

Abstract
HIV-1 Integrase (IN), one of the essential enzymes in HIV infection, has been validated as a target for HIV treatment. While more than 20 drugs have been approved by the FDA to treat HIV/AIDS, only one drug, Raltegravir (1), was approved as an IN inhibitor. The rapid mutation of the virus, which leads to multidrug resistant HIV strains, presents an urgent need to find potent compounds that can serve as second-generation IN inhibitors. The pyrazolone scaffold, predicted by a computational modeling study using GS-9137(2) as a pharmacophoric model, has shown to inhibit the IN catalytic activities in low micromolar range. We have synthesized various analogues based on the pyrazolone scaffold and performed SAR studies. This paper will showcase the up-to-date result of this scaffold as a promising HIV-1 IN inhibitor.
Acquired immunodeficiency syndrome (AIDS) is a debilitating disease affecting more than 39 million people worldwide. Since the disease was first detected in 1981, more than 25 million people have died of AIDS1. Consequently, the World Health Organization recognizes HIV as the fourth largest cause of death globally1. As the most recent advancement toward eradication of this pandemic, a highly successful combination therapy of three HIV drugs known as HAART (highly active antiretroviral therapy) is considered as the best treatment against AIDS so far. However, increasing drug resistance due to rapid mutation of this retrovirus has prompted the faster development of many reserved drugs against resistant viral strains.
HIV-1 Integrase (IN) has been validated to be a crucial target for shutting down the HIV reproductive cycle. This viral enzyme catalyzes the insertion of proviral DNA into the host genome in two-step processes: 1) 3′-processing step that removes two nucleotides from the 3′-hydroxyl end of its viral DNA and 2) strand transfer step, which joins 3′-end of the viral DNA onto the host DNA by nucleophilic addition. Additionally, the presences of divalent metals such as Mg2+ or Mn2+ are required for the catalysis of these two steps2.
Two compounds demonstrate effective inhibition of IN (Figure 1). Presently the only FDA approved drug is Raltegravir (MK-0518, 1), which selectively inhibits the strand transfer step and has shown an excellent efficacy in HIV-1 patients3. Another IN inhibitor, GS-9137 (Elvitegravir, 2), a quinolone 3-carboxylic acid, is currently in late-stage clinical trials4. GS-9137 showed highly potent antiretroviral activity in both treatmentnaive and experienced patients in the Phase II clinical trials. Like other IN inhibitors, resistant mutants against this drug have been identified in the catalytic core domain of the IN5.
Figure 1.

Examples of clinically used HIV-1 integrase inhibitors
Although the recent success in the discovery and the clinical development of IN inhibitors as novel antiretroviral agents provided us a new choice in the treatment of HIV/AIDS, multidrug resistant HIV-1 strains have emerged as a result of the dynamic nature of the HIV genome coupled with the requirement of a sustained antiretroviral treatment regimen in chronic HIV-1 patients. Once the first generation of IN inhibitors becomes commonly used in the clinic, the emergence of resistant HIV-1 virus strains containing mutations in the IN is inevitable. Therefore, alternative second generation agents with improved resistance profiles are urgently needed.
It has been our goal to design and discover novel and structurally diverse second-generation IN inhibitors that show favorable inhibition profiles against known resistant mutant strains. We quickly became interested in the work of Dayam et al.6 who reported a series of potential lead compounds for IN inhibitor by using structure-based pharmacophore model derived from quinolone 3-carboxylic acids (such as in GS-9137). Among the compounds, the pyrazolone 3 is the most potent in inhibiting IN and its IC50 is in the low micromolar range.
The preliminary study of this pyrazolone scaffold prompted us to develop a more diversified library that includes phenyl-substituted pyrazolone compounds. This pyrazolone can be modified on three possible sites (R1, R2, and R3) as shown in Figure 2. The R1 modification involves the benzylidene ring and its benzyloxy substituent of compound 3, which is believed to bind to the hydrophobic region in the active site of IN. The R2 modification will change the functional group in the proximity of the pyrazolone’s carbonyl group, which is crucial for metal chelation inside the enzyme active site. Lastly, R3 modification could tune the electronic factors on the pyrazolone core ring.
Figure 2.

Three possible sites of modification in the pyrazolone scaffold
For modification on the benzylidene ring of the pyrazolone, the following 4a-4g analogs were synthesized as depicted in Scheme 1. This modification will determine the factors needed for increasing binding affinity to the metal ion inside the IN active site. They are as follows: the position of the methoxy group, the position of the benzyloxy group, and the linker space joining two aromatic rings. Starting with 3- bromoaniline (7), the corresponding hydrazine was synthesized quantitatively via a diazotiation reaction followed by tin (II) chloride reduction. The resulting hydrazine was reacted with trifluoroacetoacetate in boiling ethanol to yield the pyrazolone 8, which was then reacted with various aromatic aldehydes (R1CHO)7a in boiling water to yield the desired analogs bearing different benzylidene tethers.
Scheme 1. Synthesis of R1 modified pyrazolone compounds.

Reagents and conditions: i) NaNO2, HCl, 0°C, then SnCl2.2H2O; ii) Ethyl trifluoroacetoacetate, EtOH, reflux; iii) R1CHO, H2O, reflux
These compounds were subjected to in vitro testing, and the results (Table 1) showed that compound 4g was the most potent compound in this R1 modified library with an IC50 of 11±1 μM for inhibition on strand transfer activity.
Table 1.
HIV-1 Integrase Inhibitory Activity of Compound 4 Analogs
| Compds | R1 | 3′-Processing IC50 (μM) | Strand Transfer IC50 (μM) |
|---|---|---|---|
| 4a |  |
89±10 | 34±2 |
| 4b |  |
>100 | >100 |
| 4c |  |
61±7 | 15±3 |
| 4d |  |
50±8 | 30±7 |
| 4e | 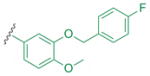 |
83±5 | 40±9 |
| 4f |  |
>100 | 22±3 |
| 4g |  |
21±1 | 11±1 |
Since the second type of alteration is crucial for the metal chelation ability of pyrazolone compounds, the choice of starting hydrazines should be important. Cyclization of various hydrazines 9a-9o with ethyl trifluoroacetoacetate in boiling ethanol yielded the corresponding pyrazolone intermediates 10a-10o, which were then treated with substituted benzaldehyde 11 to generate the R2 modified pyrazolone analogs (Scheme 2). Unfortunately, several attempts to make pyrazolones with an ortho-halophenyl R2 substituent (Cl and Br) were not successful due to decomposition of the intermediates when refluxing in boiling water. Also, the syntheses of pyrazolones with heterocyclic R2 groups (such as pyridine, pyrimidine, and tetrazole) were not as straightforward as those of their phenyl substituted pyrazolone counterparts. The last condensation step to synthesize these heterocyclic pyrazolones failed to generate the desired products even after prolonged reaction time.
Scheme 2. Synthesis of R2 Modified Pyrazolone Compounds.

Reagents and conditions: i) ethyl trifluroacetoacetate, EtOH, reflux; ii) 3-(4-fluorobenzyloxy)-4-methoxybenzaldehyde (11), H2O, reflux
The in vitro results of R2 modified pyrazolones are summarized in Table 2. The presence of larger substituents (−Ph and −I) was more favored than the smaller substituents (−F and −CF3). Meta substitution was slightly more preferred to the para substitution. The best compound in this group was 5j, which has IC50 value of 12±1 μM for the strand transfer reaction.
Table 2.
HIV-1 Integrase Inhibitory Activity of Compound 5 Analogs
| Compds | R2 | 3′-Processing IC50 (μM) | Strand Transfer IC50 (μM) |
|---|---|---|---|
| 5a | phenyl | >100 | >100 |
| 5b | m-tolyl | >100 | >100 |
| 5c | m-trifluoromethylphenyl | 90±13 | 24±4 |
| 5d | m-phenylphenyl | >100 | 16±5 |
| 5e | m-nitrophenyl | 52±13 | 19±2 |
| 5f | m-fluorophenyl | >100 | 60±0.1 |
| 5g | m-chlorophenyl | 88±24 | 30±6 |
| 5h (=4e) | m-bromophenyl | 83±5 | 40±9 |
| 5i | m-iodophenyl | >100 | 35±3 |
| 5j | 3,5-dichlorophenyl | >100 | 12±1 |
| 5k | p-bromophenyl | 82±1 | 25±6 |
| 56l | p-iodophenyl | >100 | 69±9 |
| 5m | p-carboxylphenyl | >100 | 41±12 |
| 5n | p-sulfonyamidophenyl | >100 | >100 |
| 5o | benzothiazolyl | >100 | 27±6 |
The role of the last modification is to investigate the effect of the 5-substituent of the pyrazolone ring on IN inhibition. Table 3 lists many analogs bearing different R3 substituents on the ring. We predicted that variation of the electronic nature of the pyrazolone through electron donating (NHC(=O)CF3), electron withdrawing (CF3, CO2H, CO2Et), or weakly interacting substituents (Me and Et) may assist in determination of the electronic factor necessary for binding toward the metal inside the IN active site. In general, synthesis of this analog was accomplished by cyclization of 12 with a variety of ethyl acetate derivatives to generate intermediates 13, followed by sequential condensation to form pyrazolones 6. The compounds 6a-6c which contain different aliphatic carbon R3 groups (e.g. Me, Et, and CF3) were synthesized from the reaction of mbromophenyl hydrazine with methyl acetoacetate, methyl propionoacetate, and methyl trifluoroacetoacetate, respectively, followed by subsequent condensation with 11. For the synthesis of 6d, 12 was condensed with ethyl cyanoacetate7b, acylated with trifluoroacetic anhydride to generate 13d, and then condensed further with 11 to give 6d. The similar twostep protocol was carried out for the synthesis of 6e-6h utilizing but-2-ynedioic acid diethyl ester and dimethylacetonedicarboxylate7c as depicted in Scheme 3 below.
Table 3.
HIV-1 Integrase Inhibitory Activity of Compound 6 Analogs
| Compds | R3 | 3′-Processing IC50 (μM) | Strand Transfer IC50 (μM) |
|---|---|---|---|
| 6a | methyl | >100 | >100 |
| 6b | ethyl | >100 | >100 |
| 6c (=5h) | trifluoromethyl | 83±5 | 40±9 |
| 6d | trifluoroacetamide | >100 | >100 |
| 6e | methyl homoacetate | >100 | 100 |
| 6f | homoacetate | >100 | 57 |
| 6g | ethyl carboxylate | >100 | 88±11 |
| 6h | carboxylate | 55±5 | 19±3 |
Scheme 3. Synthesis of R3 Modified Pyrazolone Compounds.

Reagents and conditions: i) (for 12a-12c), methyl acetoacetate, methyl propionacetate, or methyl trifluoroacetoacetate in boiling EtOH; (for 12d), methyl cyanoacetoacetate in EtOH, then trifluoroacetic anhydride; (for 12e), dimethylacetone-1,3-dicarboxylate in AcOH, 100°C; (for 12g), diethyl acetylenedicarboxylate, K2CO3 in EtOH; ii) 3-(4-fluorobenzyloxy)-4-methoxybenzaldehyde (11), H2O, reflux; (for 4f and 4h) LiOH, MeOH/THF/H2O
The presence of electron donating or electron neutral group did not increase the inhibitory activity against the IN. However, the presence of electron withdrawing group such as CF3 and CO2H increased the inhibitory activity. Compound 6h exhibited the most potent analog with ST IC50 of 19±3 μM.
To conclude the optimization study, we combined the best features obtained from the prior optimization studies. The combination of these features for each modification effort yielded a second prototype compound 14a, which after bioassay did not give a better inhibitory activity (ST IC50 of 11μM). Puzzled with this result, we elongated the tether of the second benzyl group as in compound 14b. Finally, switching the position of the fluorobenzyl group provided compound 14d which has the lowest ST IC50 so far (3±0.4μM).
Utilizing Autodock Vina8 to validate this experimental finding (Table 4), we began checking the lowest energy conformations of compounds 14a-14d. Compound 14d was the only compound with the anticipated conformation, where the carbonyl in the pyrazolone core was aligned closely to metal cofactor, so it would disrupt the IN activity toward cleaving DNA strand and/or transferring its viral DNA by sequestering the active site of IN (Figure 3a). On the other hand, the rest of optimized compounds (14a-14c) interacted with metal ion through their carboxylate moieties. We postulated that the absence of the methyl on the benzylidene ring gave rise to extra stabilization possibly through hydrogen bonding donor interaction with His67, while the presence of the carboxylate functional group also serves as a hydrogen bond acceptor from Asn155 as shown in Figure 3b. The nitro substituent gave an extra stabilization through hydrogen bond acceptor with Gln148.
Table 4.
HIV-1 Integrase Inhibitory Activity of Second Round Optimized Compounds
| Compds | R1 | 3′-Processing IC50 (μM) | Strand Transfer IC50 (μM) | |
|---|---|---|---|---|
| 14a |  |
 |
15 | 11 |
| 14b |  |
60±14 | 14±4 | |
| 14c |  |
95±7 | 10±1 | |
| 14d |  |
21±8 | 3±0.4 |
Figure 3.

(A) Predicted binding conformations of compound 14d inside the HIV-1 integrase active site (modified PDB1QS4). The lowest energy conformation is represented as a solid stick representation, while the rests are the other possible conformation. The docking analyses were performed around the green sphere which represents the metal active site. (B) Predicted protein-ligand interaction between compound 14d with HIV-integrase. Three residues (H67, N155, and Q148) show plausible interactions with this docked compound.
In conclusion, we have exploited SAR studies of pyrazolone scaffold for IN which was discovered through pharmacophore models. The compound 14d exhibited single-digit micromolar activity against IN strand transfer process. This work is expected to provide helpful information for the discovery of novel IN inhibitor.
Acknowledgments
The authors thank General Clinical Research Center Grant (M01-RR000043) and the National Institute of Health (S10 RR025432) for generous financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) WHO website. ( http://www.who.int/hiv/en/); (b) 2006 Report on the Global AIDS Epidemic. UNAIDS; Geneva, Switzerland: 2006. [Google Scholar]
- 2.(a) Hazuda DJ, Felock PJ, Hastings JC, Praminik B, Wolfe AL. J Virol. 1997;71:7005–7011. doi: 10.1128/jvi.71.9.7005-7011.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Marchand C, Krajewski K, Lee HF, Antony S, Johnson AA, Amin R, Roller P, Kvaratskhelia M, Pommier Y. Nucleic Acid Res. 2006;34:5157–5165. doi: 10.1093/nar/gkl667. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Semenova E, Marchand C, Pommier Y. Adv Pharmacol. 2008;56:199–228. doi: 10.1016/S1054-3589(07)56007-2. [DOI] [PubMed] [Google Scholar]
- 3.(a) Markowitz M, Morales-Ramirez JO, Nguyen BY, Kovacs CM, Steigbigel RT, Cooper DA, Liporace R, Schwartz R, Isaacs R, Gilde LR, Wenning L, Zhao J, Teppler H. J Acquir Immune Defic Syndr. 2006;43:509–515. doi: 10.1097/QAI.0b013e31802b4956. [DOI] [PubMed] [Google Scholar]; (b) Grinsztein B, Nguyen BY, Katlama C, Getell JM, Lazzatin A, Vittecoq D, Gonzalez CJ, Chen J, Harvey CM, Isaacs RD. Lancet. 2007;369:1261–1269. doi: 10.1016/S0140-6736(07)60597-2. [DOI] [PubMed] [Google Scholar]
- 4.(a) Sato M, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, Kawakami H, Matsuzaki Y, Watanabe W, Yamataka K, Ikeda S, Kodama E, Matsuoka M, Shinkai H. J Med Chem. 2006;49:1506–1508. doi: 10.1021/jm0600139. [DOI] [PubMed] [Google Scholar]; (b) DeJesus E, Berger D, Markowitz M, Cohen C, Kawkins T, Ruene P, Elion R, Farthing C, Zhong L, Cheng AK, McColl D, Kearney BP. J Acquir Immune Defic Synd. 2006;43:1. doi: 10.1097/01.qai.0000233308.82860.2f. [DOI] [PubMed] [Google Scholar]; (c) Sato M, Kawakami H, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, Matsuzaki Y, Yamataka K, Ikeda S, Shinkai H. J Med Chem. 2009;52:4869–4882. doi: 10.1021/jm900460z. [DOI] [PubMed] [Google Scholar]
- 5.Jones G, Ledford R, Yu F, Miller M, Tsiang M, McColl D. 14th Conference on Retroviruses and Opportunistic Infections, Los Angeles; Feb 25–28, 2007; Los Angeles, USA. [Google Scholar]
- 6.Dayam R, Al-Mawsawi LQ, Zawahir Z, Vitvrouw M, Debyser Z, Neamati N. J Med Chem. 2008;51:1136–1144. doi: 10.1021/jm070609b. [DOI] [PubMed] [Google Scholar]
- 7.(a) Abass M, Othman ES. Synth Commun. 2001:3361–3376. [Google Scholar]; (b) Weissberger A, Porter HD. Org Synth. 1948:87–88. [Google Scholar]; (c) Bevk D, Jakse R, Svete J, Golobic A, Golic L, Stanovnik B. Heterocycles. 2003:197–223. [Google Scholar]
- 8.Trott O, Olson AJ. J Comp Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]