androgen receptor. Castration or flutamide treatment significantly improved post-ischemic myocardial function (Figure 2A). Consistently, increased activation of Akt (Figure 2B) was noticed in castrated or flutamide-treated male hearts, suggesting that endogenous testosterone downregulates myocardial Akt activation following I/R.
Figure 2.
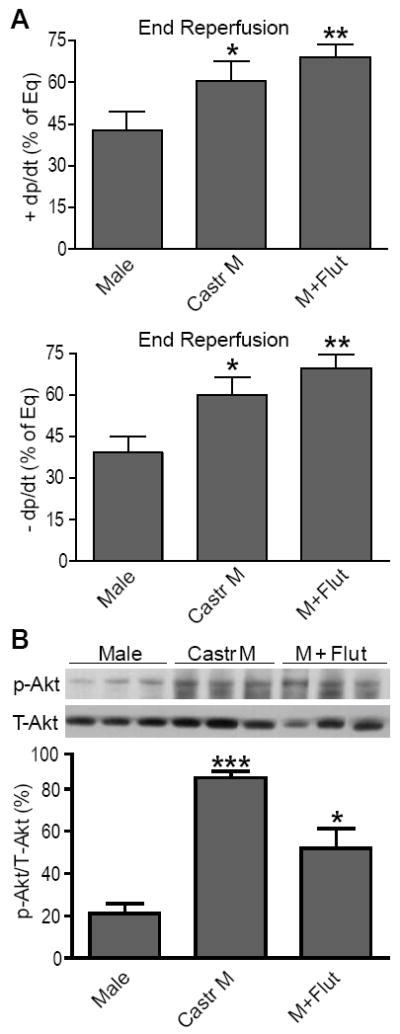
Effects of endogenous testosterone on myocardial function and activation of Akt following I/R. Improved cardiac +/- dP/dt (A) and increased myocardial activation of Akt (B) in castrated or flutamide-treated males. Shown are 3 immunoblots/group. N=4-9/group, Mean ± SEM, *p<0.05, **p<0.01, ***p<0.001 vs. Male.
We next examined downstream signals of Akt to elucidate their potential role in testosterone-mediated myocardial injury following I/R. As shown in figure 3A, sex differences existed in myocardial p-Bad, with decreased levels of p-Bad in males compared to female hearts after I/R. Castration or flutamide treatment significantly increased p-Bad (Figure 3A) in male hearts, indicating that endogenous testosterone negatively mediates myocardial Bad phosphorylation. Additionally, given the importance of Akt as a transcriptional and translational regulator of Bcl-2 (anti-apoptosis) and Bax (pro-apoptosis) (13, 14), we determined mRNA and protein levels of myocardial Bcl-2 and Bax. Male hearts exhibited lower levels of Bcl-2 mRNA and protein (Figure 3B, C), and a decreased ratio of Bcl-2/Bax (Figure 3F). Although elevated myocardial Bax was noted in castrated or flutamide-treated males (Figure 3E), endogenous testosterone depletion or androgen receptor inhibition significantly increased Bcl-2 (mRNA and protein) to a higher level in male hearts (Figure 3B, C), resulting in the increased ratio of Bcl-2/Bax in these hearts following I/R.
Figure 3.
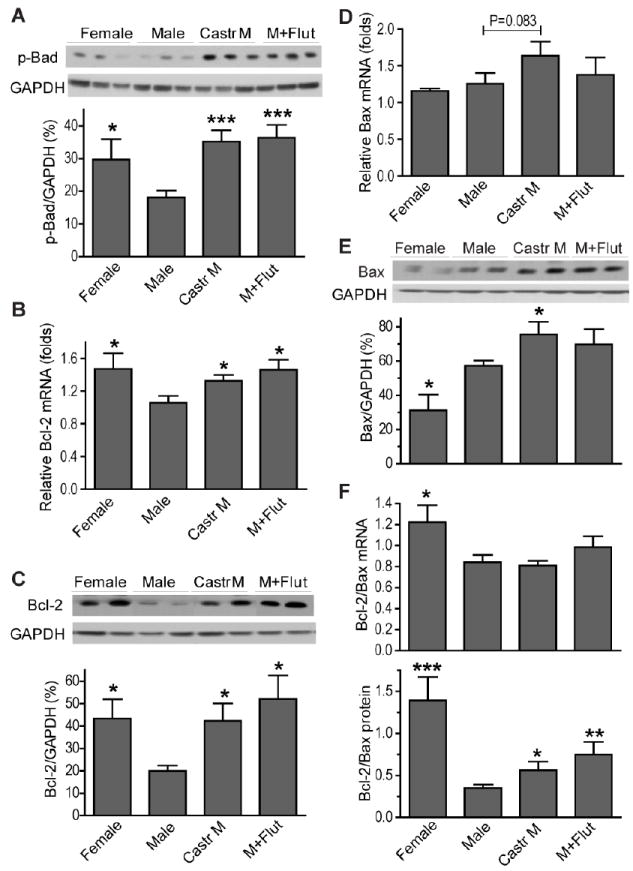
Effects of endogenous testosterone on Akt downstream signals: Bad, Bcl-2 and Bax. Higher levels of p-Bad (A), anti-apoptotic Bcl-2 mRNA (B) and protein (C) in female, castrated male and male+Flut hearts after I/R. D. Bax mRNA; E. Bax protein; F. The ratio of Bcl-2/Bax (mRNA and protein). N=4-9/group, Mean ± SEM, *p<0.05, **p<0.01, ***p<0.001 vs. Male.
FOXO3a is also an important downstream target of Akt in the heart. In figure 4A, markedly increased phosphorylation/inactivation of FOXO3a was observed in castrated or flutamide-treated male hearts subjected to I/R in accordance with the Akt results. FOXO3a promotes apoptosis by upregulating transcription of cell death-related genes including Bim and Fas-L. Therefore, protein and mRNA levels of Bim and Fas-L were measured to determine the role of these FOXO3a-regulated target genes in testosterone-mediated cardiac injury following I/R. Surprisingly, no sex difference existed in myocardial gene expression of Bim (Figure 4B, C). Additionally, castration or flutamide treatment did not affect Bim mRNA levels (Figure 4B) but did increase protein levels (Figure 4C). Interestingly, higher mRNA and protein levels of Fas-L were observed in male hearts after I/R (Figure 4D, E). Furthermore, castration or flutamide treatment markedly reduced Fas-L protein expression (Figure 4E), but not mRNA levels (Figure 4D).
Figure 4.
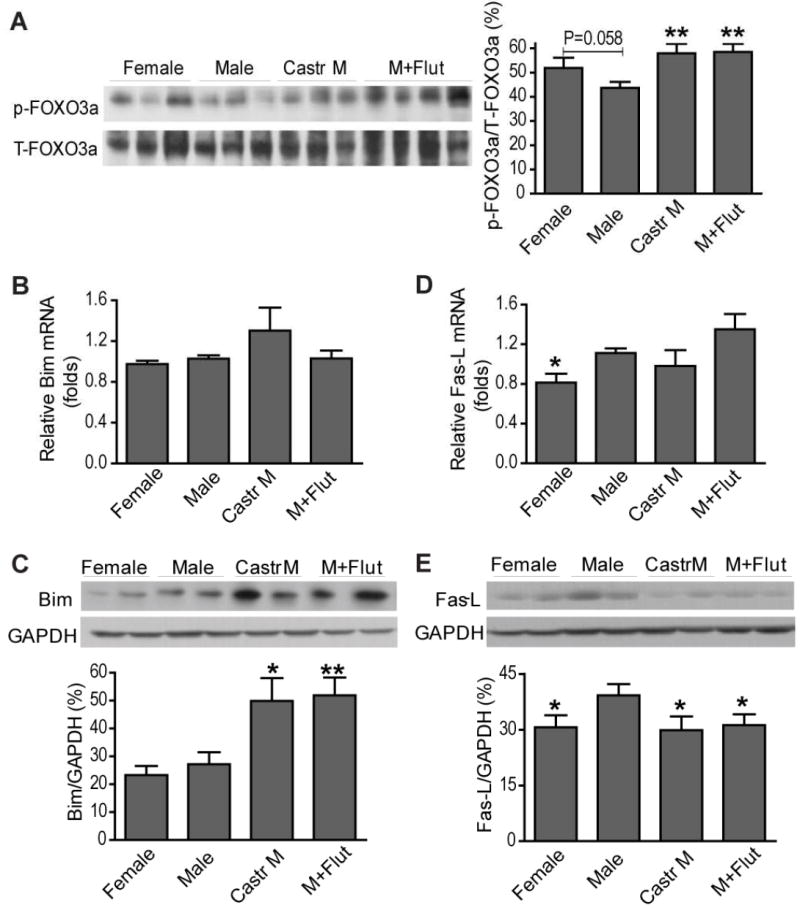
Effects of endogenous testosterone on myocardial p-FOXO3a and its regulated target genes following I/R. Myocardial p-FOXO3a levels (A), Bim mRNA (B) and protein (C), as well as Fas-L mRNA (D) and protein (E) are shown from female, male, castrated male and flutamide treated male hearts. N=4-9/group, Mean ± SEM, *p<0.05, **p<0.01 vs. Male.
Role of exogenous testosterone in post-ischemic cardiac dysfunction and myocardial Akt signaling
We next utilized exogenous testosterone administration to further confirm the deleterious effects of testosterone on myocardium in response to I/R by using chronic DHT supplement or ATI treatment in castrated males. Surprisingly, chronic administration of DHT neither worsened nor improved cardiac function in castrated males after I/R (Figure 5A). DHT supplementation did not significantly affect myocardial p-Akt (Figure 5B), p-Bad (Figure 6A) or p-FOXO3a (Figure 7A) following I/R. Consistently, no marked changes in FOXO3a-regulated genes (Bim and Fas-L) were noted in DHT-treated castrated males (Figure 7B-E). Similarly, DHT supplementation did not affect Bcl-2 gene expression, but decreased mRNA and protein levels of Bax (Figure 6D, E), leading to the increased ratio of Bcl-2/Bax (Figure 6F) in castrated males. In contrast, ATI treatment significantly exacerbated post-ischemic cardiac dysfunction in castrated males (Figure 5A). In line with these observations, markedly reduced p-Akt (Figure 5B) and p-/inactive-FOXO3a (Figure 7A) were observed in ATI-treated castrated males following I/R. In addition, ATI significantly decreased Bcl-2 protein levels and the ratio of Bcl-2/Bax (Figure 6C, F). However, protein levels of p-Bad (Figure 6A) and FOXO3a target genes (Figure 7B-E) were not affected after administration of ATI in castrated males after I/R.
Figure 5.
Effects of exogenous testosterone on myocardial function and activation of Akt following I/R. Myocardial +/- dP/dt (A) and activation of Akt (B) are shown in DHT or ATI-treated castrated males compared to untreated counterparts. N=6-9/group, Mean ± SEM, *p<0.05, ***p<0.001 vs. castrated male.
Figure 6.
Effects of exogenous testosterone on Akt downstream signals: Bad, Bcl-2 and Bax. Shown are p-Bad (A), anti-apoptotic Bcl-2 mRNA (B) and protein (C), Bax mRNA (D) and protein (E), and the ration of Bcl-2/Bax (F) in castrated males with or without DHT or ATI treatment after I/R. N=4-9/group, Mean ± SEM, *p<0.05 vs. castrated male.
Figure 7.
Effects of exogenous testosterone on myocardial p-FOXO3a and its regulated target genes following I/R. Myocardial p-FOXO3a levels (A), Bim mRNA (B) and protein (C), as well as Fas-L mRNA (D) and protein (E) are shown from castrated male with or without DHT or ATI treatment. N=4-9/group, Mean ± SEM, **p<0.01 vs. castrated male.
Effects of testosterone on MnSOD expression following I/R
FOXO3a upregulates expression of MnSOD (15, 16), an antioxidant-encoding gene in the heart. Given the increased phosphorylation/inactivation of FOXO3a in castrated or flutamide-treated male hearts, we investigated whether testosterone regulates myocardial MnSOD expression through FOXO3a in males following I/R. Male hearts exhibited lower levels of MnSOD (mRNA and protein) compared to females after I/R (Figure 8A, B). Castration or flutamide treatment increased MnSOD expression (mRNA and protein), however, mRNA levels of MnSOD were decreased in castrated males treated with DHT or ATI (Figure 8A).
Figure 8.
Effect of testosterone on myocardial antioxidant MnSOD mRNA (A) and protein (B) after I/R. N=4-9/group, Mean ± SEM, *p<0.05, **p<0.01 vs. male, #p<0.05, ##p<0.01 vs. castrated male. C. Schematic demonstrating the testosterone-regulated PI3K/Akt pathway and downstream signals in response to ischemia/reperfusion. AR: androgen receptor
DISCUSSION
In order to further elucidate the intrinsic biological differences between male and female responses to myocardial I/R, we conducted the present study which provided the initial evidence showing the effects of testosterone on myocardial Akt pathway in response to I/R. Here, we report that following acute I/R: 1) sex differences existed in myocardial Akt activation and its downstream signals: p-Bad, Bcl-2, p-FOXO3a, and Fas-L; 2) castration or flutamide treatment significantly increased activation of the myocardial Akt pathway, including p-Bad, Bcl-2, p-FOXO3a, and reduced Fas-L in male hearts; 3) ATI administration, but not chronic DHT treatment, reversed the effects of Akt signaling in castrated male hearts; and 4) lower levels of MnSOD expression were observed in male hearts, and castration or flutamide treatment restored myocardial MnSOD gene expression to the levels of females. The present study together with our previous findings (4), demonstrates that testosterone plays a negative role in myocardial function and Akt signaling, and may have pro-oxidant properties following I/R (Figure 8C).
Akt is an important molecular kinase that mediates myocardial protection during I/R. Accumulated evidence has shown that female hearts have higher levels of Akt activity resulting in improved cardiac functional recovery after I/R compared to males (7). In this study, we further confirmed lower levels of Akt activity in male hearts associated with worse post-ischemic cardiac function. Sex differences in Akt activity could be due to the beneficial effects of estrogen with respect to decreased myocardial Akt activation in ovariectomized rats (9). Additionally, estrogen is able to activate the Akt pathway and thus, suppress cardiomyocyte apoptosis during myocardial infarction (8, 9). However, it is possible that sex differences in myocardial Akt activation could be explained in part by negative role of testosterone given its effects on male myocardium susceptible to I/R. In fact, we found that endogenous testosterone depletion by castration or androgen receptor inhibition by flutamide increased myocardial activation of Akt in male hearts and was associated with restored cardiac function following I/R, whereas ATI treatment reversed the myocardial Akt signaling in castrated males. Notably, Bai et al. reported that testosterone increases Akt activity in cardiomyocytes (17), which is opposite to our finding that testosterone negatively regulates myocardial Akt activation here. These disparate findings are likely due to the use of different experimental models. Bai and colleagues utilized cardiomyocyte cultures subjected to ten-minute incubation with testosterone under normoxia, whereas we utilized the whole heart perfusion system with I/R injury lasting for 65 minutes. I/R injury is able to induce production of reactive oxygen species (ROS) and causes oxidant stress, both of which have been shown to activate the Akt pathway (18, 19). Therefore, we elucidated the role of testosterone in cardiac Akt activation with a higher baseline in an injured model in this study.
Akt improves cell survival by phosphorylating several negative modulators of cell death, including Bad (13). Bad, a member of the Bcl-2 family, normally binds to the Bcl-2/Bcl-X complex and triggers apoptosis. However, phosphorylated Bad will dissociate from this complex resulting in release of Bcl-2/Bcl-X and suppression of apoptosis. Here, in line with our Akt data, castration or flutamide treatment significantly increased myocardial levels of p-Bad and Bcl-2, whereas ATI reversed this effect in castrated male hearts following I/R. On the other hand, Akt also suppresses apoptosis by controlling the balance of Bcl-2 and Bax. In the present study, an elevated ratio of Bcl-2/Bax existed in castrated and flutamide-treated male hearts indicating an anti-apoptotic state. However, higher levels of Bax protein were noticed in all male groups compared to females, suggesting the possibility of that testosterone may not be the sole mediator for the potentiated cardiac injury following I/R in normal males. Notably, the chronic administration of testosterone slightly reduced myocardial Akt activation compared to untreated males, but significantly decreased Bax protein levels and elevated the ratio of Bcl-2/Bax. This discrepancy indicates that the effect of chronic DHT supplement on myocardium following I/R is different from ATI and that the chronic treatment may provide some level of cardioprotection. This discrepancy also implies that the role of testosterone in cardiac response to I/R is complex and requires further investigation.
FOXO, another important downstream target of Akt, has been shown to downregulate Akt-promoted cell survival by inducing cell death-related genes. The nucleo-cytoplasmic shuttling of FOXO factors controls their transcriptional activity. Akt-increased FOXO phosphorylation allows their translocation from the nucleus to the cytoplasm and inactivates their ability to regulate transcriptional targets in the nucleus. Previous studies have demonstrated that Akt is able to phosphorylate/inactivate FOXO3a (16), leading to a general depression of FOXO3a targets such as Bim and Fas-L in the heart (10). Herein, we found that testosterone downregulates myocardial p-FOXO3a (inactive form) following I/R, which is in agreement with Akt results. However, there was no downregulation of gene expression of Bim in castrated or flutamide treated male hearts. Additionally, ATI treatment did not increase Bim expression (mRNA and protein). These results indicate that the testosterone-downregulated Akt-FOXO3a pathway did not influence Bim expression in male hearts subjected to I/R. Furthermore, castration or blockade of androgen receptor did not change gene levels of Fas-L, but significantly decreased Fas-L protein, suggesting that endogenous testosterone-induced upregulation of myocardial Fas-L likely occurs at the translational and/or post-translational levels, but not at the transcription stage following I/R. Taking together, these findings demonstrate that testosterone-mediated Bim and Fas-L gene expression does not occur at the transcription process by FOXO3a in male hearts during I/R.
In this study, our experiment period (a total of 65 minutes for I/R) was too brief to detect significant apoptosis, which limited us to observe the end-point of an influence of testosterone on programmed cell death after I/R. However, our previous study has clearly indicated that castration or androgen receptor inhibition reduced pro-apoptotic protein caspase-3 compared to male hearts after I/R (4). Therefore, in conjunction with these findings here, it is possible that testosterone decreases myocardial Akt activation, leading to reduced Bad phophorylation, decreased Bcl-2 levels, increased Fas-L protein and elevated caspase-3 activity, which consequently result in worsened I/R-depressed myocardial function. Admittedly, activated Akt may also mediate myocardial protection via additional mechanisms. Indeed, delivery of a constitutively active Akt mutant gene protects hypoxia-induced cardiomyocyte dysfunction through preventing hypoxia-induced abnormalities in calcium transients and shortening (5). In addition, Akt activation has been reported to protect cardiomyocytes from I/R injury through the upregulation of endothelial nitric oxide synthase (20). Furthermore, activation of the Akt pathway has been linked to a lower incidence of arrhythmias after myocardial infarction (21). Therefore, in addition to elevation of apoptotic signaling, testosterone-decreased Akt activation in males may worsen myocardial function following I/R through other molecules.
The antioxidant/free radical scavenging gene MnSOD has recently been shown to be a target gene of FOXO3a in the heart (10, 15, 16). Of note, sex-specific differences exist in the oxidant-antioxidant systems with higher levels of baseline oxidation in males compared to females (22, 23). In addition, endogenous testosterone depletion has been shown to accelerate MnSOD activation and reduce the production of ROS, thereby leading to decreased kidney damage after I/R (22). In this study, increased mRNA and protein levels of MnSOD were observed in castrated or flutamide-treated male hearts following I/R, whereas testosterone “added back” by DHT or ATI decreased MnSOD transcription in castrated male hearts. This result is in parallel with previous studies showing the negative role of testosterone in MnSOD (24, 25). Therefore, it is possible that testosterone decreases MnSOD, increases I/R-induced ROS production, and finally, accelerates I/R-depressed myocardial function in males. However, the findings of decreased FOXO3a activity but increased MnSOD in castrated or flutamide-treated male hearts do not support a notion that testosterone downregulates MnSOD through FOXO3a following I/R (Figure 8C). Therefore, the detailed mechanisms regarding how testosterone mediates the oxidant-antioxidant systems in the heart subjected to I/R require further extensive investigation.
Acknowledgments
Thanks to Dr. Tim Lahm (Division of Pulmonary/Allergy/Critical Care and Occupational Medicine, IUSM) for reviewing and editing this manuscript.
GRANTS This work was supported by NIH R00 HL0876077 (MW).
Footnotes
DISCLOSURES None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang M, Crisostomo PR, Markel TA, Wang Y, Meldrum DR. Mechanisms of sex differences in TNFR2-mediated cardioprotection. Circulation. 2008;118:S38–45. doi: 10.1161/CIRCULATIONAHA.107.756890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang M, Baker L, Tsai BM, Meldrum KK, Meldrum DR. Sex differences in the myocardial inflammatory response to ischemia-reperfusion injury. Am J Physiol Endocrinol Metab. 2005;288:E321–326. doi: 10.1152/ajpendo.00278.2004. [DOI] [PubMed] [Google Scholar]
- 3.Cavasin MA, Sankey SS, Yu AL, Menon S, Yang XP. Estrogen and testosterone have opposing effects on chronic cardiac remodeling and function in mice with myocardial infarction. Am J Physiol Heart Circ Physiol. 2003;284:H1560–1569. doi: 10.1152/ajpheart.01087.2002. [DOI] [PubMed] [Google Scholar]
- 4.Wang M, Tsai BM, Kher A, Baker LB, Wairiuko GM, Meldrum DR. Role of endogenous testosterone in myocardial proinflammatory and proapoptotic signaling after acute ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2005;288:H221–226. doi: 10.1152/ajpheart.00784.2004. [DOI] [PubMed] [Google Scholar]
- 5.Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330–335. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
- 6.Fan GC, Zhou X, Wang X, Song G, Qian J, Nicolaou P, Chen G, Ren X, Kranias EG. Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ Res. 2008;103:1270–1279. doi: 10.1161/CIRCRESAHA.108.182832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camper-Kirby D, Welch S, Walker A, Shiraishi I, Setchell KD, Schaefer E, Kajstura J, Anversa P, Sussman MA. Myocardial Akt activation and gender: increased nuclear activity in females versus males. Circ Res. 2001;88:1020–1027. doi: 10.1161/hh1001.090858. [DOI] [PubMed] [Google Scholar]
- 8.Patten RD, Pourati I, Aronovitz MJ, Baur J, Celestin F, Chen X, Michael A, Haq S, Nuedling S, Grohe C, Force T, Mendelsohn ME, Karas RH. 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ Res. 2004;95:692–699. doi: 10.1161/01.RES.0000144126.57786.89. [DOI] [PubMed] [Google Scholar]
- 9.Ren J, Hintz KK, Roughead ZK, Duan J, Colligan PB, Ren BH, Lee KJ, Zeng H. Impact of estrogen replacement on ventricular myocyte contractile function and protein kinase B/Akt activation. Am J Physiol Heart Circ Physiol. 2003;284:H1800–1807. doi: 10.1152/ajpheart.00866.2002. [DOI] [PubMed] [Google Scholar]
- 10.Guo J, Gertsberg Z, Ozgen N, Steinberg SF. p66Shc links alpha1-adrenergic receptors to a reactive oxygen species-dependent AKT-FOXO3A phosphorylation pathway in cardiomyocytes. Circ Res. 2009;104:660–669. doi: 10.1161/CIRCRESAHA.108.186288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin H, Chao L, Chao J. Kallikrein/kinin protects against myocardial apoptosis after ischemia/reperfusion via Akt-glycogen synthase kinase-3 and Akt-Bad.14-3-3 signaling pathways. J Biol Chem. 2005;280:8022–8030. doi: 10.1074/jbc.M407179200. [DOI] [PubMed] [Google Scholar]
- 12.Crisostomo PR, Wang M, Wairiuko GM, Morrell ED, Meldrum DR. Brief exposure to exogenous testosterone increases death signaling and adversely affects myocardial function after ischemia. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1168–1174. doi: 10.1152/ajpregu.00833.2005. [DOI] [PubMed] [Google Scholar]
- 13.Chakir K, Daya SK, Tunin RS, Helm RH, Byrne MJ, Dimaano VL, Lardo AC, Abraham TP, Tomaselli GF, Kass DA. Reversal of global apoptosis and regional stress kinase activation by cardiac resynchronization. Circulation. 2008;117:1369–1377. doi: 10.1161/CIRCULATIONAHA.107.706291. [DOI] [PubMed] [Google Scholar]
- 14.Matsui T, Nagoshi T, Rosenzweig A. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2003;2:220–223. [PubMed] [Google Scholar]
- 15.Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 16.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bai CX, Kurokawa J, Tamagawa M, Nakaya H, Furukawa T. Nontranscriptional regulation of cardiac repolarization currents by testosterone. Circulation. 2005;112:1701–1710. doi: 10.1161/CIRCULATIONAHA.104.523217. [DOI] [PubMed] [Google Scholar]
- 18.Jamnicki-Abegg M, Weihrauch D, Pagel PS, Kersten JR, Bosnjak ZJ, Warltier DC, Bienengraeber MW. Isoflurane inhibits cardiac myocyte apoptosis during oxidative and inflammatory stress by activating Akt and enhancing Bcl-2 expression. Anesthesiology. 2005;103:1006–1014. doi: 10.1097/00000542-200511000-00015. [DOI] [PubMed] [Google Scholar]
- 19.Wang B, Shravah J, Luo H, Raedschelders K, Chen DD, Ansley DM. Propofol protects against hydrogen peroxide-induced injury in cardiac H9c2 cells via Akt activation and Bcl-2 up-regulation. Biochem Biophys Res Commun. 2009;389:105–111. doi: 10.1016/j.bbrc.2009.08.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao ZH, Wojcik KR, Dossumbekova A, Hsu C, Mehendale SR, Li CQ, Qin Y, Sharp WW, Chang WT, Hamann KJ, Yuan CS, Hoek TL. Grape seed proanthocyanidins protect cardiomyocytes from ischemia and reperfusion injury via Akt-NOS signaling. J Cell Biochem. 2009;107:697–705. doi: 10.1002/jcb.22170. [DOI] [PubMed] [Google Scholar]
- 21.Ravingerova T, Matejikova J, Neckar J, Andelova E, Kolar F. Differential role of PI3K/Akt pathway in the infarct size limitation and antiarrhythmic protection in the rat heart. Mol Cell Biochem. 2007;297:111–120. doi: 10.1007/s11010-006-9335-z. [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Kil IS, Seok YM, Yang ES, Kim DK, Lim DG, Park JW, Bonventre JV, Park KM. Orchiectomy attenuates post-ischemic oxidative stress and ischemia/reperfusion injury in mice. A role for manganese superoxide dismutase. J Biol Chem. 2006;281:20349–20356. doi: 10.1074/jbc.M512740200. [DOI] [PubMed] [Google Scholar]
- 23.Actis-Goretta L, Carrasquedo F, Fraga CG. The regular supplementation with an antioxidant mixture decreases oxidative stress in healthy humans. Gender effect. Clin Chim Acta. 2004;349:97–103. doi: 10.1016/j.cccn.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 24.Sastre J, Borras C, Garcia-Sala D, Lloret A, Pallardo FV, Vina J. Mitochondrial damage in aging and apoptosis. Ann N Y Acad Sci. 2002;959:448–451. doi: 10.1111/j.1749-6632.2002.tb02114.x. [DOI] [PubMed] [Google Scholar]
- 25.Jacob MH, Janner Dda R, Bello-Klein A, Llesuy SF, Ribeiro MF. Dehydroepiandrosterone modulates antioxidant enzymes and Akt signaling in healthy Wistar rat hearts. J Steroid Biochem Mol Biol. 2008;112:138–144. doi: 10.1016/j.jsbmb.2008.09.008. [DOI] [PubMed] [Google Scholar]