

Abstract
We took a discovery approach to explore the actions of cAMP and two of its analogs, one a cAMP mimic ((Sp)-adenosine cyclic 3′:5′-monophosphorothioate ((Sp)-cAMPS)) and the other a diastereoisomeric antagonist ((Rp)-cAMPS), on a model system of the type Iα cyclic AMP-dependent protein kinase holoenzyme, RIα(91–244)·C-subunit, by using fluorescence spectroscopy and amide H/2H exchange mass spectrometry. Specifically, for the fluorescence experiments, fluorescein maleimide was conjugated to three cysteine single residue substitution mutants, R92C, T104C, and R239C, of RIα(91–244), and the effects of cAMP, (Sp)-cAMPS, and (Rp)-cAMPS on the kinetics of R-C binding and the time-resolved anisotropy of the reporter group at each conjugation site were measured. For the amide exchange experiments, ESI-TOF mass spectrometry with pepsin proteolytic fragmentation was used to assess the effects of (Rp)-cAMPS on amide exchange of the RIα(91–244)·C-subunit complex. We found that cAMP and its mimic perturbed at least parts of the C-subunit interaction Sites 2 and 3 but probably not Site 1 via reduced interactions of the linker region and αC of RIα(91–244). Surprisingly, (Rp)-cAMPS not only increased the affinity of RIα(91–244) toward the C-subunit by 5-fold but also produced long range effects that propagated through both the C- and R-subunits to produce limited unfolding and/or enhanced conformational flexibility. This combination of effects is consistent with (Rp)-cAMPS acting by enhancing the internal entropy of the R·C complex. Finally, the (Rp)-cAMPS-induced increase in affinity of RIα(91–244) toward the C-subunit indicates that (Rp)-cAMPS is better described as an inverse agonist because it decreases the fractional dissociation of the cyclic AMP-dependent protein kinase holoenzyme and in turn its basal activity.
Cyclic AMP-dependent protein kinase (PKA)1 plays a crucial role in a plethora of cellular functions. All isoforms of PKA are composed of two catalytic (C) subunits and homodimeric regulatory (R) subunits (1–3). As the name implies, cAMP is a major PKA regulator (4). Much progress has been made in the last decade in delineating the molecular basis of action of cAMP. An important tactic in this endeavor has been through the comparison of the effects of cAMP with those of two phosphorothioate cAMP analogs: (Sp)-cAMPS (a cAMP mimic) and (Rp)-cAMPS (an antagonist and a diastereoisomer of (Sp)-cAMPS). Although the importance of geometry of the sulfur substitution is critical in determining the pharmacological properties of the two phosphorothioate cAMP analogs, the molecular basis for this behavior is not fully understood. To date, these comparisons have only been made using either wild-type or truncated mutants of the type Iα regulatory subunit (RIα) that are free in solution, not complexed to the C-subunit. X-ray spectroscopic examination of ligand-bound RIα(92–379) complexes reveals few differences between ligand-bound complexes, but the (Rp)-cAMPS complex is structurally “looser” with higher thermal factors than complexes formed with either cAMP or (Sp)-cAMPS (5). This is consistent with the observation that both cAMP and (Sp)-cAMPS, but not (Rp)-cAMPS, raise the urea concentration required for wild-type RIα unfolding (6). Further insight into the structural basis of cAMP action stems from NMR spectroscopic comparison of the effects of (Rp)-cAMPS, cAMP, and (Sp)-cAMPS on chemical shifts and 15N relaxation of the RIα(119–244) mutant (7). In addition to producing fewer significant chemical shift changes than either cAMP or (Sp)-cAMPS, (Rp)-cAMPS binding is associated with enhanced millisecond to microsecond time scale backbone motions of a β-turn (β2,3 loop) and around the phosphate-binding cassette (PBC) (7).
Further insight into the molecular basis of actions of cAMP and its analogs should come from the analysis of ligand-bound R·C complexes. Unfortunately, the large size of even the heterodimeric R·C complex (∼95 kDa) and the difficulty of preparing (Rp)-cAMPS·R·C-subunit crystals currently preclude the use of both NMR spectroscopy and x-ray crystallography. Consequently, we took two alternative lower resolution approaches to this issue. One approach involves the use of site-directed labeling combined with fluorescence spectroscopy to examine both the effects of cAMP and its analogs on R-C subunit binding kinetics and on the conformational dynamics of RIα(91–244). RIα(91–244) includes the “A” cyclic nucleotide binding (CNB) domain, the pseudosubstrate, and linker domains and represents the minimal segments necessary for high affinity C-subunit binding (Fig. 1) (8). The other approach involves an examination of the effects of cAMP and its analogs on solvent exposure/conformational flexibility of RIα(91–244)·C-subunit complex using H/2H amide exchange measured with a combination of mass spectrometry (ESI-Q-TOF) and proteolytic fragmentation. In the first approach, fluorescein maleimide (FM) was conjugated to three cysteine substitution mutants with the substitution sites located near or within the pseudosubstrate sequence, the linker domain, or αC (R92C, T104C, and R239C, respectively) of RIα(91–244) (Fig. 1). The time-resolved fluorescence anisotropy results suggest that cAMP and (Sp)-cAMPS reduce the interaction of the RIα linker domain and αC with the two peripheral R-C interaction sites on the C-subunit (so-called Sites 2 and 3) without affecting the interaction of the pseudosubstrate sequence with the active site cleft (so-called Site 1). Because of limitations of the amide H/2H exchange experiments, only the effects of (Rp)-cAMPS on H/2H amide exchange in RIα(91–244)·C-subunit complex could be investigated. The results showed that (Rp)-cAMPS induces a relatively widespread increase in amide exchange, indicating limited unfolding and/or enhanced conformational flexibility that is propagated almost globally through the C-subunit and, at least, part of RIα. These conformational changes were accompanied by a 5-fold increase in the affinity of RIα(91–244) toward C-subunit, suggesting that, at least, some of the (Rp)-cAMPS effects are mediated by an increase in internal entropy. Finally, the (Rp)-cAMPS-induced increase in R-C affinity indicates that (Rp)-cAMPS is better described as an inverse agonist because the basal activity of the PKA holoenzyme should be decreased by (Rp)-cAMPS.
Fig. 1.

Overview of PKA structure and cAMP analogs. A, domain organization of RIα showing the domain boundaries of RIα(91–244) where the pseudosubstrate in green is connected to CNB-A domain in blue by a linker segment. B, structure of RIα(91–244) in the C-subunit-bound conformation (Protein Data Bank code 1U7E (23)) showing the pseudosubstrate in green, linker in yellow, and helical subdomain comprising helices αN, αA, αB, and αC in blue and β-subdomain in tan. The PBC is in red. C, structure of the C·RIα(91–244) holoenzyme showing the C-subunit in tan and RIα(91–244) in blue. Sites for introduction of cysteines by site-directed mutagenesis are represented by red circles. The cAMP binding site (PBC) is in red. D, structure of cAMP showing the 2′-OH group and 3′–5′ phosphodiester bond. The exocyclic oxygens upon replacement with sulfur atoms to generate the (Sp)-cAMPS and (Rp)-cAMPS diastereomers are highlighted.
EXPERIMENTAL PROCEDURES
Materials
ATP, cAMP, and MOPS were obtained from Sigma-Aldrich. (Sp)-cAMPS and (Rp)-cAMPS triethylamine were obtained from Biolog (Bremen, Germany). Nickel-nitrilotriacetic acid resin was obtained from Qiagen (Chatsworth, CA). Immobilized pepsin cartridges (Poroszyme) were obtained from Applied Biosystems (Redwood City, CA). Immobilized cobalt affinity columns (Talon, Clontech) were used for purification of hexahistidine-tagged PKA C-subunit following the manufacturer's specifications. Deuterium oxide (99.9% deuterium) was obtained from Cambridge Isotopes (Andover, MA). Trifluoroacetic acid (TFA) and acetonitrile were from Fisher Scientific. PD10 columns were from GE Healthcare. All other chemicals were at least reagent grade.
Preparation of RIα(91–244) Mutants and C-subunit
Three cysteine substitution mutants (R92C, T104C, and R239C) of RIα(91–244) subunit were prepared as described previously (9). Hexahistidine-tagged murine PKA Cα-subunit was expressed in Escherichia coli and purified as described previously (10). Isozyme I was used for all experiments. The holoenzyme of PKA was prepared as described earlier using 50 mm MOPS, pH 7.0, 1 mm DTT, 0.2 mm ATP, and 2 mm MgCl2 (9).
Fluorescein Maleimide Labeling
The R-subunit samples (12.5–25 nmol) were initially buffer-exchanged by elution through a G-25 column (1.5 × 7 cm) equilibrated with buffer (50 mm MOPS, 50 mm NaCl, pH 7.0). The protein fractions were pooled, and the concentrations of the pooled samples were determined with the Bradford assay (11). The protein concentrations of the reaction mixtures ranged between 3 and 6 μm. The reactions were allowed to proceed for 1 h at room temperature, protected from light, and then eluted through a PD10 column (2.5 × 6 cm) followed by a Sephacryl S-200 column (2.5 × 7 cm), both equilibrated with buffer A (50 mm MOPS, 50 mm NaCl, 1 mm DTT, pH 7.0) at room temperature. Fluorescein emission (excitation at 470 nm and emission at 525 nm) from the column fractions was measured, and the fluorescent fractions with retention times that corresponded to unmodified R-subunit were pooled. Aliquots of the pooled fractions were subjected to gel electrophoresis under denaturing conditions (12% SDS-PAGE), and the fluorescent bands were visualized with a mineral lamp to assess the presence of unconjugated fluorescein in the samples.
Phosphotransferase Assay
The activity of the labeled R mutants was evaluated by assessing their ability to inhibit the phosphotransferase activity of recombinant C-subunit using the method of Cook et al. (12) with Kemptide as a substrate.
Determination of Stoichiometry of Labeling
The stoichiometry of FM-labeled R was determined spectrophotometrically by substitution of the measured absorbance values at 497 nm (A497) and the protein concentration [R] determined with the Bradford assay (11) into the following expression.
Formation of RIα(91–244)·C Complexes
The labeled and unlabeled RIα mutants (1 μm) were combined with a 1.2-fold molar excess of recombinant C-subunit and then dialyzed (cutoff, 30 kDa) overnight at 4 °C against 3 × 1-liter changes in buffer B (50 mm MOPS, 50 mm NaCl, 1 mm DTT, 2 mm MgCl2, 0.2 mm ATP, pH 7.0). Free C- and R-subunits were separated from the RIα(91–244)·C complexes by elution through a Sephacryl S-200 column (1.5 × 25 cm) using buffer B.
Stopped-flow Kinetic Measurements
Stopped-flow experiments were performed with an Applied Photophysics SX.18MV (Leatherhead, UK) stopped-flow spectrofluorometer. The samples were excited at 460 nm, and an Omega interference filter (510DF23) was used to select the fluorescence signal. The second-order association rate constants for binding of C-subunit to the RIα(91–244) conjugate was determined from the slope of plots of the observed rate of fluorescence change versus final C-subunit concentration. The first-order rate constant of R·C dissociation was determined by mixing the preformed FM-R92C RIα(91–244) complexes with a 10-fold excess of unlabeled RIα(91–244) and fitting the tracings to a single exponential equation. All samples were prepared in buffer containing MgATP (50 mm MOPS, 50 mm NaCl, 1 mm DTT, 2 mm MgCl2, 0.2 mm ATP, pH 7.0). Equilibrium dissociation constants (Kd values) were determined as a ratio of the dissociation to association rate constants.
Time-resolved Fluorescence Anisotropy
Emission anisotropy was determined as described previously (13). Unless stated otherwise, emission anisotropy decay was analyzed with the impulse reconvolution method implemented in the DAS6TM software package from HORIBA Jobin Yvon IBH Ltd. (Glasgow, UK) described elsewhere (14). Briefly and simply, this approach splits the analysis into two steps: analysis of the total emission decay, S(t), from the vertically, I‖(t), and orthogonally, I⊥(t), polarized emission components followed by analysis of the vertical/perpendicular difference emission decay, D(t). S(t), free of anisotropy effects, is given by the expression
and is analyzed as a biexponential function. G is a measure of the instrumental polarization bias (0.995). D(t), which includes both fluorescence and anisotropy parameters, is given by the following expression.
D(t) is deconvolved with the results from the S(t) analysis as a constraint, yielding the following expression.
Here, β1 and β2 are the amplitudes of the anisotropy at time 0 for the fast and slow anisotropy decay processes, respectively. φfast and φslow are the fast and slow rotational correlation times of the anisotropy decay, respectively. φslow usually yields an estimate of the whole-body rotational correlation time when it is less than about 5 times the emission lifetime and when the rate of the segmental motions around the site of reporter group conjugation greatly differ from the rate of the whole-body diffusion. A nonassociative model was assumed where the emission relaxation times are common to all the rotational correlation times. Goodness of fit was evaluated from the values of the reduced χr2 and by visual inspection of the weighted residual plots. All time-resolved anisotropy measurements were performed with samples suspended in buffer B at 22 °C.
Preparation of RIα(91–244)·C-subunit Complexes for H/2H Exchange Analysis
RIα(91–244)·C-subunit complexes were concentrated to ∼150 μm using concentrators (Amicon, Millipore) (molecular mass cutoff, 10 kDa), and where appropriate, (Rp)-cAMPS was added to a concentration of 10 mm. This enabled the final concentration of (Rp)-cAMPS to be 1 mm under deuterium exchange conditions.
Amide Hydrogen-Deuterium Exchange ESI-Q-TOF Mass Spectrometry
The C·RIα(91–244) R92C protein complexes (109 μm) in the absence and presence of 1 mm (Rp)-cAMPS were allowed to exchange with deuterated buffer solutions by 10-fold dilutions of protein samples in deuterated 50 mm MOPS, 50 mm NaCl, 2 mm MgCl2, 0.2 mm ATP, 1 mm DTT, pHread = 7.0 (deuterated buffer B). The reactions were then quenched after timed intervals of 30 s, 1 min, 2 min, 5 min, and 10 min by placement on ice and the addition of 180 μl of 0.2% trifluoroacetic acid, pHread = 2.5. Undeuterated samples were included as a negative control. One sample was allowed to exchange with deuterated buffer B for 24 h to allow for complete deuteration of solvent-exposed regions of the protein. This was used to calculate back-exchange under our experimental conditions. 50 μl of the reaction samples were digested through an on-line Poroszyme immobilized pepsin cartridge (Applied Biosystems) before passing through an ACQUITY UPLC BEH C18 column. The reaction samples were then analyzed on a Waters Synapt high definition mass spectrometry (HDMS) system (Waters). Both the pepsin cartridge and C18 column were housed in a refrigerated module at 2 °C to minimize deuterium back-exchange during analysis (15, 16).
Peptide identification was achieved by Protein Lynx Global Server (PLGS) (v2.4) search software (Waters) using a database containing a primary sequence of PKA RIα(91–244) and C-subunits. Cleavage peptides by nonspecific proteases with a mass tolerance of 10 ppm were used to sequence the peptides. Continuous instrument calibration was carried out with Glu-fibrinogen peptide at 100 fmol/μl. Deuterium exchange quantitation was by HX-Express software (17). A control experiment was carried out to calculate the deuterium back-exchange loss during the experiment by incubating ligand-free RIα(91–244) with deuterated buffer A for 24 h at room temperature (20 °C). Even after extended deuteration, RIα(91–244) still showed some solvent-inaccessible and ordered regions that were not completely deuterated. For an accurate measurement of back-exchange loss, we therefore focused only on peptides from within highly solvent-exchangeable regions of the protein, identified as those regions that show greater relative exchange at shorter time points (10-min exchange). The region in RIα(91–244) spanning residues 111–130 is a highly solvent-exchangeable region (10), and all five overlapping peptides used for calculations of back-exchange span this region and showed nearly complete exchange in ligand-free RIα(91–244) following 10-min deuterium exchange and would represent fully deuterated samples following 24-h exchange. An example of the effects of deuteration following 24-h exchange on a fragment spanning residues 111–126 (m/z = 632.70) is illustrated in supplemental Fig. 9. The average deuteration back-exchange of 32.8 ± 1.6% was calculated from average back-exchange values for five peptides: RIα(111–123) (m/z = 547.65) (back-exchange, 34.4%), RIα(111–119) (m/z = 559.34) (back-exchange, 33.7%), RIα(111–126) (m/z = 632.701) (back-exchange, 30.4%), RIα(111–119) (m/z = 595.34) (back-exchange, 33.7%), and RIα(112–126) (m/z = 578.35) (back-exchange, 32.0%). All deuterium exchange values reported were corrected for a 32.7% back-exchange by multiplying the raw centroid values by a multiplication factor of 1.49.
RESULTS
Preparation, Labeling, and Characterization of RIα(91–244) Mutants
The three cysteine substitution mutants were overexpressed and purified to greater than 95% as assessed by visual inspection of Coomassie-stained SDS-PAGE gel and FPLC profiles. Mass spectrometry of each mutant was consistent with the expected fragmentation masses by peptide mass fingerprinting by MALDI-TOF analysis of the desired mutant (data not shown), and each mutant was as equally effective as RIα(91–244) at inhibiting the phosphotransferase activity of the wild-type C-subunit (data not shown) (9). Denaturing SDS-PAGE analysis of the fluorescein conjugates showed no detectable nonconjugated probe, and the stoichiometry of fluorescein conjugation was ∼28, ∼24, and ∼40% for the FM-R92C, FM-T104C, and FM-R239C RIα(91–244) conjugates, respectively.
Effects of cAMP, (Rp)-cAMPS, and (Sp)-cAMPS on Kinetics of C-subunit Binding to RIα(91–244)
To gain a better understanding of the effects of (Rp)-cAMPS on R-C interactions and to determine the reactant concentrations required to maintain R- and C-subunits in a heterodimeric state in the presence of cAMP or (Sp)-cAMPS, the association and dissociation rate constants for R-C binding were determined in the presence and absence of cAMP, (Rp)-cAMPS, and (Sp)-cAMPS. Because FM-R92C conjugate binding to C-subunit is associated with a relatively large and unambiguous change in fluorescence (∼40% decrease) compared with the other conjugated mutants (9), the FM-R92C conjugate was utilized to assess the effects of cAMP and two phosphorothioate analogs on the kinetics of C-subunit binding. Representative stopped-flow tracings associated with the rapid mixing of the FM-R92C conjugate with various concentrations of C-subunit in the absence and presence of cAMP (10 μm), (Rp)-cAMPS (100 μm), and (Sp)-cAMPS (100 μm) are illustrated in Fig. 2. The fluorescence tracings were well fit to a single exponential equation, and the bimolecular association rate constants were determined from the slopes of the plots of the observed rates for fluorescence change versus final concentration of C-subunit. The values of the association rate constants were essentially the same, ranging between 1.7 and 2.3 × 107 m−1 s−1 (Table I), indicating that the binding of cAMP and its analogs does not affect the association process. The effects of cAMP and its analogs on the time course of dissociation of the FM-R92C·C-subunit complex on the other hand were analog-dependent. Fig. 3, A and B, illustrate representative stopped-flow tracings associated with the rapid mixing of preformed FM-R92C·C-subunit complexes (100 nm) with excess unlabeled RIα(91–244) (1 μm) in the absence and presence of cAMP (10 μm), (Rp)-cAMPS (100 μm), or (Sp)-cAMPS (100 μm). Fitting the experimental tracings to a single exponential equation yielded a unimolecular dissociation rate constant for the control sample of 3.8 × 10−3 s−1, essentially identical to what we observed previously (9). As expected, the presence of cAMP or (Sp)-cAMPS was associated with an increase in the dissociation rate constant (150–200-fold; Table I). The presence of (Rp)-cAMPS was associated with an unexpected result. Because (Rp)-cAMPS is generally regarded as a competitive antagonist, we expected it to have no significant effect on FM-R92C·C-subunit dissociation, but surprisingly, (Rp)-cAMPS slowed the dissociation rate by 5-fold, increasing the affinity of FM-R92C for C-subunit by an equal amount (Table I).
Fig. 2.
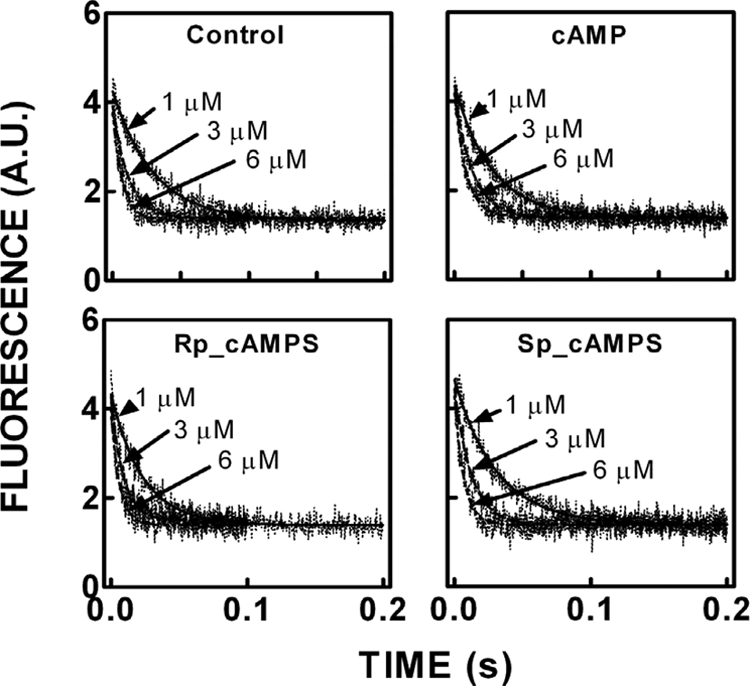
Effects of cAMP, (Rp)-cAMPS, and (Sp)-cAMPS on the stopped-flow kinetics of FM-R92C RIα(91–244) binding to the C-subunit. Top left, control. Top right, in the presence of 10 μm cAMP. Bottom left, in the presence of 100 μm (Rp)-cAMPS. Bottom right, in the presence of 100 μm (Sp)-cAMPS. Stopped-flow traces of FM-R92C-RIα(91–244) rapidly mixed with C-subunit in the absence of cAMP are shown. The final concentration of FM-RIα(91–244) R92C was 100 nm, and the concentration of the C-subunit was varied from 1 to 6 μm. The decreasing emission follows the time course of binding. The solid lines denote the best fit of the data to a single exponential equation. Samples were suspended in a buffer containing 50 mm MOPS, pH 7.0, 50 mm NaCl, 1 mm DTT, 2 mm MgCl2, and 0.2 mm ATP at 22 °C. A.U., absorbance units.
Table I. Effect of cAMP, (Sp)-cAMPS, and (Rp)-cAMPS on association and dissociation rate constants for C-subunit binding to FM-R92C RIα(91–244).
| k1a | k−1b | Kdc | |
|---|---|---|---|
| m−1s−1 | s−1 | m | |
| Control | 2.0 ± 012 × 107 | 3.8 ± 0.1 × 10−3 | 1.9 × 10−10 |
| cAMP | 1.7 ± 0.11 × 107 | 0.55 ± 0.05 | 3.2 × 10−8 |
| (Rp)-cAMPS | 1.9 ± 0.17 × 107 | 7.5 ± 0.3 × 10−4 | 4.0 × 10−11 |
| (Sp)-cAMPS | 1.9 ± 0.16 × 107 | 0.75 ± 0.02 | 4.0 × 10−8 |
a Bimolecular association rate constants determined from rapid mixing of FM-labeled RIα(91–244) mutants (100 nm) with various concentrations of C-subunit in the absence and presence of cAMP (10 μm), (Rp)-cAMPS (100 μm), or (Sp)-cAMPS (100 μm) under pseudo first-order conditions (n = 4).
b Unimolecular dissociation rate constants determined by rapidly mixing excess unlabeled RIα(91–244) with preformed RIα(91–244)·C complex (100 nm) of FM-labeled RIα(91–244) conjugates and C-subunit in the absence and presence of cAMP (10 μm), (Rp)-cAMPS (100 μm), or (Sp)-cAMPS (100 μm) (n = 3).
c Equilibrium dissociation constants determined by dividing the unimolecular dissociation by the bimolecular association rate constants.
Fig. 3.

Effects of cAMP, (Rp)-cAMPS, and (Sp)-cAMPS on stopped-flow kinetics of FM-R92C RIα(91–244) dissociating from C-subunit. A, control and in the presence of 100 μm (Rp)-cAMPS. B, in the presence of 10 μm cAMP and 100 μm (Sp)-cAMPS. Stopped-flow traces of FM-R92C-RIα(91–244)·C-subunit complexes rapidly mixed with excess unlabeled RIα(91–244) are shown. The final concentration of FM-RIα(91–244) R92C was 100 nm, and the concentration of the unlabeled RIα(91–244) was 1 μm. The increasing emission follows the time course of dissociation of the labeled RIα(91–244). The solid lines denote the best fit of the data to a single exponential equation. Please see the legend to Fig. 2 for additional details. A.U., absorbance units.
Anisotropy Decay of FM Conjugates Free and Bound to C-subunit
To lay a foundation for analyzing the effects of cAMP and its analogs on solution structure and dynamics of C-subunit-bound RIα(91–244), the time-resolved anisotropy of FM conjugated to substituted cysteines at residues Arg92, Thr104, and Arg239 was studied. These residues were chosen to be near but not part of the C-subunit binding surface (Fig. 1). Representative anisotropy decays of the unliganded conjugated mutants free in solution and complexed to the C-subunit are illustrated in Fig. 4, and the fitting parameters are summarized in Table II. Although the interpretation of anisotropy decay is frequently problematic, in the present case, the results are largely consistent with crystallographic and NMR structural results. The typical anisotropy decay from a reporter group conjugated to a side chain of a globular protein fits reasonably well to a biexponential equation with the time zero anisotropy (β1 + β2) at a value less than the fundamental anisotropy for the reporter group due to rapid, unresolvable depolarization of torsional tether arm motion. (The fundamental anisotropy of FM equals 0.34.) The slowest rotational correlation time (φslow) is usually attributable to global whole-body rotational diffusion. The amplitude of this slow depolarization process (β2) is a complex function of the maximum angular excursion of diffusion in a cone associated with both tether arm and α-carbonyl backbone motion adjacent to the site of conjugation and is inversely related to the maximum excursion angle of the reporter group (18, 19). The faster correlation time (φfast) is a complex function of both diffusion rate and the maximum angular excursion (assuming diffusion in a cone) of primarily α-carbonyl backbone diffusion around the site of conjugation. Unresolved anisotropy decay, i.e. the difference between the fundamental and time zero anisotropy, most likely reflects subnanosecond tether arm torsional motions.
Fig. 4.

Time-resolved anisotropy decay of FM-R92C, FM-T104C, and FM-R239C RIα(91–244)·C-subunit free in solution (A) and complexed to C-subunit (B). The smooth line through each decay plot was generated with best fit parameters for a biexponential function (Equation 4). Measurements were made with the samples suspended in 50 mm MOPS, pH 7.0 with 50 mm NaCl, 1 mm DTT, 0.2 mm ATP, and 2 mm MgCl2 at 22 °C. IRF, instrument response function.
Table II. Time-resolved anisotropy decay parameters of FM-R92C, FM-T104C, and FM-R239C RIα(91–244) mutants free and complexed to the C-subunit in absence and presence of cAMP, (Rp)-cAMPS, or (Sp)-cAMPS.
The concentrations of the FM-RIα(91–244)conjugates, C-subunit, and cyclic nucleotides were 0.4, 4, and 100 μm, respectively.
| Mutant state | Condition | β1a | β2b | φfastc | φslowd | χr2e | 〈τ〉 |
|---|---|---|---|---|---|---|---|
| FM-R92C | |||||||
| Holo | Control | 0.080 ± 0.002 | 0.176 ± 0.015 | 1.5 ± 0.1 | 38 | 1.6 | 2.6 ± 0.1 |
| Holo | cAMP | 0.082 ± 0.001 | 0.198 ± 0.015 | 1.5 ± 0.2 | 38 | 1.6 | 2.7 ± 0.1 |
| Holo | (Rp)-cAMPS | 0.078 ± 0.003 | 0.180 ± 0.010 | 1.6 ± 0.1 | 38 | 1.5 | 2.6 ± 0.1 |
| Holo | (Sp)-cAMPS | 0.076 ± 0.002 | 0.190 ± 0.007 | 1.7 ± 0.1 | 38 | 1.5 | 2.7 ± 0.1 |
| Apo | Control | 0.089 ± 0.011 | 0.130 ± 0.007 | 0.9 ± 0.1 | 7.6 ± 1.1 | 1.6 | 3.6 ± 0.1 |
| Apo | cAMP | 0.087 ± 0.016 | 0.132 ± 0.001 | 0.9 ± 0.1 | 7.5 ± 1.8 | 1.5 | 3.6 ± 0.1 |
| Apo | (Rp)-cAMPS | 0.088 ± 0.005 | 0.133 ± 0.005 | 0.9 ± 0.1 | 7.1 ± 0.4 | 1.4 | 3.6 ± 0.1 |
| Apo | (Sp)-cAMPS | 0.081 ± 0.002 | 0.132 ± 0.005 | 0.9 ± 0.1 | 7.1 ± 0.1 | 1.9 | 3.6 ± 0.1 |
| FM-T104C | |||||||
| Holo | Control | 0.040 ± 0.002 | 0.241 ± 0.014 | 3.0 ± 0.6 | 38 | 1.4 | 3.5 ± 0.1 |
| Holo | cAMP | 0.031 ± 0.004 | 0.199 ± 0.018 | 3.8 ± 0.3 | 38 | 1.5 | 3.7 ± 0.3 |
| Holo | (Rp)-cAMPS | 0.041 ± 0.004 | 0.237 ± 0.013 | 2.9 ± 0.3 | 38 | 1.5 | 3.5 ± 0.1 |
| Holo | (Sp)-cAMPS | 0.047 ± 0.002 | 0.194 ± 0.016 | 3.2 ± 0.2 | 38 | 1.6 | 3.5 ± 0.1 |
| Apo | Control | 0.096 ± 0.010 | 0.154 ± 0.004 | 2.1 ± 0.1 | 11.0 ± 2.2 | 1.4 | 3.6 ± 0.3 |
| Apo | cAMP | 0.092 ± 0.019 | 0.165 ± 0.007 | 2.1 ± 0.3 | 10.7 ± 3.2 | 1.5 | 3.7 ± 0.2 |
| Apo | (Rp)-cAMPS | 0.091 ± 0.006 | 0.153 ± 0.017 | 1.9 ± 0.1 | 9.8 ± 1.0 | 1.4 | 3.6 ± 0.1 |
| Apo | (Sp)-cAMPS | 0.097 ± 0.009 | 0.152 ± 0.008 | 2.1 ± 0.1 | 10.8 ± 2.2 | 1.5 | 3.6 ± 0.1 |
| FM-R239C | |||||||
| Holo | Control | 0.052 ± 0.003 | 0.215 ± 0.006 | 1.7 ± 0.1 | 38 | 1.2 | 3.6 ± 0.1 |
| Holo | cAMP | 0.057 ± 0.004 | 0.203 ± 0.013 | 2.3 ± 0.2 | 38 | 1.2 | 3.5 ± 0.1 |
| Holo | (Rp)-cAMPS | 0.053 ± 0.002 | 0.197 ± 0.010 | 1.7 ± 0.2 | 38 | 1.2 | 3.6 ± 0.1 |
| Holo | (Sp)-cAMPS | 0.054 ± 0.002 | 0.196 ± 0.008 | 2.3 ± 0.3 | 38 | 1.2 | 3.5 ± 0.1 |
| Apo | Control | 0.081 ± 0.001 | 0.173 ± 0.008 | 2.4 ± 0.3 | 13.1 ± 1.2 | 1.5 | 3.3 ± 0.3 |
| Apo | cAMP | 0.068 ± 0.002 | 0.175 ± 0.004 | 1.6 ± 0.1 | 10.2 ± 0.6 | 1.4 | 3.3 ± 0.2 |
| Apo | (Rp)-cAMPS | 0.073 ± 0.011 | 0.167 ± 0.004 | 2.0 ± 0.2 | 11.0 ± 2.5 | 1.4 | 3.3 ± 0.1 |
| Apo | (Sp)-cAMPS | 0.065 ± 0.005 | 0.168 ± .006 | 1.7 ± 0.1 | 9.4 ± 0.7 | 1.5 | 3.3 ± 0.1 |
a Amplitude of the fast anisotropy decay processes.
b Amplitude of the slow anisotropy decay processes.
c Rotational correlation time of fast anisotropy decay processes.
d Rotational correlation time of slow anisotropy decay processes.
e Reduced χr2 of the anisotropy decay analysis.
For the FM-conjugated mutants free in solution, the slow rotational correlation times ranged between 7.6 and 13.0 ns, which is about what is predicted from the Stokes-Einstein equation for a 15-kDa sphere (5.9 ns) or that estimated by the Hydropro computer program (version 7.c) (20) (10.9 ns) using the atomic coordinates of the x-ray structure of RIα(91–244). The amplitudes of the slow depolarization processes (β2) were in the rank order R92C < T104C < R239C and ranged between 0.130 and 0.173. This rank order is consistent with what would be expected from visual inspection of the crystal structure of RIα(91–244) with the unstructured N-terminal R92C position being the most unhindered followed by the T104C at 17 residues N-terminal from the structured αN (starting at Lys121) followed by residue R239C near the C terminus of αC. The fast rotational correlation times are similarly ranked order with the R92C having the fastest correlation times (0.9 ns) followed by the T104C (2.1 ns) and R239C (2.4 ns). Thus, alone and free in solution the area around the N-terminal R92C residue is the most flexible of the FM conjugates, near the C-terminal R239C residue is the least flexible, and a few residues before the αN have intermediate flexibility.
For the C-subunit-bound conjugates, parameter fitting was performed with the values of the slow correlation times (φslow) fixed at 38 ns, the value predicted by the Hydropro computer program (version 7.c) (20) using the atomic coordinates of the RIα(91–244)·C-subunit complex (Protein Data Bank code 1U7E). Fixing φslow was performed to reduce the variability caused by the short lifetime of the reporter groups relative to the whole-body rotational correlation time (Table II). The fitting parameters generated by this procedure are summarized in Table II.
For all three conjugates, C-subunit binding was associated with relatively large increases in the amplitudes of the slower depolarization processes (β2), which is consistent with a reduced range (angular excursion) of local (tether arm and/or adjacent α-carbonyl backbone) motions of the reporter groups. Specifically, C-subunit binding increased β2 from 0.130 to 0.176 for FM-R92C, 0.154 to 0.241 for FM-T104C, and 0.173 to 0.215 for FM-R239C (Table II). The effects of C-subunit binding on the amplitudes (β1) and rates (φfast) of the fast decay processes were more complex. For the FM-R92C conjugate, C-subunit binding decreased the fast correlation time (φfast) from 1.5 to 0.9 ns with no change in β1 but an increase in the time 0 anisotropy (β1 + β2) from 0.219 to 0.259 (Table II), which is consistent with a restriction of subnanosecond local tether arm motions and faster local α-carbonyl backbone motions. For the FM-T104C conjugate, C-subunit binding increased φfast from 2.1 to 3.0 ns, decreased β1 from 0.096 to 0.040, and increased time zero anisotropy from 0.251 to 0.281 (Table II), which is consistent with reduced motions of both α-carbonyl backbone and tether arm motions. For the FM-R239C conjugate, C-subunit binding decreased φfast from 2.4 to 1.7 ns and decreased β1 from 0.081 to 0.052 without changing significantly the time zero anisotropy (Table II), suggesting a decrease in the range but not rate of adjacent α-carbonyl backbone flexibility without constraining tether arm motions.
Effects of cAMP, (Rp)-cAMPS, and (Sp)-cAMPS on Time-resolved Anisotropy Decay of FM-RIα(91–244) Conjugates
In the absence of C-subunit, the only ligand effects observed were with the reporter group attached to residue 239 near the end of αC. Here, cAMP and (Sp)-cAMPS (and perhaps (Rp)-cAMPS as well) reduced the “fast” rotational correlation time (φfast) from 2.4 to 1.6–1.7 ns and reduced the amplitude of the fast decay processes (β1) (from 0.081 to 0.65–0.68) (Table II). This would be consistent with an increased interaction of the end of αC with the PBC and the protein core, which would reduce the range of motions. (Note that φfast is a complex function of both the range of motions and rate of diffusion, so a decrease in the range of motions will produce a decrease in φfast.)
In studies of ligand effects on C-subunit-bound FM-RIα(91–244) conjugates, C-subunit was added in 10-fold excess of the FM-RIα(91–244) conjugates (0.4 μm) to minimize cAMP- or (Sp)-cAMP-induced dissociation. Under these conditions, the concentration of C-subunit was at least 50-fold greater than the Kd for R-C binding in the presence of either cAMP or (Sp)-cAMPS (Table I), so no more than 1% dissociation would be expected to occur upon addition of either cAMP or (Sp)-cAMPS. For C-subunit-bound FM-conjugates, ligand-dependent effects were observed with the FM-T104C and the FM-R239C conjugates and here only with added cAMP and (Sp)-cAMPS. Specifically, for the reporter group attached to residue 104 near the C-terminal side of the linker domain, cAMP and (Sp)-cAMPS but not (Rp)-cAMPS reduced the amplitude of the slow decay processes (β2) from 0.241 to 0.194–0.199, which was associated with a reduction of the time zero anisotropy from 0.280 to 0.230–0.240 without a significant change in φfast (Table II). This effect would be consistent with enhanced angular excursions of the local motions without significant changes in the rate of diffusion. For the FM-R239C conjugate, cAMP and (Sp)-cAMPS but not (Rp)-cAMPS increased the fast rotational correlation time (φfast) from 1.7 to 2.3 ns without affecting the decay amplitudes (Table II). This effect would be consistent with enhanced constraints on the motions of the end of αC due to increased interactions with the RIα(91–244) core and/or C-subunit; the former is more likely. Together, these results suggest that cAMP and (Sp)-cAMPS decrease the interaction of the linker domain and αC of RIα with the C-subunit without affecting the interaction of the pseudosubstrate sequence with the catalytic cleft.
Effects of (Rp)-cAMPS on H/2H Exchange in RIα(91–244)·C-subunit Complexes
The actions of cAMP and the two phosphorothioate analogs on the structure and dynamics of PKA were investigated further by amide H/2H exchange analysis measured by ESI-Q-TOF mass spectrometry in combination with proteolytic fragmentation by pepsin as described under “Experimental Procedures.” In total, 21 proteolytic fragments from RIα(91–244) that covered 84% of its primary sequence and 50 proteolytic fragments from the C-subunit that covered 85% of its primary sequence were identified. The extent of deuteration of the fragments from RIα(91–244) and the C-subunit within 5 min is summarized in Tables III and IV. An example of the effects of deuteration and (Rp)-cAMPS on a fragment spanning the PBC (residues 204–221, m/z = 664.3) is illustrated in Fig. 5.
Table III. (Rp)-cAMPS effects on amide H/2H exchange in RIα(91–244) complexed to the C-subunit.
Non-overlapping 95% confidence intervals relative to control values were determined by the PrizmTM computer program.
| Fragment of R-subunit (m/z) (region) | Number of exchangeable amides | Charge (z) | Maximum amides exchanged (mean ± S.E.)a |
|
|---|---|---|---|---|
| Control | Plus (Rp)-cAMPS | |||
| 102–106 (640.29) (linker) | 4 | 1 | 3.0 ± 0.01 | 2.9 ± 0.07 |
| 111–126 (632.70) (linker) | 14 | 3 | 8.7 ± 0.26 | 9.0 ± 0.34 |
| 112–119 (477.81) (linker) | 6 | 2 | 4.2 ± 0.09 | 4.6 ± 0.13 |
| 126–134 (493.29) (αXn) | 8 | 2 | 3.4 ± 0.05 | 3.6 ± 0.10 |
| 136–143 (976.40) (loop-αA) | 7 | 1 | 1.8 ± 0.12 | 2.4 ± 0.14 |
| 136–148 (797.86) (loop-αA) | 12 | 2 | 1.3 ± 0.08 | 1.7 ± 0.10 |
| 144–148 (637.34) (αA) | 4 | 1 | 0.3 ± 0.24 | 0.1 ± 0.02 |
| 149–156 (913.42) (αA) | 6 | 1 | 1.2 ± 0.03 | 1.2 ± 0.23 |
| 157–172 (846.89) (β2-β3) | 15 | 2 | 5.7 ± 0.18 | 4.8 ± 0.22 |
| 161–172 (1322.58) (β2-β3) | 11 | 1 | 4.8 ± 0.17 | 3.9 ± 0.21 |
| 162–172 (1225.54) (β2-β3) | 10 | 1 | 4.7 ± 0.16 | 3.9 ± 0.20 |
| 172–180 (1101.49) (β4) | 8 | 1 | 1.1 ± 0.02 | 1.1 ± 0.04 |
| 183–187 (638.28) (β4) | 4 | 1 | 2.0 ± 0.02 | 2.0 ± 0.04 |
| 188–198 (1097.49) (β5-β6) | 10 | 1 | 4.0 ± 0.10 | 4.0 ± 0.15 |
| 188–201(1396.64) (β5-β6-αB′) | 13 | 1 | 6.4 ± 0.22 | 4.5 ± 0.16 |
| 202–212 (567.32) (αB′) | 9 | 2 | 3.7 ± 0.14 | 1.9 ± 0.18 |
| 202–221 (705.76) (αB′-β7) | 18 | 3 | 4.1 ± 0.18 | 2.4 ± 0.14 |
| 204–221(644.3) (αB′-β7) | 16 | 3 | 4.0 ± 0.13 | 2.0 ± 0.14 |
| 213–221 (500.83) (β7) | 8 | 2 | 0.8 ± 0.01 | 1.0 ± 0.03 |
| 222–229 (506.23) (β8-αB) | 6 | 2 | 0.6 ± 0.05 | 0.6 ± 0.07 |
| 230–238 (523.81) (αB-αC) | 8 | 2 | 2.6 ± 0.05 | 2.6 ± 0.11 |
a Average number of deuterons exchanged determined from fitting plots of the time course of deuteration during a 10-min exposure to deuterium oxide to a single exponential equation. Values reported are the mean and S.E. of the amplitude term of fits, and results are from at least two independent experiments. Peptides showing significant differences upon (Rp)-cAMPS binding are in bold. All deuterium exchange values reported were corrected for a 32.7% back-exchange by multiplying the raw centroid values by a multiplication factor of 1.49.
Table IV. (Rp)-cAMPS effects on amide H/2H exchange in the C-subunit complexed to RIα(91–244).
Non-overlapping 95% confidence intervals relative to control values were determined by the Prizm computer program.
| Fragment of C-subunit (m/z) (region) | Number of exchangeable amides | Charge (z) | Maximum amides exchanged (mean ± S.E.)a |
|
|---|---|---|---|---|
| Control | Plus (Rp)-cAMPS | |||
| 7–18 (700.32) (αA) | 11 | 2 | 8.2 ± 0.05 | 9.7 ± 0.19 |
| 14–31 (732.72) (αA) | 17 | 3 | 4.3 ± 0.11 | 5.3 ± 0.11 |
| 19–26 (461.25) (αA) | 7 | 2 | 4.2 ± 0.10 | 5.3 ± 0.37 |
| 19–27 (534.79) (αA) | 8 | 2 | 4.4 ± 0.13 | 4.9 ± 0.18 |
| 21–36 (953.5) (αA-β1) | 14 | 2 | 8.4 ± 0.19 | 9.5 ± 0.22 |
| 27–40 (822.44) (αA-β1) | 12 | 2 | 7.7 ± 0.17 | 8.3 ± 0.20 |
| 28–40 (765.90) (αA-β1) | 11 | 2 | 7.6 ± 0.12 | 8.2 ± 0.19 |
| 42–55 (792.90) (β1) | 13 | 2 | 1.9 ± 0.07 | 2.0 ± 0.13 |
| 45–55 (597.83) (β1) | 10 | 2 | 3.0 ± 0.11 | 3.0 ± 0.14 |
| 60–71 (700.84) (β1-β2) | 11 | 2 | 0.4 ± 0.02 | 0.9 ± 0.04 |
| 83–100 (727.74) (αB-αC) | 17 | 3 | 3.3 ± 0.09 | 3.2 ± 0.13 |
| 92–100 (544.83) (αC) | 8 | 2 | 1.7 ± 0.06 | 1.6 ± 0.09 |
| 98–103 (736.403) (αC) | 4 | 1 | 0.9 ± 0.02 | 0.8 ± 0.04 |
| 98–104 (807.44) (αC) | 5 | 1 | 1.6 ± 0.03 | 1.6 ± 0.09 |
| 104–108 (635.38) (β4) | 4 | 1 | Solvent-inaccessible | Solvent-inaccessible |
| 104–110 (869.48) (β4) | 6 | 1 | 1.0 ± 0.02 | 1.2 ± 0.07 |
| 105–110 (770.41) (β4) | 5 | 1 | 1.0 ± 0.05 | 1.1 ± 0.08 |
| 106–115 (1200.55) (β4) | 9 | 1 | 1.7 ± 0.06 | 1.7 ± 0.11 |
| 109–116 (924.44) (β4) | 7 | 1 | 1.0 ± 0.02 | 1.1 ± 0.06 |
| 119–126 (825.38) (β5) | 7 | 1 | 0.2 ± 0.02 | 0.4 ± 0.02 |
| 122–128 (726.32) (β5-αD) | 6 | 1 | 1.0 ± 0.17 | 0.8 ± 0.08 |
| 122–129 (855.36) (β5-αD) | 7 | 1 | 1.0 ± 0.14 | 1.0 ± 0.29 |
| 133–145 (570.29) (αD-αE) | 12 | 3 | 1.2 ± 0.03 | 1.3 ± 0.01 |
| 144–151 (470.26) (αE) | 7 | 2 | 3.0 ± 0.10 | 2.9 ± 0.03 |
| 156–163 (989.49) (αE-β6) | 7 | 1 | 0.3 ± 0.02 | 0.3 ± 0.01 |
| 162–173 (743.94) (β6-β7) | 10 | 2 | 0.5 ± 0.04 | 0.3 ± 0.3 |
| 173–178 (673.35) (β7) | 5 | 1 | 1.3 ± 0.01 | 0.9 ± 0.05 |
| 180–185 (722.36) (β8) | 5 | 1 | 0.3 ± 0.24 | 0.2 ± 0.19 |
| 180–187 (926.46) (β8) | 7 | 1 | Solvent-inaccessible | Solvent-inaccessible |
| 182–187 (685.32) (β8) | 5 | 1 | Solvent-inaccessible | Solvent-inaccessible |
| 188–212 (981.52) (β9-activation loop) | 22 | 3 | 3.5 ± 0.08 | 3.5 ± 0.08 |
| 189–212(932.50) (β9-activation loop) | 21 | 3 | 3.3 ± 0.08 | 3.2 ± 0.09 |
| 198–211 (1418.73) (β9-activation loop) | 10 | 1 | Solvent-inaccessible | Solvent-inaccessible |
| 205–211 (768.49) | 5 | 1 | Solvent-inaccessible | Solvent-inaccessible |
| 222–227 (658.40) (αF) | 5 | 1 | Solvent-inaccessible | Solvent-inaccessible |
| 231–245 (826.90) (αF-αG) | 11 | 2 | 5.3 ± 0.12 | 5.0 ± 0.19 |
| 232–239 (869.43) (αF) | 5 | 1 | 1.1 ± 0.03 | 1.2 ± 0.02 |
| 241–247 (784.42) (αF-αG) | 5 | 1 | 3.0 ± 0.10 | 2.9 ± 0.27 |
| 262–269 (890.48) (αH) | 7 | 1 | Solvent-inaccessible | Solvent-inaccessible |
| 262–273 (693.90) (αH) | 11 | 2 | Solvent-inaccessible | Solvent-inaccessible |
| 268–273 (741.50) (αH) | 5 | 1 | Solvent-inaccessible | Solvent-inaccessible |
| 268–274 (869.56) (αH) | 6 | 1 | Solvent-inaccessible | Solvent-inaccessible |
| 278–302(748.40) (αH-αI) | 24 | 4 | 7.0 ± 0.20 | 8.1 ± 0.20 |
| 291–303 (831.41) (αH-αI) | 12 | 2 | 3.1 ± 0.16 | 3.3 ± 0.22 |
| 298–303 (740.33) (αH-αI) | 5 | 1 | 2.4 ± 0.08 | 2.6 ± 0.15 |
| 303–326(669.87) (C-terminal tail) | 19 | 4 | 9.5 ± 0.12 | 9.9 ± 0.20 |
| 303–327(706.64) (C-terminal tail) | 20 | 4 | 9.0 ± 0.17 | 9.4 ± 0.17 |
| 305–327(623.85) (C-terminal tail) | 18 | 4 | 10.0 ± 0.13 | 10.5 ± 0.25 |
| 306–327 (632.34) (C-terminal tail) | 17 | 4 | 8.1 ± 0.06 | 8.5 ± 0.13 |
| 335–346 (728.36) (C-terminal tail) | 11 | 2 | 7.0 ± 0.10 | 7.9 ± 0.11 |
a Average number of deuterons exchanged determined from fitting plots of the time course of deuteration during a 10-min exposure to deuterium oxide to a single exponential equation. Values reported are the mean and S.E. of the amplitude term of fits, and results are from at least two independent experiments. All deuterium exchange values reported were corrected for a 32.7% back-exchange by multiplying the centroid values by a multiplication factor of 1.49. Peptides showing significant differences upon (Rp)-cAMPS binding are in bold.
Fig. 5.

ESI-TOF spectra of one of the peptides spanning PBC (residues 204–221) in RIα(91–244). The spectra are expanded to show the isotopic distribution for the peptide of interest (m/z = 644.39). The higher mass peaks in the envelope are caused by naturally occurring isotopes. The isotopic envelope for the same peptide after 10 min of deuteration from C·RIα(91–244) bound to (Rp)-cAMPS (A), (Rp)-cAMPS ligand-free (B), and undeuterated sample (C).
Plots of the time course of deuteration of fragments associated with significant (Rp)-cAMPS-induced changes in deuteration are illustrated in Figs. 6 and 7. The maximum number of deuterons exchanged during a 5-min deuterium oxide exposure was determined from fitting plots of the time course of deuteration to a single exponential equation. Results obtained by this method of data analysis are comparable with the results obtained by using the average number of deuterons exchanged following a 5-min incubation from three independent experiments (10, 21, 22).
Fig. 6.

Time course of deuterium incorporation into backbone amides in regions of RIα(91–244) subunit in C·RIα(91–244) in absence (○) and presence of (Rp)-cAMPS (●) for peptides. A, m/z = 797.86, z = 2 (residues 136–148). B, m/z = 846.89, z = 2 (residues 157–172). C, m/z = 567.32, z = 2 (residues 202–212). D, m/z = 523.81, z = 2 (residues 230–238). The solid lines denote the best fit of the data to a single exponential equation. Error bars were calculated from 2 replicate experiments.
Fig. 7.

Effects of (Rp)-cAMPS on time course of deuterium incorporation into backbone amides in regions of C-subunit complexed to RIα(91–244). Data are plotted in the absence (○) and presence of (Rp)-cAMPS (●) for the peptides. A, m/z = 700.32, z = 2 (residues 7–18). B, m/z = 732.72, z = 3 (residues 14–31); C, 673.35, z = 5 (173–178). D, m/z = 728.36, z = 2 (residues 335–346). The solid lines denote the best fit of the data to a single exponential equation. Error bars were calculated from 2 replicate experiments.
Decreased exchange in the R-subunit was observed in two regions. The first region included the PBC spanned by five overlapping peptides (m/z = 1396.64, residues 188–201; m/z = 567.32, residues 202–212; m/z = 705.76, residues 202–221; m/z = 664.30, residues 204–221; and m/z = 500.83, residues 213–221) that showed a decrease in deuterium exchange (1–2 deuterons) (Table III). Subtractive analysis with these overlapping peptides enabled further localization of the site of decreased exchange upon (Rp)-cAMPS binding. For instance, the peptide 188–198 showed no difference in exchange, but the larger peptide 188–201 showed a protection of two deuterons upon (Rp)-cAMPS binding, indicating that residues 199–201 reflected (Rp)-cAMPS binding. Similarly, subtractive analysis of overlapping peptides (202–212, 204–212, 202–221, and 213–221) allowed narrowing down of the site of decreased exchange to residues 205–212 of the PBC. The decreased exchange seen in this region is consistent with the roles of Glu200 and Arg209 in coordinating the 2′-OH and phosphodiester moieties of cAMP, respectively (23). In the absence of any ligand, this region is highly shielded from solvent and/or structured because a large proportion of its residues do not exchange within 5 min (10). (Rp)-cAMPS binding into this pocket likely provides additional shielding, and this accounts for the observed decrease in deuterium exchange.
The second region of the R-subunit showing decreased exchange upon binding of (Rp)-cAMPS is a region spanning the critical Asp170 residue with three nested peptides spanning residues 157–172 (m/z = 846.89), 161–172 (m/z = 1322.58), and 162–172(m/z = 1225.54). Subtractive analysis allowed narrowing down of the sites of decreased exchange of one deuteron to residues 162–172. One region showed increased exchange in amide exchange at the αA helix (residues 136–143, m/z = 976.4) and is part of the peripheral interaction surface with the C-subunit (Table III).
For the C-subunit, (Rp)-cAMPS binding resulted in no difference in deuterium exchange (Table IV) in a majority (42 of 50) of all peptides analyzed. Seven peptides showed increased exchange at sites distal to the R-C intersubunit interface. Interestingly, a majority of the peptides showing increased exchange spanned regions of the protein outside the core kinase domain; these included the N-terminal αA helix and the C-terminal tail. Subtractive analysis of the five overlapping fragments that covered residues 237–267 localized a one-deuteron increase in residues 247–250 in αG and a one-deuteron increase in residues 251–261 in the loop connecting the αG and αH in the C-terminal strand. Thus, (Rp)-cAMPS induces limited unfolding and/or enhanced conformational flexibility that is spread almost globally through the C-subunit and, at least, a portion of RIα(91–244) (Fig. 8).
Fig. 8.

Effects of (Rp)-cAMPS binding to PKA holoenzyme (RIα(91–244)·C-subunit by amide H/2H exchange displayed on structure of C·RIα(91–244) complex (Protein Data Bank code 1U7E). Segments are colored based on the effects of (Rp)-cAMPS binding on amide exchange: segments showing increased exchange are red, segments showing no differences are tan, and segments showing decreased exchange are blue. Segments missing in the analysis are gray.
We had hoped to analyze the effects of (Sp)-cAMPS and cAMP on H/2H exchange in holoenzyme complexes by performing the experiments under conditions that would minimize holoenzyme dissociation (high protein concentrations and an excess of either free C-subunit or RIα(91–244)). Unfortunately, the presence of (Sp)-cAMPS and excess subunit decreased the resolvability of the deuterated fragments and made meaningful analysis problematic, so the effects of (Sp)-cAMPS and cAMP could not be determined.
DISCUSSION
The most significant result of our efforts to explore the actions of cAMP and two phosphorothioate analogs on the type Iα PKA holoenzyme is that (Rp)-cAMPS acts by increasing the affinity of RIα(91–244) toward the C-subunit and thus is better described as an inverse agonist because it decreases the fractional dissociation of the holoenzyme and, in turn, its basal activity. Less exciting but still important is the finding that cAMP and its mimic appear to reduce the interaction of RIα(91–244) with the C-subunit “contact” Sites 2 and 3 but probably not Site 1. Additionally, (Rp)-cAMPS binding to RIα(91–244)·C-subunit complex produces long range effects that propagate through both the R- and C-subunits to induce limited unfolding and/or enhanced conformational flexibility. To put these results in perspective, it is of value to review the conformational states of RIα, the structural basis for R-C interaction, the major cAMP-dependent intramolecular signaling pathways that spread out from the PBC, and the physical consequences of sulfur for oxygen substitution into cAMP.
RIα exists in two major conformations: one with high affinity toward the C-subunit and one with low affinity. The low affinity conformation is relatively compact with the two CNB domains in close proximity and with distinct αB and αC helixes at an angle of ∼120° angle to each other. The high affinity conformation is highly extended, almost barbell-shaped, with the two CNB domains at opposite ends of the molecule and linked by a combined, single αB and αC helix (αB/C). Cyclic AMP binding to the holoenzyme shifts the conformational equilibrium of RIα from the high affinity to the low affinity conformation. Cyclic AMP binding to RIα also reduces the overall conformational flexibility of the free RIα, which is observed as lower thermal factors in the x-ray structure (5) and an elevation of the urea concentration required for denaturation (6).
With regard to the structural basis of R-C subunit interaction, the minimal structural elements of RIα necessary for high affinity C-subunit binding and noncooperative cAMP regulation are contained in the doubly truncated mutant RIα(91–244) (8). The crystal structure of the RIα(91–244)·C-subunit complex (24) reveals the interacting segments of RIα and C-subunit. For RIα(91–244), the directly interacting segments include the pseudosubstrate sequence (residues 94–97) and elements of the linker region (residues 98–120), the 310 loop (residues 133–140), the PBC (residues 198–211), and the extended αB plus αC helix (residues 226–244). The complementary interface of the C-subunit divides into three regions or sites. Site 1 includes elements of the glycine-rich loop (residues 50–55), αF (residues 218–233), and P + 1 loop (residues 198–205). Site 2 includes all or elements of the APE linker (residues 206–208), αF, and αG (residues 243–252). Site 3 includes elements of the activation loop (residues 193–197). Site 1 of the C-subunit interacts with the pseudosubstrate sequence of RIα (P − 3 to P + 1 residues); Site 2 interacts with the 310 loop, PBC, αB, and αC of RIα; and Site 3 interacts with elements of the RIα linker region and the extended αB plus αC helix.
Based on the x-ray and NMR structural analyses of isolated cAMP-bound and free RIα mutants, the effects of cAMP binding appear to be mediated through both direct and indirect intramolecular non-covalent bonding pathways that spread out from the PBC. Particularly noteworthy are the pathways from the PBC to αB and αC. The critical indirect pathway includes the relay point Asp170 at the N terminus of β3 that links to the equatorial phosphate oxygen of bound cAMP through Arg209 in the PBC to Arg226 at the N-terminal end of αB (7, 25). Substitution of Lys for Arg at residue 209 blocks binding to the CNB-A site (26) and transforms (Rp)-cAMPS into an agonist (27). A direct pathway connects the exocyclic equatorial phosphate oxygen of bound cAMP to the amide nitrogens of Leu201 and Gly199 in or next to the PBC. Additionally, secondary pathways link the PBC, αA, and αB. Specifically, αA and αB are linked through Asp146 and Arg230, whereas PBC and αB are linked through Leu203, Ile204, and Arg230. Mutational analysis (28) and inference from the x-ray structure of RIα(91–244) indicate a link between the 2′-OH of cAMP to Arg241 in αC via a Glu200 bridge in the PBC.
Functionally, the binding of cAMP to the PBC can be considered as a phosphorylation event that acts as an electrostatic switch. Arguably, the most important moiety of cAMP involved in the activation process is its phosphate oxygen in the equatorial position (Rp) anchored by a critical, conserved Arg209 (29). As discussed above, cAMP binding activates this electrostatic switch. Substitution of sulfur into the Rp position of cAMP yields an inverse agonist, whereas substitution of sulfur into the Sp position yields a cAMP mimetic. In the case of (Rp)-cAMPS, the sulfur is bulkier than oxygen, and although the S–P bond is more electronegative than the O–P bond, the phosphorothioate sulfur is more polarizable, and its negative charge density is less than that of the corresponding phosphonate. The phosphorothioates are thus stronger acids and therefore less capable of hydrogen bonding (30–32). The combination of the larger size and reduced polarity of the S–P bond yields an entity that is unable to activate the Arg209-Asp170-Arg226 switch and stabilize RIα into a low affinity state toward the C-subunit.
The present fluorescence anisotropy results show that with RIα(91–244)·C-subunit complexes cAMP and (Sp)-cAMPS decrease the mobility of, at least, a portion of αC, presumably by activation of the above mentioned networks that would enhance the interactions of αB and αC with the PBC and the core of RIα(91–244). The anisotropy results also show that cAMP and (Sp)-cAMPS increase the mobility of, at least, a portion of the linker region (around residue 104), which can be explained if there is a decrease in the interaction of the linker region with αC of RIα(91–244). From the crystal structure of the RIα(91–244)·C-subunit complex (Protein Data Bank code 1U7E), this latter interaction is mediated by a non-covalent bond network connecting the linker region (via Glu101, Tyr103, Glu105, and Asp107) to αB and αC (via Asp227, Arg230, Arg231, and Met234). No evidence was found for cAMP or (Sp)-cAMPS perturbation of the interaction of the pseudosubstrate sequence with C-subunit Site 1. Taken together, these results are consistent with cAMP and (Sp)-cAMPS enhancing the interaction of αB and αC with the PBC and the core of RIα while decreasing the links between αB and αC with the linker region of RIα(91–244). All this leads to a reduction of the interaction of RIα with Sites 2 and 3 of the C-subunit probably without a direct effect on Site 1.
In the absence of C-subunit, the flexibility of the pseudosubstrate sequence and the linker regions, as indicated by the low electron densities of these regions in the x-ray crystal structures of cAMP-bound RIα(91–244), render intramolecular signaling pathways between the PBC and these regions unlikely. Highly flexible regions are poor conduits for intramolecular signaling because the dwell time of their conformational states is much shorter than the time required to achieve the large scale conformational changes associated with functional states. The anisotropy decay results reported here also supports the flexible character of this region. Specifically, the amplitude of the slow decay processes (β2; an inverse measure of the maximum diffusional excursion angle of the reporter group) is the smallest for FM-R92C (0.130; 38% of total decay) and somewhat larger for FM-T104C (0.154; 45% of total decay). Indeed, the fast rotational correlation time of FM-R92C is near the detection limit of our instrument (φfast = 0.9 ns) and is consistent with rapid α-carbonyl backbone motions. In this light, the failure to detect any effects of cAMP or its analogs on the N-terminal region of the pseudosubstrate or linker regions is not surprising. In the case of the end of αC, both cAMP and (Sp)-cAMPS appeared to decrease both the amplitude (β1) and the correlation time (φfast) of the fast anisotropy decay processes of the reporter group at residue 239, suggestive of a greater interaction of αB and αC with the core of RIα(91–244) upon cAMP or (Sp)-cAMPS binding.
Although the anisotropy measurements reveal little about the actions of (Rp)-cAMPS, results from H/2H amide exchange analysis were more illuminating. (Rp)-cAMPS induces large conformational changes in the RIα(91–244)·C-subunit complex. In RIα(91–244), amide exchange increased in both αA and αC and decreased by one or two deuterons in the PBC, presumably through steric shielding effects of (Rp)-cAMPS binding to this site. It is important that the results confirm the importance of the Arg209/Asp170 relay in propagating the effects of cAMP binding to the R-C interface. This is one region that showed decreased exchange upon (Rp)-cAMPS binding and strongly suggests that the basis for the inability of (Rp)-cAMPS to facilitate dissociation of the C-subunit is the uncoupling of the charge relay linking Arg209 and Asp170 and is consistent with previous mutagenesis data (26, 29). In the C-subunit, changes in amide exchange occurred almost globally with single deuteron increases in fragments associated with the αA-β1 loop, αF, αG, αG-αH loop, αH-αI loop, and the C-terminal tail. Reduced amide exchange was observed in only two segments between residues 262 and 264 at the C-terminal end of αH and residues 173–178, both distal to the R-C interface.
These changes are remarkable for at least two reasons. First, the near global increases in amide exchange were accompanied by a 5-fold increase in affinity of RIα(91–244) toward C-subunit, equivalent to a small change in relative free energy of binding (∼0.9 kcal/mol). The increased amide exchange indicates increase solvent accessibility and/or conformational flexibility, which could be due to enhanced internal entropy and, in turn, provide an explanation for the increased affinity. (An enhancement in enthalpy would primarily produce stabilization.) It must be pointed out that some of the regions that showed increased exchange are within the N terminus of the C-subunit and span helix αA and the C terminus. The αA in PKA is very critical for thermostability and more importantly for proper orientation of the two subdomains to maintain the substrate-binding cleft (33). The increased exchange in this region and other regions distal to the intersubunit interface corresponding to one to two additional deuterons exchanged potentially reflects the increased conformational mobility of both lobes of the C-subunit. A similar effect has been seen in p38α mitogen-activated protein kinase where the dephosphorylated protein shows increased exchange throughout the molecule, reflecting increased relative conformational mobility of the unphosphorylated protein (34). The enhanced mobility is likely responsible for enhanced binding to the R-subunit upon binding of (Rp)-cAMPS. Also, one needs to consider that (Rp)-cAMPS shifts slightly the equilibrium from a state with low affinity toward the C-subunit to one with high affinity. Second, the fact that these near global conformational changes occur in the absence of the activation of the electrostatic phosphate oxygen switch discussed above point to the importance of other components of cAMP such as the 2′-OH and 3′-O of the ribose and/or the adenine ring to produce long range conformational changes albeit with limited functional significance.
It should be noted that Gesellchen et al. (35) previously reported that (Rp)-cAMPS increased the affinity of RIα toward C-subunit using a non-quantitative bioluminescence resonance energy transfer assay. Here, we have put numbers to this effect and have described (Rp)-cAMPS more accurately as an inverse agonist.
Supplementary Material
Acknowledgments
We thank Mark Ritchie, Waters Pacific Private Ltd., Singapore and Michael Eggertson, Martha Stapels, and Keith Fadgen, Waters Corp., Milford, MA for assistance with peptide sequencing and identification and amide exchange mass spectrometry.
* This work was supported, in whole or in part, by National Institutes of Health Grant GM34921 (to S.S.T.) from the USPHS. This work was also supported by a grant from the Howard Hughes Medical Institute (to S. S. T.) and a grant from the Agency for Science, Technology and Research (A*STAR)-Biomedical Research Council, Singapore (to G. S. A.).
 This article contains supplemental Fig. 9.
This article contains supplemental Fig. 9.
1 The abbreviations used are:
- PKA
- cyclic AMP-dependent protein kinase
- C-subunit
- catalytic subunit of cAMP-dependent protein kinase
- R-subunit
- regulatory subunit of cAMP-dependent protein kinase
- RIα
- type Iα regulatory subunit
- RIα(91–244)
- deletion mutant of type Iα isoform of the R subunit
- CNB
- cyclic nucleotide binding
- FM
- fluorescein maleimide
- PBC
- phosphate-binding cassette
- cAMPS
- adenosine cyclic 3′:5′-monophosphorothioate.
REFERENCES
- 1.Beebe S. J., Corbin J. D. (1986) Cyclic nucleotide-dependent protein kinases. Enzymes 17, 43–111 [Google Scholar]
- 2.Johnson D. A., Akamine P., Radzio-Andzelm E., Madhusudan M., Taylor S. S. (2001) Dynamics of cAMP-dependent protein kinase. Chem. Rev. 101, 2243–2270 [DOI] [PubMed] [Google Scholar]
- 3.Shabb J. B. (2001) Physiological substrates of cAMP-dependent protein kinase. Chem. Rev. 101, 2381–2411 [DOI] [PubMed] [Google Scholar]
- 4.Walsh D. A., Perkins J. P., Krebs E. G. (1968) An adenosine 3′,5′-monophosphate-dependent protein kinase from rabbit skeletal muscle. J. Biol. Chem. 243, 3763–3765 [PubMed] [Google Scholar]
- 5.Wu J., Brown S., Xuong N. H., Taylor S. S. (2004) RIalpha subunit of PKA: a cAMP-free structure reveals a hydrophobic capping mechanism for docking cAMP into site B. Structure 12, 1057–1065 [DOI] [PubMed] [Google Scholar]
- 6.Dostmann W. R. G. (1995) (RP)-cAMPS inhibits the cAMP-dependent protein kinase by blocking the cAMP-induced conformational transition. FEBS Lett. 375, 231–234 [DOI] [PubMed] [Google Scholar]
- 7.Das R., Melacini G. (2007) A model for agonism and antagonism in an ancient and ubiquitous cAMP-binding domain. J. Biol. Chem. 282, 581–593 [DOI] [PubMed] [Google Scholar]
- 8.Huang L. J., Taylor S. S. (1998) Dissecting cAMP binding domain A in the RIalpha subunit of cAMP-dependent protein kinase. Distinct subsites for recognition of cAMP and the catalytic subunit. J. Biol. Chem. 273, 26739–26746 [DOI] [PubMed] [Google Scholar]
- 9.Anand G., Taylor S. S., Johnson D. A. (2007) Cyclic-AMP and pseudosubstrate effects on type-I A-kinase regulatory and catalytic subunit binding kinetics. Biochemistry 46, 9283–9291 [DOI] [PubMed] [Google Scholar]
- 10.Anand G. S., Hughes C. A., Jones J. M., Taylor S. S., Komives E. A. (2002) Amide H/2H exchange reveals communication between the cAMP and catalytic subunit-binding sites in the R(I)alpha subunit of protein kinase A. J. Mol. Biol. 323, 377–386 [DOI] [PubMed] [Google Scholar]
- 11.Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 12.Cook P. F., Neville M. E., Jr., Vrana K. E., Hartl F. T., Roskoski R., Jr. (1982) Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry 21, 5794–5799 [DOI] [PubMed] [Google Scholar]
- 13.Hibbs R. E., Johnson D. A., Shi J., Hansen S. B., Taylor P. (2005) Structural dynamics of the alpha-neurotoxin-acetylcholine-binding protein complex: hydrodynamic and fluorescence anisotropy decay analyses. Biochemistry 44, 16602–16611 [DOI] [PubMed] [Google Scholar]
- 14.Birch D. J. S., Imhof R. E. (1991) Time-domain fluorescence spectroscopy using time-correlated single photon counting, in Topics in Fluorescence Spectroscopy: Techniques (Lakowicz J. R. ed) Vol. 1, pp. 1–95, Plenum, New York [Google Scholar]
- 15.Engen J. R. (2009) Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal. Chem. 81, 7870–7875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Y., Engen J. R., Hobbins W. B. (2006) Ultra performance liquid chromatography (UPLC) further improves hydrogen/deuterium exchange mass spectrometry. J. Am. Soc. Mass. Spectrom. 17, 163–167 [DOI] [PubMed] [Google Scholar]
- 17.Weis D. D., Engen J. R., Kass I. J. (2006) Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass. Spectrom. 17, 1700–1703 [DOI] [PubMed] [Google Scholar]
- 18.Gangal M., Cox S., Lew J., Clifford T., Garrod S. M., Aschbaher M., Taylor S. S., Johnson D. A. (1998) Backbone flexibility of five sites on the catalytic subunit of cAMP-dependent protein kinase in the open and closed conformations. Biochemistry 37, 13728–13735 [DOI] [PubMed] [Google Scholar]
- 19.Kinosita K., Jr., Kawato S, Ikegami A. (1977) A theory of fluorescence polarization decay in membranes. Biophys. J. 20, 289–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.García De La Torre J., Huertas M. L., Carrasco B. (2000) Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 78, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hughes C. A., Mandell J. G., Anand G. S., Stock A. M., Komives E. A. (2001) Phosphorylation causes subtle changes in solvent accessibility at the interdomain interface of methylesterase CheB. J. Mol. Biol. 307, 967–976 [DOI] [PubMed] [Google Scholar]
- 22.Mandell J. G., Falick A. M., Komives E. A. (1998) Measurement of amide hydrogen exchange by MALDI-TOF mass spectrometry. Anal. Chem. 70, 3987–3995 [DOI] [PubMed] [Google Scholar]
- 23.Su Y., Dostmann W. R., Herberg F. W., Durick K., Xuong N. H., Ten Eyck L., Taylor S. S., Varughese K. I. (1995) Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science 269, 807–813 [DOI] [PubMed] [Google Scholar]
- 24.Kim C., Xuong N. H., Taylor S. S. (2005) Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science 307, 690–696 [DOI] [PubMed] [Google Scholar]
- 25.Kim C., Cheng C. Y., Saldanha S. A., Taylor S. S. (2007) PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 130, 1032–1043 [DOI] [PubMed] [Google Scholar]
- 26.Bubis J., Neitzel J. J., Saraswat L. D., Taylor S. S. (1988) A point mutation abolishes binding of cAMP to site A in the regulatory subunit of cAMP-dependent protein kinase. J. Biol. Chem. 263, 9668–9673 [PubMed] [Google Scholar]
- 27.Dostmann W. R., Taylor S. S. (1991) Identifying the molecular switches that determine whether (Rp)-cAMPS functions as an antagonist or an agonist in the activation of cAMP-dependent protein kinase I. Biochemistry 30, 8710–8716 [DOI] [PubMed] [Google Scholar]
- 28.Vigil D., Lin J. H., Sotriffer C. A., Pennypacker J. K., McCammon J. A., Taylor S. S. (2006) A simple electrostatic switch important in the activation of type I protein kinase A by cyclic AMP. Protein Sci. 15, 113–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abu-Abed M., Das R., Wang L., Melacini G. (2007) Definition of an electrostatic relay switch critical for the cAMP-dependent activation of protein kinase A as revealed by the D170A mutant of RIalpha. Proteins 69, 112–124 [DOI] [PubMed] [Google Scholar]
- 30.Frey P. A., Sammons R. D. (1985) Bond order and charge localization in nucleoside phosphorothioates. Science 228, 541–545 [DOI] [PubMed] [Google Scholar]
- 31.Liang C., Allen L. C. (1987) Sulfur does not form double bonds in phosphorothioate anions. J. Am. Chem. Soc. 109, 6449–6453 [Google Scholar]
- 32.Basch H., Krauss M., Stevens W. J. (1991) Comparison of the electronic structure of the P-O and P-S bonds. J. Mol. Struct. 81, 277–291 [Google Scholar]
- 33.Herberg F. W., Zimmermann B., McGlone M., Taylor S. S. (1997) Importance of the A-helix of the catalytic subunit of cAMP-dependent protein kinase for stability and for orienting subdomains at the cleft interface. Protein Sci. 6, 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sours K. M., Kwok S. C., Rachidi T., Lee T., Ring A., Hoofnagle A. N., Resing K. A., Ahn N. G. (2008) Hydrogen-exchange mass spectrometry reveals activation-induced changes in the conformational mobility of p38α MAP kinase. J. Mol. Biol. 379, 1075–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gesellchen F., Prinz A., Zimmermann B., Herberg F. W. (2006) Quantification of cAMP antagonist action in vitro and in living cells. Eur. J. Cell. Biol. 85, 663–672 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.