

Abstract
We have designed and synthesized analogues of compound C, a non-specific inhibitor of 5’-AMP-activated protein kinase (AMPK), using a computational fragment-based drug design (FBDD) approach. Synthesizing only twenty-seven analogues yielded a compound that was equipotent to compound C in the inhibition of the human AMPK (hAMPK) α2 subunit in the heterotrimeric complex in vitro, exhibited significantly improved selectivity against a subset of relevant kinases, and demonstrated enhanced cellular inhibition of AMPK.
Keywords: Fragment-based drug design, FBDD, 5’-AMP-activated protein kinase, AMPK, compound C
5’-AMP-activated protein kinase (AMPK) is a major regulator of normal cellular energy metabolism [1]. AMPK activity is sensitive to the cellular AMP:ATP ratio, which is increased by physiological stresses such decreased oxygen and glucose concentrations (hypoxia and hypoglycemia). AMPK activity is also associated with the growth of solid tumors, which commonly contain regions of pathologic hypoxia and hypoglycemia [2]. AMPK cooperates with hypoxia-inducible factor-1 (HIF-1), the primary transcriptional regulator of the mammalian response to hypoxia and other metabolic stresses that lower cellular ATP levels, in pathophysiological tumor microenvironments [3, 4]. Thus, in the context of microenvironmental stress, AMPK may represent a novel target for the treatment of solid tumors. The successful design of potent and selective inhibitors of AMPK that exhibit activity is an important first step toward establishing the proof-of-concept for the therapeutic value of AMPK inhibition in solid tumor microenvironments.
This Letter describes our efforts to design a Type I inhibitor of AMPK with a superior biological profile than that of compound C (Figure 1), which is a commonly used experimental direct inhibitor of the enzyme [5], through the use of our proprietary fragment-based drug design (FBDD) software [6–9] and Imagiro™ [10], our proprietary torsion-space molecular mechanics software package. Compound C ([4-(2-piperidin-1-yl-ethoxy)-phenyl])-3-pyridin-4-yl-pyrrazolo[1,5-a]-pyrimidine) is considered to act as a competitive inhibitor of ATP binding to the catalytic α subunit of AMPK [5, 11]. However, compound C has significantly limited specificity as an AMPK inhibitor [5].
Figure 1.

Chemical structure of compound C.
We discuss the SAR of a series of pyrazolopyrimidine and aminooxazole analogues of compound C and present their general binding mode obtained through the use of homology modeling and Grand Canonical Monte Carlo (GCMC) fragment simulations [9]. A focused kinase selectivity profile for the most active subset of these compounds is presented, as well as the cellular AMPK inhibition data for the most promising pyrazolopyrimidine.
We generated a model of the catalytic α2 subunit of AMPK with the DFG-loop in the ‘in’ conformation using MOE [12]. The crystal structure of the AMPK α2 subunit (RCSB ID# 2H6D, 1.85 Å) [13] was used as the overall template for the protein model. The activation loop (A-loop) was not crystallographically resolved in this structure and the DFG loop is in the ‘out’ conformation. Residues 157–179 of the microtubule affinity regulating kinse-1 (MARK-1) crystal structure (RCSB ID# 2HAK, 2.60 Å) [14] were used as a template for the ‘in’ conformation of the DFG-loop and the missing residues for the A-loop. MARK-1 was selected as the template for the A-loop conformation of the AMPK α2 subunit due to the 50% sequence identity between the AMPK and MARK-1 kinase domains. Compound C, which inhibits AMPK with reported IC50 values ranging from 0.1 to 0.2 µM [15] was used to model the inhibitor-bound protein complex. The three fragments comprising the core structure of compound C (benzene, pyridine, and pyrazolo[1,5-a]pyrimidine, (Figure 1) were simulated for binding to the AMPK homology model using our Grand Canonical Monte Carlo (GCMC) simulation approach [6–8]. Compound C was assembled from the resulting fragment simulation data and found to bind in the ATP site. The resulting complex was subjected to energy minimization, which was terminated when the energy gradient reached 0.001, followed by 259 ps of molecular dynamics simulations in torsion space using Imagiro [10] to ensure that the kinetic and potential energies of the system were equilibrated, arriving at the optimized homology model (Figure 2A).
Figure 2.

Minimized predicted lowest-energy binding pose of compound C (A) and compound 5 (B) (carbons in magenta) in complex with the hAMPK α2 subunit without their respective ethylpiperidinyl or ethylmorpholino tails. The hydrogen-bonds between the pyridinone oxygen and the zeta nitrogen of Lys-45; the pyridinone nitrogen and the delta oxygen of Asp-157 (B) and between the pyrazolopyrimidine core and the backbone nitrogen of Val-96 in the hinge region (A, B) are shown in green. Note that the DFG loop (orange) is in the ‘in’ conformation.
In the final model, the pyrazolo[1,5-a]pyrimidine core was observed to present one ring nitrogen to act as a hydrogen bond acceptor with the backbone nitrogen of residue Val-96 in the hinge region, while the 4-pyridine fragment forms a hydrogen bond with the side chain of Lys-45. The piperidine fragment is oriented towards the solvent and does not appear to form specific interactions with AMPK. We hypothesize that this fragment performs the function of a solubilizing group. The P-loop (residues 23 – 30) is moved slightly towards the N-terminal lobe of the kinase in the final model relative to the 2H6D crystal structure of AMPK to allow for better steric contact with the pyridinyl moiety of compound C. The overall RMSD of the Cα atoms between the two structures is 2.4 Å.
In order to design inhibitors of AMPK, the binding of 1785 small molecular fragments [16] was simulated against the homology model using the GCMC approach. Hypothetical compounds were designed using the results of the fragment simulations to find replacements for the pyridine fragment that maximized interactions with Lys-45 as well as Asp-157 in the DFG-loop as shown in Figure 2B. Replacements for the pyrazolo[1,5-a]pyrimidine fragment were explored, with the most successful being aminooxazole. All designed compounds were evaluated using computational ADME filters [17] and prioritized for synthesis on the basis of their calculated binding affinities and ADME properties.
The synthesis of designed substituted pyrazolo[1,5-a]pyrimidine derivatives was accomplished according to Scheme 1. The bromide intermediate 2 was readily obtained from the commercially available 1 [18] via an SN2 type reaction with 4-(2-chloroethyl) morpholine hydrochloride salt under basic conditions. The bromide 2 was then reacted with selected commercial available or readily synthesized boronates or boronic acids in microwave palladium catalyzed Suzuki cross-coupling reactions with good to moderate yields to give compounds 3–10 [19] Compound 11 was obtained after a subsequent Suzuki coupling reaction between intermediate 15 and bromide 2 followed by deprotection of the PMB groups. The boronate 15 was prepared after a PMB protection of commercially available 4-bromobenzene-1,2-diol 14 followed by typical boronate formation in good yield.
Scheme 1.

Synthesis of pyrazolo[1,5-a]pyrimidine compounds. Reagents and conditions: (a) K2CO3, DMF, 80° C, 90%, 4-(2-chloroethyl)morpholine HCl; (b) 10 mol% PdCl2(PPh3)2, Na2CO3 2 M, ArB(OH)2; (c) PMBCl, cat. NaI, DCM, rt, 80%; (d) 10% PdCl2 dppf.dcm, Bis (pinacolato) diborane, NaOAc, DMF, 80°, 75%.
Two synthetic routes were developed for analog 5. The overall reaction in Scheme 1 was followed. The boronate 13 was prepared after PMB protection of compound 12, followed by standard boronate formation in good yield. The product was then reacted with bromide 2 under Suzuki cross coupling conditions to give the desired analog 5 after deprotection. Inhibitor 9 was also converted to analog 5 via a one pot two step synthesis consisting of diazotization and hydrolysis in moderate yield as outlined in Scheme 2 [20].
Scheme 2.

Alternative synthetic route to 5.
The inhibitory activities of all analogs were tested against the hAMPK α2 subunit in complex with the heterotrimer (hAMPK α2,β1,γ1). The structure-activity relationship of the pyrazolo[1,5-a]pyrimidines is summarized in Table 1. Synthetic intermediates 1 and 2 were included to test the contribution of each fragment on the binding of the pyrazolo[1,5-a]pyrimidines to AMPK. The addition of the morpholinoethyl resulted in a two-fold improvement in potency observed in 2 relative to 1. Replacement of the piperidine tail in compound C (Figure 1) with the morpholinoethyl group in 4 resulted in a seven-fold loss in potency. Additionally, the ADME filters used to prioritize the designed compounds for synthesis predicted that compounds with the morpholino fragment would have better human intestinal absorption [17]. The enhanced potency of 4 relative to 3 was attributed to the ability of the nitrogen on the pyridine ring to form a hydrogen bond with the side chain of Lys-45. We analyzed the results of the fragment simulation data to identify fragments that could form an additional hydrogen bond with the side chain of Asp-157 on the DFG-loop while retaining contact with Lys-45. Compounds 5, 6, 7, 8, 9, and 11 were designed using this approach.
Table 1.
Inhibition of hAMPK (α2, β1, γ1) by pyrazolopyrimidines
 | |||
|---|---|---|---|
| No. | R1 | R2 | IC50(µM)a |
| 1 | 7.7 | ||
| 2 | 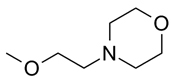 |
3.5 | |
| 3 |  |
 |
1.8 |
| 4 |  |
 |
0.28 |
| 5 |  |
 |
0.06 |
| 6 |  |
 |
0.27 |
| 7 |  |
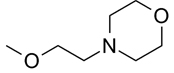 |
0.14 |
| 8 |  |
 |
0.22 |
| 9 |  |
 |
2.1 |
| 10 |  |
 |
2.2 |
| 11 |  |
 |
1.1 |
values were measured using the Hot Spot™ filtration binding 33P Kinase assay, Reaction Biology, Malvern, PA. Compounds were tested in ten-dose IC50 mode with three-fold dilutions starting at 10 µM. All reactions were carried out in the presence of 10 µM ATP.
The pyridinone fragment of compounds 5 (Figure 2) and 6 showed favorable interactions with the Lys-45 / Asp-157 pair and two plausible attachment points to the pyrazolo[1,2-a]pyrimidine core. Both variants were made to test the preference for the geometry of the Lys-45 / Asp-157 interaction. Compound 6 showed no improvement over 4, while 5 displayed equipotent activity compared to compound C. Compound 7 displayed two fold improvement in activity relative to 4. As with the pyridinone fragment, the GCMC simulations in indicated that the 2-aminopyridine fragment would form profitable interactions with the Lys-45 / Asp-157 pair, and two compounds could be designed with different attachment points to the pyridine ring that preserve the hydrogen bonds. Compound 8 was found to be equipotent with 4, while 9 was ten-fold weaker. The catechol was also identified as a potentially favorable fragment that might preserved the key interactions, so 11 was synthesized but found to be four fold weaker than 4. The advanced intermediate 10, lacking a hydrogen bond donor, was two fold less potent than the target compound 11. Another reason for the reduced potency of 10 relative to 11 is likely due to steric clashes between the methoxy groups and the side chain atoms of either Lys-45 or Asp-157. Taken together, the resulting SAR for the pairs of 5 and 6 as well as 8 and 9 demonstrate a clear preference for the trajectory from the core for the hydrogen bond acceptor and donor atoms, while compounds 5 and 7 show that the addition of a hydrogen bond donor to exploit the interaction with Asp-157 can result in improved potency against hAMPK.
We investigated the replacement of the pyrazolo[1,5-a]pyrimidine core by searching for fragments that would form favorable interactions with the hinge region while preserving the orientation of fragments that were believed to form favorable contacts with Lys-45 and Asp-157. Several core replacements suggested by the fragment simulations were used to design new compounds with reduced molecular weight that preserved the hydrogen bonding interaction with the backbone nitrogen of Val-96. These included 2-aminooxazoles (Table 2), pyridines, pyrimidines, 2-aminothiazoles, 2-aminooxazoles, pyrazolo[1,5-a]pyrimidine-7-amines, and pyrazolo[1,5-a]pyrimidin-2-amines. Due to synthetic feasibility concerns, only the designs containing pyridine, 2-aminooxazole, and pyrazolo[1,5-a]pyrimidine-7-amine cores were synthesized and tested. The pyridine and pyrazolopyrimidine-7-amine cores showed weak or no activity (> 10 µM) in the APMK assay (data not shown). The most active of these core replacements were the 2-aminooxazole series. The 2-aminooxazoles are predicted to bind to AMPK with the oxazole ring nitrogen forming the hydrogen bond with the backbone of Val-96. The 2-amino group is predicted to form an additional hydrogen bond contact with the backbone carbonyl of Ser-97. Replacements for the 2-(piperidin-1-yl)ethoxy fragment were also investigated and the most promising one identified from the GCMC simulations was benzene sulfonamide. The oxygen of the sulfonamide was predicted for form a hydrogen bond with the backbone nitrogen of Glu-100, while the nitrogen acts as a hydrogen bond donor to a side chain oxygen of the same residue.
Table 2.
Inhibition of hAMPK α2 by aminooxazoles.
 | ||
|---|---|---|
| No. | R | IC50(µM)a |
| 21 |  |
2.9 |
| 22 |  |
5.0 |
as in Table 1.
The synthesis of the 2-aminooxazole compounds using an iminophosphorane-mediated cyclization is outlined in Scheme 3 [21]. The reaction of an isothiocyanate with an acyl azide and triphenylphosphine in dioxane at 95° C or in dichloromethane at room temperature provided the 2-aminooxazole [22]. The acyl azide 20 was obtained from the available 2-chloro-1-(3,4-dihydrophenyl)ethanone via a Finkelstein reaction with sodium azide [23, 24]. The acyl azide intermediate 19 was synthesized from the corresponding acyl bromide 18 as shown in Scheme 4. Compound 17 was generated via Stille cross-coupling from 16 (see also Scheme 1) [25, 26]. Compounds 19 and 20 were converted to the intermediate iminophosphorane upon treatment with PPh3, which was followed by rearrangement and deprotection to yield 21 and 22.
Scheme 3.

General method for the synthesis of 2-aminooxazoles.
Scheme 4.

Synthesis of 2-aminooxazoles. Reagents and Conditions: (a) 10 mol% PdCl2(PPh3)2, LiCl, vinyltin reagent; (b) NBS THF/water; (c) NaN3 acetone/water; (d) PPh3, ArNSC; (e) TFA, 100° C, µwave.
Compound C has previously been reported to be a potent inhibitor of a number of kinases in addition to AMPK, including Eph-A2, c-Src, Lck, KDR, and MNK-1 [15]. A kinase selectivity screen was carried out using these six kinases plus the relevant cancer kinases Flt1, Flt3, and Rsk1. We tested the most potent pyrazolo[1,5-a]pyrimidines in addition to two 2-aminooxazoles along with compound C. As seen in Table 3, compound C is non-selective against the kinases in screen, inhibiting seven of the eight kinases more potently than AMPK. Compound 5, which is equipotent with compound C for AMPK, is substantially more selective. With the exception of Flt3, 5 showed improvements in selectivity between 4.5-fold for Mnk1 to 60-fold for Flt1 as compared to compound C.
Table 3.
Near kinase selectivity panel of pyrazolopyrimidines and aminooxazoles.
| IC50 (µM)a | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | hAMPKα2 | c-Src | EphA2 | Flt1 | Flt3 | KDR | Lck | Mnk1 | Rsk1 |
| Compound | 0.041 | 0.002 | 0.011 | 0.011 | <0.001 | 0.004 | 0.016 | 0.011 | 0.21 |
| C | |||||||||
| 4 | 0.27 | 0.050 | 0.25 | 0.21 | 0.001 | 0.056 | 0.30 | 0.13 | 2.1 |
| 5 | 0.061 | 0.061 | 0.26 | 0.47 | <0.001 | 0.10 | 0.43 | 0.045 | 2.1 |
| 7 | 0.14 | 3.3 | 0.30 | 0.004 | <0.001 | 0.010 | 0.094 | 0.027 | 0.22 |
| 8 | 0.22 | 0.021 | 0.84 | 0.040 | 0.001 | 0.058 | 0.44 | 0.164 | 1.1 |
| 11 | 1.1 | 0.096 | 0.74 | 0.002 | 0.001 | 0.001 | 0.02 | 0.95 | 0.56 |
| 21 | 2.8 | 3.2 | >10 | 0.074 | 0.13 | 0.100 | 0.97 | 2.2 | 3.4 |
| 22 | 5.0 | 8.2 | 5.6 | >10 | 0.12 | >10 | >10 | 1.10 | >10 |
as in Table 1.
Figure 3 compares the effect of 5 and compound C on basal AMPK activity (detected by immunoblotting for specific phosphorylation of the AMPK substrate acetyl CoA carboxylase 1 (ACC) on Ser79; Phospho-ACC [3, 27]) in a mouse embryonic fibroblast (MEF) cell line. Transformed derivatives of these cells were previously used to investigate the contribution of AMPK to experimental tumor growth [3]. Both 5 and compound C inhibited cellular AMPK activity (P-ACC levels), but 5 was a more potent AMPK inhibitor than compound C (e.g., ≤5 µM, 6 h) in MEF cells. It is noteworthy that both 5 and compound C appeared to decrease specific phosphorylation of the AMPKα catalytic subunit (P-AMPK levels; results not shown) in these cells; it is not clear, however, whether this effect is direct or indirect [28]. In summary, the results shown in Figure 3 demonstrate that 5 inhibited endogenous AMPK activity in cultured mammalian cells.
Figure 3.

Effects of compound 5 and compound C on AMPK-dependent ACC phosphorylation (P-ACC) and phosphorylation of AMPKα on Thr172 (P-AMPK) in MEFs cultured in complete medium (DMEM + 25 mM HEPES [pH 7.4] + 10% fetal bovine serum [FBS]). A Representative immunoblots of total protein from MEFs harvested following exposure to compound 5 or compound C (1, 5, or 10 µM; 6 h). Replicate blots were probed for the relative levels of P-ACC or total ACC. B Corresponding histogram of densitometry measurements from multiple immunoblots of total protein from untreated (control) MEFs and MEFs treated with compound 5 or compound C; data were normalized to the control data points. Error bars, ±SD from independent cultures/data point (n=3). Statistical comparisons were made between control and treated cells (2-tailed unpaired t test; p≤0.05 was considered a significant difference): only significant differences between control and treated cells are shown (*p=0.03, 5 µM compound 5; **p=0.03, 10 µM compound 5; ***p=0.04, 10 µM compound C). Immunoblotting protocols have been described in detail. [28]
In conclusion, pyrazolo[1,5-a]pyrimidine and 2-aminooxazole inhibitors of the human AMPKα2 subunit were identified through the use of FBDD and medicinal chemistry efforts. A binding model was obtained from the results of GCMC fragment simulations in to a homology model of the protein and is consistent with the observed SAR. The most potent pyrazolo[1,5-a]pyrimidine is equipotent with compound C in the in vitro AMPK assay and shows improved kinase selectivity. Compound 5 also shows greater inhibition against AMPK activity in a cellular assay relative to compound C.
Acknowledgements
This work was supported by the grants CA132529 and CA73807 from the NIH, National Cancer Institute. We would like to thank Dr. Kevin Moriarty and Dr. Martha Kelly for helpful discussions and critical review of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mishra R, Cool BL, Laderoute KR, Foretz M, Viollet B, Simonson MS. J. Biol. Chem. 2008;283:10461. doi: 10.1074/jbc.M800902200. [DOI] [PubMed] [Google Scholar]
- 2.Pang T, Zhang Z-S, Gu M, Qiu B-Y, Yu L-F, Cao P-R, Shao W, Su M-B, Li J-Y, Nan F-J, Li J. J. Biol. Chem. 2008;283:16051. doi: 10.1074/jbc.M710114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. Mol. Cell. Biol. 2006;26:5336. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papandreou I, Lim AL, Laderoute K, Denko NC. Cell Death Differ. 2008;15:1572. doi: 10.1038/cdd.2008.84. [DOI] [PubMed] [Google Scholar]
- 5.Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, Giri S, Andreelli F. Crit. Rev. Biochem. Mol. Biol. 2010;45:276. doi: 10.3109/10409238.2010.488215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark M, Meshkat S, Wiseman JS. J. Chem. Inf. Model. 2009;49:934. doi: 10.1021/ci8004397. [DOI] [PubMed] [Google Scholar]
- 7.Clark M, Guarnieri F, Shkurko I, Wiseman J. J. Chem. Inf. Model. 2006;46:231. doi: 10.1021/ci050268f. [DOI] [PubMed] [Google Scholar]
- 8.Clark M, Meshkat S, Talbot G, Carnevali P, Wiseman JS. J. Chem. Inf. Model. 2009;49:1901. doi: 10.1021/ci900132r. [DOI] [PubMed] [Google Scholar]
- 9.Guarnieri F, Mezei M. J. Am. Chem. Soc. 1996;118:8493. [Google Scholar]
- 10.Carnevali P, Toth G, Toubassi G, Meshkat SN. J. Am. Chem. Soc. 2003;125:14244. doi: 10.1021/ja036647b. [DOI] [PubMed] [Google Scholar]
- 11.Zhou G, Myers R, Li Y, Chen YZ, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. J. Clin. Invest. 2001;108:1167. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montreal, Quebec, Canada: Chemical Computing Group Inc.; 2006. [Google Scholar]
- 13.Littler DR, Walker JR, Davis T, Wybenga-Groot LE, Finerty PJ, Jr, Newman E, Mackenzie F, Dhe-Paganon S. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66(Pt.2):142. doi: 10.1107/S1744309109052543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marx A, Nugoor C, Muller J, Panneerselvam S, Timm T, Bilang M, Mylonas E, Svergun DI, Mandelkow EM, Mandelkow E. J. Biol. Chem. 2006;281:27586. doi: 10.1074/jbc.M604865200. [DOI] [PubMed] [Google Scholar]
- 15.Bain J, Plater L, Elliot M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. Biochem. J. 2007;408:297. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore WR., Jr Curr. Opin. Drug Discov. Devel. 2005;8:355. [PubMed] [Google Scholar]
- 17.Klon AE, Lowrie JF, Diller DJ. J. Chem. Inf. Model. 2006;46:1945. doi: 10.1021/ci0601315. [DOI] [PubMed] [Google Scholar]
- 18.Churcher I, Hunt PA, Stanton MG. WO/2007/085873 2007
- 19.Cuny GD, Yu PB, Laha JK, Xing X, Liu J-F, Lai CS, Deng DY, Sachidanandan C, Bloch KD, Peterson RT. Bioorg. Med. Chem. Lett. 2008;18:4388. doi: 10.1016/j.bmcl.2008.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daab JC, Bracher F. Monatsh. Chem. 2003;134:573. [Google Scholar]
- 21.Dhar TGM, Shen Z, Guo J, Liu C, Watterson SH, Gu HH, Pitts WJ, Fleener CA, Rouleau KA, Sherbina NZ, McIntyre KW, Witmer MR, Tredup JA, Chen B-C, Zhao R, Bednarz MS, Cheney DL, MacMaster JF, Miller LM, Berry KK, Harper TW, Barrish JC, Hollenbaugh DL, Iwanowicz EJ. J. Med. Chem. 2002;45:2127. doi: 10.1021/jm0105777. [DOI] [PubMed] [Google Scholar]
- 22.Froeyen P. Phosphorus, Sulfur, Silicon Relat. Elem. 1991;60:81. [Google Scholar]
- 23.Chung F, Tisné C, Lecourt T, Dardel F, Micouin L. Angew Chem. Int. Ed. 2007;46:4489. doi: 10.1002/anie.200605201. [DOI] [PubMed] [Google Scholar]
- 24.Sutton JC, Pi Z, Rejean R, L'Heureux A, Thibeault C, Lam PYS. WO/2006/078621. US Patent. 2006
- 25.Mitchell TN. In: Metal-Catalyzed Cross-Coupling Reactions. Second Edition. Prof. Dr. Armin de Meijere PDFD, editor. 2008. p. 125. [Google Scholar]
- 26.Farina V, Roth GP. Advances in Metal-Organic Chemistry. 1996. p. 1. [Google Scholar]
- 27.Hardie DG, Pan DA. Biochem. Soc. Trans. 2002;30:1064. doi: 10.1042/bst0301064. [DOI] [PubMed] [Google Scholar]
- 28.Laderoute KR, Calaoagan JM, Madrid PB, Klon AE, Ehrlich PJ. Cancer Biol. Ther. 2010;10:68. doi: 10.4161/cbt.10.1.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]