

Abstract
Indinavir (Crivaxan®) is a potent inhibitor of the HIV (human immunodeficiency virus) protease. This enzyme has an important role in viral replication and is considered to be very attractive target for new antiretroviral drugs. However, it becomes less effective due to highly resistant new viral strains of HIV, which have multiple mutations in their proteases. For this reason, we used a lead expansion method to create a new set of compounds with a new mode of action to protease binding site. 1300 compounds chemically diverse from the initial hit were generated and screened to determine their ability to interact with protease and establish their QSAR properties. Further computational analyses revealed one unique compound with different protease binding ability from the initial hit and its role for possible new class of protease inhibitors is discussed in this report.
Keywords: protease, Indinavir, lead expansion, docking, pharmacophore
Background
The HIV-1 (human immunodeficiency virus type 1) protease is a C2 symmetric and an aspartic acid homodimeric enzyme, where aspartate 25 plays a pivotal role in binding the substrate [1]. The HIV-1 protease does not have a homologue in mammalian cells and has a function to cleave the Gag-Pol polyprotein precursor into shorter pieces to create the active protein components for viral packaging and maturation. This proteolysis occurs late in the viral life cycle and is essential for viral infectivity [2]. The protease active site is located in the buried area (tunnel), where the two subunits meet each other. Highly active antiretroviral therapy (HAART), using protease inhibitors (PI), is commonly used in management of HIV infection. These inhibitors are able to irreversibly bind the HIV-1 protease to block its function. Among these compounds, Indinavir (Crixivan, MK-639, or IDV) [3,4] is a potent and selective protease inhibitor that had approval for AIDS therapy. But genotypic analyses of the viral populations during the course of protease inhibitor therapy had shown various mutations that can occur in as many as 20 amino acids within the protease gene [5,6]. 13.7 % of IDV failure were identified for 1021 new viral genotypes from HIV patients[7]. In case of HIV-1 subtype C (African strain), it had been shown that the most common primary mutation observed in PI treated patients was L90M [8], a main course in multi-PI resistance establishment [9]. Even naturally occurring polymorphism, such as L89M and I93L, located in the hydrophobic core of the enzyme would possibly change the shape of the substrate-binding cleft and diminish the potency of IDV [10]. Only flexibility of the newly synthesized compounds seems to overcome this effect. Here, we describe the designing scheme for possible HIV-1 protease inhibitors using a lead expansion protocol with a pharmacophoric-shape similarity scoring function.
Methodology
HIV-1 subtype C protease and Indinavir structures
The three dimensional structure of the HIV-1 protease - IDV complex (PDB code: 2R5P) was retrieved from Protein Data Bank at 2.3 Å RMSD resolution [10]. The VADAR (volume, area, dihedral angle reporter) server [11], which is an improved version of the PROCHECK software, was used for stereochemical validation of HIV-1 protease. Altogether, 95 % of all residues were in φ-ψ core areas. Indinavir was obtained from PubChem database (CID: 5362440). IDV initial refinement was performed by means of the MarvinSketch program provided by ChemAxon [12].
Protease active site detection
Protein binding surfaces could have very complicated and irregular structures. Atoms could form pockets, cavities and tunnels. Solvent molecules can get into these tunnels from outer environment and move through them. Buried shape and volume of such tunnels vary in time due to protein dynamics and kinetics. Here, we analyzed buried tunnels of the protease using the CAVER module, to identify certain atomic positions of hidden binding moieties [13]. Dijkstra's algorithm was implemented in searching process and started from source node (starting point), which is located deeply in the protein pocket. Before the search procedure, 136 water molecules, chloride and sodium ions were removed from IDV-bound protease (liganded holo-structure) protein database file. All calculations were performed on 32612 grid points. Catalytic tunnel (key interaction site) was detected in the protease structure ((Figure 2(A)). The tunnel coordinates to specify the starting point in x, y and z axis are 2.5, 6.5, -7.5 respectively. These coordinates were taken from the AutoDock manual [14].IDV
Figure 2.
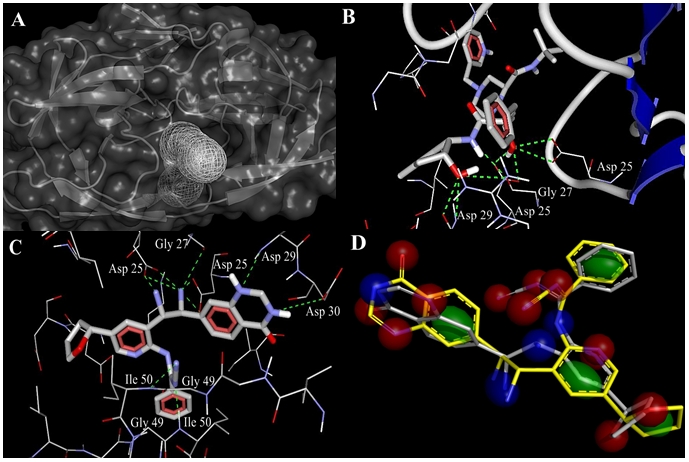
(A) HIV protease catalytic tunnel (binding site) was predicted by PyMol CAVER module. IDV (B) and novel hit (C) interactions with the HIV-1 protease are shown. H-bonds are depicted as dashed lines. IDV – protease complex was analyzed as a crystal structure. 3D alignment and ‘fuzzy’ model (D) of the hit ‘native’ conformation (gray) together with its ‘functional’ conformation (yellow) at 2.0 Å RMSD. The shown potential pharmacophore points are color-coded as follows: lipophilic areas are green, H-bond donors and acceptors are colored in blue and red respectively.
Compound library generation
Pharmacophoric ‐ shape similarity scoring function was used for compound library generation. This is build-in function integrated in the MuseTM molecular design workflow to accelerate the identification and optimization of lead candidates. This function seeks molecules that have fairly different structures but similar 3D pharmacophores and shapes. It is appropriate for lead expansion ‐ exploration around an initial hit. Indinavir compound was used as a reference molecule and all similarities were measured with respect to it. The reference structure was also used as the initial population for invention. In our case a default set of compounds was used if no any other structures were given. Such generation of multiple compounds with similar shape but different 3D structure organization would provide more interesting results in the invention and library screening afterwards. For lead expansion method we did not specified any seed structures and preserved cores. 99 undesirable and 35 questionable substructures were excluded from to be present in invented structures. Structural ranking was done by means of the Pareto-Borda method to maximize pharmacophoric-shape similarity in preferential shape similarity specified range (from 0.35 to 0.50 score units).
ADME/Tox studies
Available decision tree algorithms such as: Benigni-Rossa rulebase, Verhaar scheme, Cramer rules and START (structural alerts for reactivity in toxtree) biodegradability were used for the analysis. Benigni-Rossa rule base predicts the possibility of carcinogenicity and mutagenecity by discriminant analysis and structural rules [15]. START biodegradability estimates biodegradation potential of the chemical compound based on structural alerts compiled from the Canadian Environmental Protection Agency.
Protein-ligand docking
The Molegro Virtual Docker [16] and the AutoDock software were implemented to analyze ligand interactions with the HIV-1 protease binding site. The Molegro Virtual Docker and the AutoDock combine a rapid energy evaluation through pre-calculated grids of affinity potentials with a variety of search algorithms to find appropriate binding positions. In Molegro Virtual Docker docking experiment we used MolDock scoring function, which is based on a piecewise linear potential and a re-ranking procedure was applied to the highest ranked poses to increase docking accuracy. Affinity grid resolution was set to 0.3 Å. Ligand evaluations were based on internal energy of binding, internal H-bonds formation, Sp2-Sp2 (trigonal planar electron domain geometry) torsion angles. Binding site was previously detected by CAVER module and further measured for cavity volume (282.1 Å3), surface (681.0 Å2) and radius (15.0 Å). Customized search algorithm was set to MolDock SE (simplex evolution). Number of runs was 10. Parameter settings were set to 1500 iterations, 50 population sizes, 100.0 kcal/mol of energy threshold for pose generation, 300 simplex evolution steps and 1.0 neighbor distance factor. All dockings were performed at 1.0 Å RMSD threshold. For preparing the AutoDock docking parameter file we used default settings (genetic algorithm parameters: population size = 150, number of energy evaluations = 2500000, rate of gene mutation = 0.02, rate of crossover = 0.8, maximum number of generations = 27000, number of GA runs = 10, initial dihedrals were randomly specified, elitism value was set to 1). By default, clustering of docked results was done at 0.5 Å RMSD. Prior to docking, total Kollman and Gasteiger charges were added to the protein and the ligand.
LIQUID-fuzzy pharmacophore models
The PyMol LIQUID module was utilized to create a pharmacophore ‘fuzzy’ model. At the first model computation LIQUID used standard cluster radii to calculate cluster sizes at 4.0 Å RMSD for lipophilic areas and 1.9 Å RMSD for H-bond donors and acceptors [17]. In our experiment we set up default cluster radius (2.0 Å) for each potential pharmacophore point type ( Figure 2(D)).
Discussion
Genetic algorithm produced 1300 compounds (70 generations) of diverse chemical structure. 500 of top molecules were selected according to their shape similarity (0.7-0.9 functional scores). These molecules, which had pharmacophoric score less than 0.8 were excluded from the list. 227 out of 500 compounds satisfied all that criteria and were tested for Lipinski's Rule of Five using the LigandScout pharmacophore software [18]. Only one compound (novel hit) did not have rule violations, in contrast to IDV, which had one violation of Lipinski's rule. A useful parameter, such as topological polar surface area (TPSA), was defined for these two molecules as the surface sum over all polar atoms to evaluate parameters for the prediction of cell permeability and drug transport properties (Table 1 supplementary material). This parameter shows good drug transport, even in ability to penetrate the blood-brain barrier. In this case TPSA should not exceed 600 Å2threshold. The novel hit was analyzed for Cramer rules, carcinogenicity and biodegradability. It also showed a strong affinity (in comparison with reference molecule) to the HIV-1 protease binding site. Docking results revealed the interaction modes for this compound with the HIV-1 protease. Novel hit adopts IDV binding mode (Asp 25, 29 and Gly 27) and also has the additional interacting residues (Gly 49-Ile 50, Asp 29-Ala 28, Asp 30). Moreover, it exceeds IDV in H-bond formations (10 instead of 9). New compound's nitrile group is creating H-bonds with donor nitrogen of Gly 49 - Ile 50 amide group (Figure 2(B,C)).
Probably this acceptor nitrile group along with the tetrahydropyridine ring might have some influence on the ligand conformational shift with subsequent change in binding affinity mode. In both docking cases, the catalytic function of the Asp 25 residues is completely blocked. Subsequently, we performed the AutoDock experiments to validate the above data and revealed reduction in hydrogen bonds formation presumably due to differences in affinity grid resolution of Molegro Virtual Docker and AutoDock (0.3 vs 0.375 Å). However, consistent with previous observation, all crucial amino acids obtained from Molegro docking experiments are also present in the AutoDock docking profiles (Figure 3(A,B)).The IDV spatial conformation recruits less docking energy (-15.1 kcal/mol) in comparison to novel hit and forms H-bonds with amino acid residues of chain B. In this orientation, the amide nitrogen donor together with carboxyl group (hydroxyl moiety) of the Asp 29 residue, the Asp 25 carboxyl group (hydroxyl moiety) and the Gly 27 amide oxygen acceptor make hydrogen bonds with two hydroxyl functional groups and one amide nitrogen donor of the ligand molecule.
Figure 3.

(A) Indinavir and (B) novel hit docking profiles calculated by AutoDock software. Both ligands are gray. HIV protease chain A and B amino acids are painted in green and magenta respectively. The hydrogen bonds are shown for each of both molecules; see text for details.
Although, novel hit conformation interacts with both chains of the HIV protease but its global minimum in docked energy (-13.8 kcal/mol) with the protease binding site is more than that of IDV, resulting in decrease of binding affinity and intermolecular force between the ligand and its receptor. The Asp 29, Ile 50 amide nitrogen donors form H-bonds with N1 of the tetrahydropyrimidine ring and nitrogen acceptor of the aminoformonitrile residue. Carbonyl and hydroxyl moieties of both Asp 25 carboxyl groups are interacting with amine functional donor groups of novel hit. Asp 25, Asp 29, Gly 27 residues are located in the ‘eye’ areas, whereas Ile 50 residue is localized in the ‘flap’ region. The ‘flap’ region of the protease plays an important role in the enzyme “opening” and ligand binding to the ‘flap’ βhairpins could block “opening” process.Finally, we measured the hydrogen bond length by measuring the distance between the donor and acceptor atoms. Usually, longest H-bond length is about 3.5 Å. Anything longer of this parameter would be considered a pure dipole-dipole interaction. It is well established that hydrogen bonds have a typical length around 2.5 Å. In our case, all H-bond distances were in the range from 1.71 to 2.11 Å. Interestingly, one bond in IDV and two bonds in the hit molecule were detected as ultra-short hydrogen bonds with donor and acceptor distances of less than 2.0 Å. This observation supports further that identified the novel hit compound should have a tendency to form strong hydrogen bonds to the protease catalytic center.One drawback to IDV and novel hit is that, they both have a heterocyclic rings with complex substituents. Those features are responsible for the third class of toxicity (highly toxic). Both molecules belong to the second class of biodegradability, which means no alerts for notifying an easily degradable chemical were found and there was one alert notifying a persistent chemical the compound under investigation is declared as “persistent chemical”.
Conclusion
In the present study, we generated library of diverse chemical compounds on the bases of Indinavir and screened them for possible anti-protease activity. However, mutagenic efficiency of HIV is very high in generating new viral strains and could be bypassed by creating drugs with different binding properties and strong affinity to the target protein. The results are shown in this report, indicate that at least one compound has binding residues different from the initial hit, with improved affinity to the protease as well as its QSAR properties. Further studies on this compound will provide useful information towards the rational design of HIV-1 protease inhibitors and their effectiveness.
Supplementary material
Figure 1.

Chemical structure of IDV and its ‘derivate’- novel hit (N'-cyano-N-{3-[(1S, 2S)-1,2-diamino-2-(4-oxo-3, 4-dihydroquinazolin-7- yl)ethyl]-5-[(2R)-oxolan-2-yl]pyridin-2-yl}benzenecarboximidamide)
Acknowledgments
The authors are grateful to the DFG (SFB630/C6, Da 208/11-1) and the IZKF (Interdisziplinäres Zentrum für Klinische Forschung der Universität Würzburg) for the support of this work.
Footnotes
Citation:Shityakov & Dandekar , Bioinformation 4(7): 295-299 (2010)
References
- 1.York DM, et al. Biochemistry. 1993;32:3196. doi: 10.1021/bi00057a007. [DOI] [PubMed] [Google Scholar]
- 2.Kohl NE, et al. Proc Natl Acad Sci. 1988;85:4686. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorsey BD, et al. J Med Chem. 1994;37:3443. doi: 10.1021/jm00047a001. [DOI] [PubMed] [Google Scholar]
- 4.Vacca JP, et al. Proc Natl Acad Sci. 1994;91:4096. doi: 10.1073/pnas.91.9.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Condra JH, et al. J Virol. 1996;70:8270. doi: 10.1128/jvi.70.12.8270-8276.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Condra JH, et al. Nature. 1995;374:569. doi: 10.1038/374569a0. [DOI] [PubMed] [Google Scholar]
- 7.Poveda E, et al. J Antimicrob Chemother. 2007;60:885. doi: 10.1093/jac/dkm276. [DOI] [PubMed] [Google Scholar]
- 8.Cane PA, et al. J Clin Microbiol. 2001;39:2652. doi: 10.1128/JCM.39.7.2652-2654.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kempf DJ, et al. J Virol. 2001;75:7462. doi: 10.1128/JVI.75.16.7462-7469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coman RM, et al. Biochemistry. 2008;47:731. doi: 10.1021/bi7018332. [DOI] [PubMed] [Google Scholar]
- 11.Willard L, et al. Nucleic Acid Res. 2003;31:3316. doi: 10.1093/nar/gkg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pirok G, et al. J Chem Inf Model. 2006;46:563. doi: 10.1021/ci050373p. [DOI] [PubMed] [Google Scholar]
- 13.Petrek M, et al. BMC Bioinformatics. 2006;7:316. doi: 10.1186/1471-2105-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. http://autodock.scripps.edu/faqs-help/manual.
- 15.Cramer GM, et al. Food Cosmet Toxicol. 1978;16:255. doi: 10.1016/s0015-6264(76)80522-6. [DOI] [PubMed] [Google Scholar]
- 16.Thomsen R, Christensen MN. J Med Chem. 2006;49:3315. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 17.Tanrikuli Y, et al. Biochemistry. 2007;8:1932. [Google Scholar]
- 18.Wolber G, Langer T. J Chem Inf Model. 2005;45:160. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.