

Abstract
In the present study, comparative genome analysis between Clostridium perfringens and the human genome was carried out to identify genes that are essential for the pathogen's survival, and non-homologous to the genes of human host, that can be used as potential drug targets. The study resulted in the identification of 426 such genes. The number of these potential drug targets thus identified is significantly lower than the genome's protein coding capacity (2558 protein coding genes). The 426 genes of C. perfringens were further analyzed for overall similarities with the essential genes of 14 different bacterial species present in Database of Essential Genes (DEG). Our results show that there are only 5 essential genes of C. perfringens that exhibit similarity with 12 species of the 14 different bacterial species present in DEG database. Of these, 1 gene was similar in 12 species and 4 genes were similar in 11 species. Thus, the study opens a new avenue for the development of potential drugs against the highly pathogenic bacterium. Further, by selecting these essential genes of C. perfringens, which are common and essential for other pathogenic microbial species, a broad spectrum anti-microbial drug can be developed. As a case study, we have built a homology model of one of the potential drug targets, ABC transporter-ATP binding protein, which can be employed for in silico docking studies by suitable inhibitors.
Keywords: Clostridium perfringens, DEG, Essential genes, Drug targets, Broad-spectrum anti microbial drug
Background
The availability of the complete genome sequence information of the human genome and a large number of microbial genomes' sequences has led to the development of new approaches to understand hostpathogen interaction. Use of bioinformatics approach and comparative analysis of the genome of a pathogenic microbe allows one to identify essential genes necessary for the survival of that pathogen. The proteins encoded by these essential genes, that are not present or are nonhomologous to the host, can be used as drug targets. Such an approach has been effectively used to identify drug targets in other bacterial species such as Pseudomonas aeruginosa [1,2], Helicobacter pylori [3], Mycobacterium tuberculosis [4], Burkholderia pseudomallei [5] and Aeromonas hydrophila [6]. Clostridium perfringens is a Grampositive, rod shaped, anaerobic bacterium that is able to form spores. It is widely distributed in the environment (e.g. in soil and sewage) and is frequently found in the gastrointestinal (GI) tract of humans, many domestic and feral animals, as well as in soil and freshwater sediments [7]. In humans, it can cause gangrene and gastrointestinal disease (e.g. food poisoning and necrotic enteritis), whereas in other animals, gastrointestinal and enterotoxemic diseases occur more frequently [8]. C. perfringens does not invade healthy cells but produces various toxins and enzymes that are responsible for associated lesions and symptoms. As a species, C. perfringens is one of the most prolific producers of toxins [9]. It has five biotypes (A, B, C, D and E), which are identified by the main types of toxins they produce (alpha, beta, iota, epsilon and theta), each type of toxin being associated with a specific syndrome. C. perfringens type A is the most common toxin type in the environment, and is responsible for gas gangrene, enterocolitis, dysenteria, and enterotoxemia. In the present study, comparative genome analysis of C. perfringens type A with that of the human genome, and use of the Database of Essential Genes (DEG) compiled by Zhang et al., [10], have resulted in the identification of the essential genes of C. perfringens, that could be used as potential drug targets.
Methodology
Comparative genome analysis
The complete genomes of C. perfringens type A, strain SM101 (Accession No. NC_008262) [11] and its human host have been sequenced and were downloaded from the NCBI website [12]. The Database of Essential Genes[10] was accessed from its website[13] and sequence alignment was performed using BLASTP. In this analysis, the assumption described by Dutta et al., [3] was followed, and proteins of less then 100 amino acid residues were excluded from the analysis. The remaining proteins were subjected to BLASTP on the NCBI server against human protein sequences to identify non-homologous sequences [14]. A minimum bit score of 100 and an Expectation value (E-value) cutoff of 10-10 was selected for shortlisting genes. The shortlisted genes were subjected to BLASTP against DEG to identify essential genes. Further analysis of the essential genes using the Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway database [15], revealed the information about different biological process in which potential target genes were involved.
Structure modeling and visualization of mode
BLASTP analysis was used to identify the most suitable template for homology modeling of Clostridium perfringens ABC transporter, ATP binding protein (CpABC) (Accession No. YP_698054). Subsequent to BLASTP analysis, the sequences of the structures of ABC transporters available in the PDB were used. The available structure of ABC transporter from Methanococcus jannaschii (Mj0796) in the Protein Database (PDB entry 1f3o, resolution=2.70, R value=0.204) was used as a template. The target and the template sequences were aligned using ClustalW. MODELLER [16], an automated comparative protein modeling program, was used for homology modeling to generate the 3- D structure of CpABC. Further, the structural model generated was visualized using the Swiss PDB viewer software [17].
Validation of the generated model
Different structure verification servers such as PROCHECK [18], WHAT_CHECK [19], VERIFY3D [20] and ProSA [21]were used to evaluate the 3D-model. These verification programs validate the predicted structure by checking various parameters. While PROCHECK, a structure verification program that relies on Ramachandran plot [22], determines the quality of the predicted structure by assessing various parameters such as lengths, angles and planarity of the peptide bonds, geometry of the hydrogen bonds, and side chain conformations of protein structures as a function of atomic resolution, WHAT_CHECK, a subset of protein verification tools from the WHATIF[23], program extensively evaluates the stereochemical parameters of the residues in the model [24]. The Verify3D determines the compatibility of an atomic model (3D) with its own amino acid sequence (1D) by assigning a structural class based on its location and environment (α, α, loop, polar, nonpolar etc.) and comparing the results to valid structures [25].
Discussion
Identification of drug targets in C. perfringens
C. perfringens is the most common cause of gas gangrene in humans. Clostridial gas gangrene is a highly lethal necrotizing soft tissue infection of skeletal muscle caused by toxins secreted by C. perfringens. Although penicillin is one of the preferred antibiotics, it is only useful if the infection is diagnosed at early stages. There is no other specific drug that can be given to a patient infected with C. perfringens. Therefore, more research in this field is required to identify new drug targets and develop therapeutic agents for controlling C. perfringens infections. Since most antibiotics target essential cellular processes, essential gene products of microbial cells are promising new targets for antibacterial drugs [10]. Targetting an essential gene necessary for the bacterial cell survival may provide an effective way to control infection.
The circular genome of C. perfringens comprises 2,897,393 nucleotides with a total number of 2701 genes. Of the 2558 protein encoding genes, only 2300 genes encode proteins of greater than 100 amino acid residues. BLAST analysis of these genes against the human genome sequence revealed 1991 genes to be non-homologous to humans. Further BLASTP analysis of the 2300 protein coding genes with DEG resulted in identification of 726 genes, which had a bit score of at least 100 at an expectation cutoff value of 10-10, as similar to the essential genes required for the growth and survival of bacteria listed in the DEG. Of these, 426 were found to have no human homologue (see Table 1 insupplementary data). Pathways information for these genes was obtained from KEGG database. All these genes are involved in the production of proteins that are useful for various important functions in C. perfringens. Out of the 426 identified genes, function of 10 genes remains unknown, and 17 genes encode conserved hypothetical proteins. The percentage distribution of the genes amongst different biological process is shown in Figure 1. A large population of these genes (∽33%) is involved in metabolic pathways. The major share of these genes constitute the proteins involved in transport and translation (17% and 12%, respectively). Highly conserved genes, in theory, are more likely to be physiologically important [26]; however, they need to be experimentally validated. Therefore, the analysis of the 426 essential genes of C. perfringens for overall similarities with all 14 species present in DEG database was carried out. Results of such an analysis are shown in Figure 2. Out of the 426 essential genes, 160 genes have similar match to at least 1 species, whereas on the other end of the spectrum, only 4 genes have similar match to 11 species and only 1 gene has an identity score of more than 100 with 12 different microbial species listed in the DEG. From this analysis, it is evident that the products of 5 genes (1 gene similar in 12 species and 4 genes similar in 11 species) are essential for most of the bacterial species present in DEG. These species include Acinetobacter baylyi ADP1, Bacillus subtilis, Escherichia coli MG1655, Francisella novicida U112, Haemophilus influenzae, Helicobacter pylori 26695, Helicobacter pylori J99, Mycobacterium tuberculosis H37Rv, Mycoplasma genitalium G37, Mycoplasma pulmonis UAB CTIP , Salmonella typhimurium, Staphylococcus aureus and Streptococcus pneumoniae. Therefore, these 5 genes can be used as potential drug targets for more than 10 highly pathogenic bacterial species, in addition to C. perfringens. These 5 target genes, thus identified, are ABC transporter- ATP-binding protein, cell division protein FtsZ, RNA polymerase sigma factor RpoD, 50S ribosomal protein L13, and 30S ribosomal protein S5. A drug designed against these targets can be effectively used to treat other bacterial infections as well. Since the number of thus identified potential candidate genes is relatively small, these can be experimentally validated to develop broad-spectrum antimicrobial drugs. Since ABC transporter-ATP-binding protein is one of the 5 potential drug targets identified, an attempt has been made to predict its structure for effective drug design.
Figure 1.
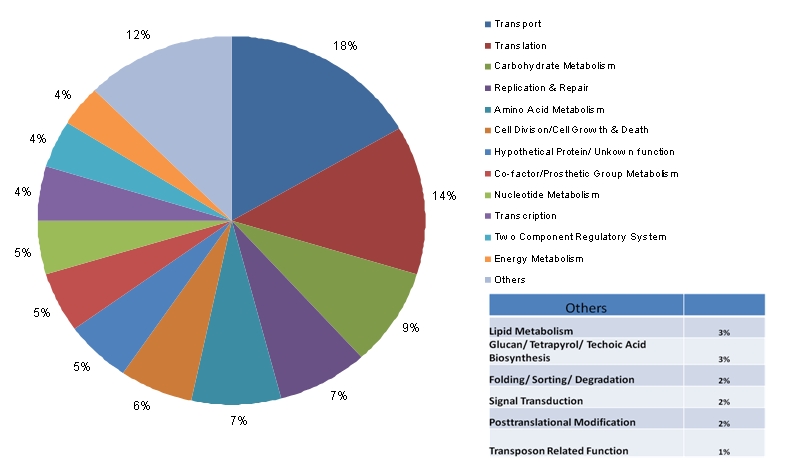
Percentage distribution of 426 target genes encoding different classes of proteins in Clostridium perfringens. Around 2% gene encode the proteins of unknown function.
Figure 2.
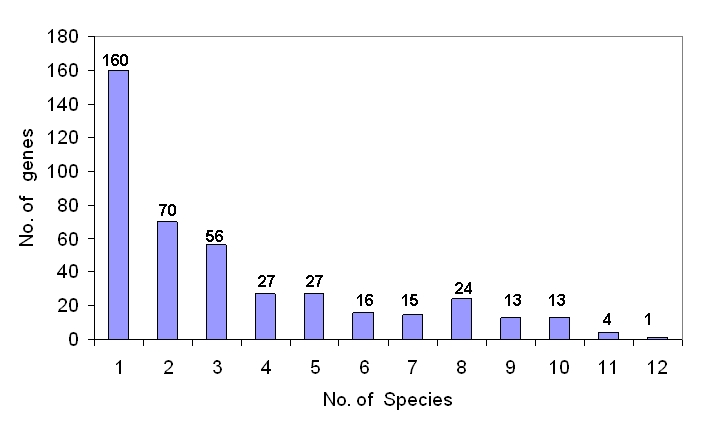
Graphical representation of the number of target genes aving similarity with those of bacterial species present in DEG. The number of genes of C. perfringens having similar match to different number of bacterial species is shown on top of the respective bars.
ABC transporter ‐ a potential broad spectrum target
ABC (ATP-binding cassette) transporters are ubiquitously present ATPdependent transmembrane solute pumps and ion channels. These superfamilies contain both uptake and efflux transport systems and form one of the largest transporters [27]. The ABC transporters couple hydrolysis of ATP to the translocation of various substrates across cell membranes. Members of this superfamily recognize substrates ranging from single ions to entire protein toxins. ABC transporters have a number of highly conserved ABC cassette motifs, many of which are involved in the binding and hydrolysis of ATP. It is generally assumed that all ABC cassettes bind and hydrolyze ATP in a similar way and use a common mechanism to provide energy for substrate transport through the membrane-spanning domains [28]. When the substrate has traversed the membrane, the transporter returns to the resting state through dissociation of ADP and inorganic phosphate. Fluoxetine and omeprazole, few of the most widely prescribed drugs in the world, have a transporter protein as site of action. Therefore, ABC transporter structures have potential value in drug designing.
Structural model and overall architecture of ABC transporter (CpABC)
Sequence alignment of the C. perfringens CpABC and ABC transporters from other species revealed Mj0796 to be the best template for homology modeling of the target sequence as the CpABC and Mj0796 shared 41% identity (Figure 3). Mj0796 is a member of the o228/LolD transporter family, involved in the export of lipoproteins via the Lol system. LolD contains a characteristic sequence called the LolD motif, which is highly conserved among LolD homologs, but not in other ABC transporters, and is located between the Walker A (GSGKST, boxed) and ABC signature (LSGGQ, marked as bold overline) motifs (Figure 3). Comparative sequence analyses, motif search, and secondary structure prediction indicated that CpABC is structurally similar to the monomer structure of the Mj0796, a lipid transporter. The crystal structure of Mj0796 (PDB entry 1f3o) was used as a template to predict the structure of CpABC and the predicted 3D structure model of CpABC was generated by Modeller, a homologymodeling program. Figure 4 shows the predicted structure in the form of ribbons as a Swiss PDB viewer representation.
Figure 3.

CLUSTALW Multiple sequence alignment of CpABC with Mj0796. Single fully conserved residues are represented by (*), conservation of strong and weak groups is denoted by (:) and (.), respectively. The boxed sequence represents the Walker A motif, whereas the ABC signature sequence is marked by bold overline.
Figure 4.

(A): Homology modeled structure of the C. perfringens ABC transporter, ATP binding protein based on template Mj0796. Model is represented in ribbon form as Swiss PDB viewer representation in secondary structure succession color scheme. N and C termini of modeled structure are represented as NH3+ and –OOC. (B) Superimposed image of the modeled structure of CpABC onto Mg-ADP bound Mj0796 (PDB entry 1f3o). CpABC is represented in red color, Mg-ADP (shown in green color) bound Mj0796 is represented in blue color. Signature sequence LSGG with three conserved residues, Q90, E171, H204 form Mj0796 and Q92, E164, H197 from model CpABC are mentioned. Arrow indicates the deletion of seven amino acids (RKRALEC) in CpABC, which forms an α-helix in the Mj0796.
Validation of generated model of CpABC
The quality of the model was evaluated using the PROCHECK program and assessed using the Ramachandran plot. It is evident from the Ramchandran plot that the predicted model has 91.4%, 8.1%, and 0.5% residues in the most favorable regions, the allowed regions, and the disallowed regions, respectively. Such a percentage distribution of the protein residues determined by Ramachandran plot shows that the predicted model is of good quality. All Ramachandrans show 6 labelled residues out of 220 whereas chi1-chi2 plots show 2 labelled residues out of 140. The model shows all the main chain and side chain parameters to be in the ’better‘ region. Another factor that is important for the predicted model to be reliable is G-factor, which is a log odds score based on the observed distribution of stereochemical parameters. For a reliable model, the score for G-factor should be above -0.50. The observed G-factor score for the present model was -0.05 for dihedrals bonds, -0.31 for covalent bonds, and -0.15 overall. The distribution of the main chain bond lengths and bond angles were 98.5% and 93.2% within limits, respectively. Also, all the planar groups were within the limits. The quality of the generated model of CpABC as evaluated by ProSA provided a z-score of -7.2, which falls within the range of values observed for the experimentally determined structures of similar lengths. The validity of the predicted model of CpABC was also verified by employing the structure verification servers WHAT_CHECK and Verify-3D. Superposition of the predicted structure of CpABC and the Mg-ADP bound Mj0796 (template, PDB entry 1f3o) is shown in Figure 4B. It is evident from the figure that the Mg-ADP binding core of the ABC subunit and all the structural motifs are highly conserved in both structures. The two superposed structures match 214 Cα atoms with an rms distance of 0.47Å. Three residues Q90, E171, and H204, important for activity of Mj0796, superposed very well with conserved residues Q92, E164 and H197 from model CpABC. However, a deletion of seven amino acids (RKRALEC), which forms an α-helix in the Mj0796 (indicated by arrow), and an insertion of three amino acids (PIS) at the C-terminal end of CpABC, is observed. Thus, the predicted model structure of C. perfringens ABC transporter, ATP binding protein, and CpABC is comparable to the structurally resolved Mj0796.
Conclusion
Comparative genome analysis is a highly efficient approach for idendifing potential proteins that can be used as potential targets for effective drug designing against pathogenic organisms. It allows restricting the potential pool of genes to a much smaller number, compared to the whole genome capacity, which can be experimentally validated. In the present study, around 426 drug targets in C. perfringens were identified by comparative genome analysis with DEG. Further, by using the subtractive genomic approach five essential genes were identified that are conserved in more than 10 other pathogenic organisms. Since the number of these conserved genes is very small, these can be experimentally tested for the development of a broadspectrum anti-microbial drug. The drug thus developed is likely to inhibit other bacterial infections, which share high sequence similarity with the five essential genes of C. perfringens SM101.
Supplementary material
Acknowledgments
Financial assistance from the Department of Biotechnology, New Delhi, India is acknowledged. The Council of Scientific and Industrial Research, New Delhi, India, and the University Grants Commission, New Delhi, India is gratefully acknowledged for senior research fellowships to AA and KG, respectively.
Footnotes
Citation:Chhabra et al, Bioinformation 4(7): 278-289 (2010)
References
- 1.Sakharkar KR, et al. In Silico Biology. 2004;4:355. [PubMed] [Google Scholar]
- 2.Perumal D, et al. In Silico Biology. 2007;7:453. [PubMed] [Google Scholar]
- 3.Dutta A, et al. In Silico Biology. 2006;6:43. [PubMed] [Google Scholar]
- 4.Anishetty S, et al. Computational Biology and Chemistry. 2005;29:368. doi: 10.1016/j.compbiolchem.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Chong CE , et al. In Silico Biology. 2006;6:341. [PubMed] [Google Scholar]
- 6.Sharma V , et al. In Silico Biology. 2008;8:331. [PubMed] [Google Scholar]
- 7.Rood JI , Cole ST. Microbiology Review. 1991;55:621. doi: 10.1128/mr.55.4.621-648.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Songer JG. Clin Microbiology Review . 1996;9:216–234. doi: 10.1128/cmr.9.2.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rood JI . Annual Review of Microbiology . 1998;52:333. doi: 10.1146/annurev.micro.52.1.333. [DOI] [PubMed] [Google Scholar]
- 10.Zhang R , et al. Nucleic Acids Research. 2004;32:D271. doi: 10.1093/nar/gkh024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Myers GS, et al. Genome Research. 2006;16:1031. doi: 10.1101/gr.5238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. http://www.ncbi.nlm.nih.gov/
- 13. http://tubic.tju.edu.cn/deg/
- 14.Altschul SF, et al. Journal of Molecular Biology . 1990;215:403. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 15.Ogata H , et al. Nucleic Acids Research. 1999;27:29. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiser A , Sali A . Methods in Enzymology. 2003;374:461. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 17. http://www.expasy.org/spdbv.
- 18.Laskowski RA, et al. Journal of Applied Crystallography . 1993;26:283. [Google Scholar]
- 19.Hooft RWW, et al. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 20.Eisenberg D, et al. Enzymology. 1997;277:396. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 21.Wiederstein M , Sippl MJ. Nucleic Acids Research. 2007;35(Web Server issue):W407. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramachandran GN , Sasisekharan V. Advances in Protein Chemistry. 1968;23:283. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 23.Vriend G. Journal of Molecular Graphics. 1990;8:52. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 24. http://swift.cmbi.ru.nl/gv/whatcheck/
- 25. http://nihserver.mbi.ucla.edu/Verify_3D/
- 26.Arigoni F . Nature Biotechnology . 1998;16:851. doi: 10.1038/nbt0998-851. [DOI] [PubMed] [Google Scholar]
- 27.Saier MH . Jr Microbiology . 2000;146:1775. doi: 10.1099/00221287-146-8-1775. [DOI] [PubMed] [Google Scholar]
- 28.Locher KP , et al. Science. 2002;296:1091. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.