

Abstract
The use of the chemotherapeutic drug cisplatin is limited in part by nephrotoxicity. Cisplatin causes renal DNA adducts and oxidative stress in rodents. The transcription factor Nrf2 (nuclear factor E2-related factor 2) induces expression of cytoprotective genes, including Nqo1 (NADPH:quinone oxidoreductase 1), Ho-1 (heme oxygenase-1), and Gclc (glutamate cysteine ligase catalytic subunit), in response to electrophilic and oxidative stress. In the present study, plasma and kidneys from wild-type and Nrf2-null mice were collected after receiving cisplatin for evaluation of renal injury, inflammation, mRNA, and protein expression. Compared with wild types, more extensive nephrotoxicity was observed in Nrf2-null mice after cisplatin treatment. Kidneys from Nrf2-null mice treated with cisplatin had more neutrophil infiltration accompanied by increased p65 nuclear factor κB binding and elevated inflammatory mediator mRNA levels. Cisplatin increased renal mRNA and protein expression of cytoprotective genes (Nqo1, Ho-1, Gclc) and transporters Mrp2 and Mrp4 in wild-type but not in Nrf2-null mice. Lastly, the Nrf2 activator, CDDO-Im [2-cyano-3,12-dioxooleana-1,9-dien-28-oic imidazolide], increased Nrf2 signaling in kidneys from wild-type mice and protected them from cisplatin toxicity. Collectively, these data indicate that the absence of Nrf2 exacerbates cisplatin renal damage and that pharmacological activation of Nrf2 may represent a novel therapy to prevent kidney injury. Coordinated regulation of detoxification enzymes and drug transporters and suppression of inflammation by Nrf2 during cisplatin nephrotoxicity are probable defense mechanisms to eliminate toxic mediators and promote proximal tubule recovery.
Introduction
Cisplatin (cis-diamminedichloroplatinum(II)) is an effective antineoplastic drug for the treatment of solid tumors, although its use is often limited by impairment of renal function. Nephrotoxicity is observed in 32 to 38% of patients after a single dose of cisplatin (Shord et al., 2006). This side effect often delays or precludes subsequent chemotherapy cycles, thereby reducing overall antineoplastic efficacy. Prior research has investigated mechanisms involved in cisplatin-induced nephrotoxicity (Pabla and Dong, 2008). Upon uptake into the cell, cisplatin undergoes nonenzymatic hydrolysis to form aquated and electrophilic products through chloride ligand-exchange reactions (Mistry et al., 1989). Loss of labile chloride ligands results in nucleophilic substitution reactions with DNA and proteins, generation of oxidative stress, inflammation, increased cytosolic free calcium, and ultimately cell death (Pabla and Dong, 2008).
A number of signaling pathways, most notably those controlled by the nuclear factor E2-related factor 2 (Nrf2) transcription factor, are activated to counteract accumulating reactive oxygen species and electrophiles (Aleksunes and Manautou, 2007). Under basal conditions, Nrf2 is sequestered in the cytoplasm by the repressor protein Keap1 (Kelch-like ECH-associated protein 1) and targeted for proteasomal degradation (Itoh et al., 1999). Exposure to pharmacological activators, such as oltipraz or CDDO-Im (2-cyano-3,12-dioxooleana-1,9-dien-28-oic imidazolide) or generation of oxidative stress, triggers Nrf2 to translocate to the nucleus where it transactivates a battery of genes by binding to antioxidant-response elements (ARE) in upstream promoter regions (Friling et al., 1990; Rushmore et al., 1991). Targets of Nrf2 transcription include proteins involved in drug metabolism, efflux transporters (such as multidrug resistance-associated proteins, Mrps), antioxidant enzymes, heat shock responses, and proteasomal degradation.
Phenotypic characterization of Nrf2-null mice has yielded interesting findings. Electron paramagnetic resonance imaging showed that liver and kidneys from female Nrf2-null mice tended to have lower free radical-reducing abilities (Hirayama et al., 2003). Likewise, male Nrf2-null mice exhibit lower constitutive mRNA expression of the heat shock protein Ho-1 and the detoxification enzyme Nqo1 in their kidneys (Tanaka et al., 2008). Therefore, it is thought that Nrf2-null mice have an impaired capacity to quench free radicals and electrophiles in kidneys (Tanaka et al., 2008). Furthermore, it was reported that aged Nrf2-null female mice have accumulation of renal lipid peroxides and develop lupus-like autoimmune glomerulonephritis (Yoh et al., 2001; Li et al., 2004). Because of these findings and the knowledge that Nrf2 is a cytoprotective factor in various pathological processes, the purpose of this study was to comprehensively evaluate the susceptibility of Nrf2-null mice to cisplatin nephrotoxicity, with particular attention to renal apoptosis and necrosis, inflammation, adaptive gene response, and compensatory proliferation. Furthermore, it was determined whether pretreatment with the Nrf2 activator CDDO-Im protects kidneys from cisplatin toxicity.
Materials and Methods
Animals.
Wild-type and Nrf2-null mice were obtained from Dr. Jefferson Chan (University of California Irvine, Irvine, CA). The Institutional Animal Care and Use Committee approved the following studies.
Cisplatin Administration.
Cisplatin was dissolved in saline after heating to 50°C and cooled to room temperature before injection. Groups of adult male mice were injected with vehicle (10 ml/kg i.p.) or cisplatin (18 or 25 mg/kg i.p.) after overnight feed-deprivation. Doses of cisplatin used in this study are similar to those used clinically (Shord et al., 2006). Feed was returned to cages 4 h after cisplatin treatment. Mice were euthanized with an overdose of pentobarbital (50 mg/kg i.p.). Kidneys and plasma (in heparinized tubes) were collected at 1 and 4 h as well as 3, 4, 5, and 6 days after cisplatin administration. To determine urine flow rate, mice treated with vehicle or cisplatin (18 mg/kg i.p.) were placed into metabolism cages on day 4, and urine was collected for 6 h. Due to a limited number of metabolism cages, mice from each group were put together in the same cage, and urine volume was adjusted for time and animal weight. Portions of each kidney were fixed in 10% formalin. The remaining tissue was snap-frozen.
CDDO-Im Protection Study.
CDDO-Im (a gift of Dr. Michael Sporn, Dartmouth Medical School, Hanover, NH) was dissolved in dimethyl sulfoxide and diluted in sesame oil (final concentration of dimethyl sulfoxide, 2%). Vehicle (10 ml/kg) or CDDO-Im (3 and 10 mg/kg per day) was administered by oral gavage for 2 days. Tissues were collected 24 h after the last dose of CDDO-Im. Additional CDDO-Im-pretreated wild-type and Nrf2-null mice were feed-deprived overnight and administered cisplatin (20 mg/kg i.p.), and tissues were collected 4 days later.
Urea Nitrogen.
Blood urea nitrogen levels were determined as an indicator of renal injury (Thermotrace, Melbourne, VIC, Australia).
Histopathology.
Paraffin-embedded kidney sections (5 μm) were stained with hematoxylin and eosin and examined for histopathologic changes by a board-certified veterinary pathologist according to a published grading scale (Manautou et al., 1998). Neutrophils were counted from five to nine mice per group in three nonoverlapping fields at 40× magnification.
RNA Isolation and Messenger RNA Quantification.
Total RNA was isolated using RNA-Bee reagent (Tel-Test, Inc., Friendswood, TX). Renal mRNA expression was determined by the Quantigene Plex 1.0 and 2.0 Reagent System (Affymetrix Inc., Santa Clara, CA). Panomics Plex sets were used: oxidative stress (2.0 panel 21076) and inflammation (1.0 panel 2045). Samples were analyzed by using a Bio-Plex System Array reader (Bio-Rad Laboratories, Hercules, CA). Five (panel 2045) or 1 μg (panel 21076) of total RNA was used. Subsequent steps have been reported previously (Aleksunes et al., 2009).
Branched DNA Signal Amplification Assay.
The mRNA expression of mouse Kim-1 (kidney injury molecule-1), PCNA (proliferating cell nuclear antigen), c-Myc, Ki67, Topo2a (topoisomerase 2a), Nqo1, Ho-1, Gclc, Mrp2, Mrp4, and Mdr1b (multidrug resistance protein 1b) were quantified using the branched DNA 1.0 signal amplification assay (Affymetrix, Inc.) (Hartley and Klaassen, 2000). Novel oligonucleotide probe sets are provided in Supplemental Table 1.
Western Blot Analysis.
Nrf2 protein expression was determined in nuclear extracts prepared with the NE-PER nuclear protein extraction kit (Thermo Fisher Scientific, Rockford, IL). Cytosolic (Nqo1, Gclc) and membrane (Ho-1, Mrp2, Mrp4, Mdr1b) kidney fractions were prepared as described previously (Aleksunes et al., 2008).
Proteins (50 μg/lane) were electrophoretically resolved. Staining conditions and sources of antibodies are provided in Supplemental Table 2 and as described previously (Aleksunes et al., 2008). Equal protein loading was confirmed: β-actin protein for cytosolic and membrane proteins and histone H3 for nuclear proteins. The Discovery Series Quantity One 1-D software (Bio-Rad Laboratories) was used to quantify protein bands.
Immunohistochemical Staining.
Indirect immunofluorescence staining of Mrp2 and Mrp4 on frozen mouse kidney sections has been reported previously (Aleksunes et al., 2008). PCNA and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining were performed on paraffin-embedded and frozen kidney sections using the Zymed PCNA kit (Invitrogen, Carlsbad, CA) and Trevigen Apoptotic Cell System In Situ Apoptosis Detection Kit (R&D Systems, Minneapolis, MN), respectively. PCNA-positive and TUNEL-positive nuclei were counted from three to seven mice per group in three nonoverlapping fields at 40× magnification. Platinum-(GG) DNA adducts were quantified in frozen kidney sections and quantified as reported previously and expressed in arbitrary fluorescence units (Liedert et al., 2006).
Transcription Factor Binding Assays.
Nuclear extracts were used to quantify DNA binding of Nrf2 and the p65 subunit of nuclear factor κB (NFκB) using TransAm Transcription Factor Assay Kits (Kits 50296 and 40096; Active Motif, Inc., Carlsbad, CA). For DNA binding of Nrf2 to ARE in Mrp2, the TransAm Kit 50296 was customized to include a streptavidin-coated plate, and the TransAm Flexi Kit protocol was followed (Kit 43298; Active Motif, Inc.). A biotin-labeled duplex oligonucleotide for the mouse Mrp2 (5′-ACTGGGATGACATAGCATTCATC-3′) ARE was synthesized according to published sequences (IDT Technologies, Coralville, IA) (Vollrath et al., 2006; Maher et al., 2007). Unlabeled oligonucleotides as well as a mutant sequence (5′-ACTGGGAGTCAGACGCATTCATC-3′) were used for 40× competition experiments.
Statistical Analysis.
The software program Prism version 4 (GraphPad Software, Inc., La Jolla, CA) was used for statistical analysis. Differences among groups were evaluated by one-way analysis of variance followed by Newman-Keuls multiple-range test. Histopathological data were rank-ordered before statistical analysis. Differences were considered statistically significance at p < 0.05.
Results
Nephrotoxicity.
Cisplatin caused dose- and time-dependent renal injury in adult male wild-type and Nrf2-null mice (Fig. 1). Blood urea nitrogen levels increased similarly in both genotypes on day 3 (Fig. 1A). Four days after cisplatin treatment, urea nitrogen levels in Nrf2-null mice were higher than those in wild types, and the increase was dose-dependent (Fig. 1, A and B). On day 5, urea nitrogen levels returned to normal in cisplatin-treated wild-type mice but remained elevated in Nrf2-null mice (normal levels by 6 days). Higher blood urea nitrogen levels in Nrf2-null mice were in agreement, with a greater deficit in urinary flow rate (pooled values from multiple mice) (Fig. 1C) and marked up-regulation of renal Kim-1 mRNA expression, compared with wild-type mice (Fig. 1D).
Fig. 1.

Blood urea nitrogen, urinary rate, and kidney injury molecule-1 mRNA expression in wild-type and Nrf2-null mice after cisplatin treatment. A, blood urea nitrogen levels in wild-type and Nrf2-null mice 3 through 6 days after cisplatin (18 mg/kg i.p.) treatment (n = 4–15). B, blood urea nitrogen levels in wild-type and Nrf2-null mice 4 days after vehicle or cisplatin (18 or 25 mg/kg i.p.) treatment (n = 8–14). C, urine flow rate of control and cisplatin (18 mg/kg)-treated wild-type and Nrf2-null mice on day 4. Pooled urine volume was quantified from five to six mice per group in a metabolic cage for 6 h and normalized to body weight and time. D, messenger RNA expression of Kim-1 was quantified using total kidney RNA from control and cisplatin (18 mg/kg)-treated wild-type and Nrf2-null mice on day 4 (n = 4–5). Data are presented as means ± S.E. Messenger RNA data were normalized to wild-type control mice. Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. * represents statistically significant differences (p < 0.05) compared with genotype control mice. † represents a statistically significant difference (p < 0.05) from cisplatin-treated wild-type mice.
Histopathologic evaluation of kidneys from cisplatin-treated mice demonstrated cellular degeneration, necrosis, apoptosis, and sloughing of proximal tubule epithelium that was more severe in Nrf2-null mice (Fig. 2 and Table 1). Necrotic tubules containing eosinophilic amorphous material and pyknotic and karyorrhectic debris were more numerous at the higher dose of cisplatin (25 mg/kg), particularly in Nrf2-null mice.
Fig. 2.

Kidney histopathology in wild-type and Nrf2-null mice after cisplatin treatment. Wild-type and Nrf2-null mice were treated with cisplatin (18 or 25 mg/kg i.p.), and kidneys were collected on day 4. Samples were fixed in formalin before routine processing and paraffin embedding. Sections (5 μm) of kidneys were stained with hematoxylin and eosin and examined by light microscopy for the presence and severity of proximal tubule degeneration, apoptosis, and necrosis as well as renal cast formation and neutrophil infiltration. * denotes representative areas of protein casts (eosinophilic amorphous material); ↑ represents apoptotic cells; + represents tubular degeneration; and ^ represents epithelial cell loss.
TABLE 1.
Histopathological analysis of kidneys from wild-type and Nrf2-null mice after cisplatin
Kidneys were removed 4 days after cisplatin (18 or 25 mg/kg i.p.) or vehicle injection and fixed in zinc formalin before paraffin embedding and staining with hematoxylin and eosin. Kidney slices were evaluated for the severity of degeneration and necrosis in proximal tubule segments. Histopathology scoring of renal proximal tubule degeneration and necrosis by a veterinary pathologist: no injury = grade 0; minimal injury (less than 10% of cells with degeneration or necrosis) = grade 1; mild injury involving 10–25% of cells = grade 2; moderate injury involving 25–40% of cells = grade 3; marked injury involving 40–50% of cells = grade 4; severe injury involving greater than 50% of cells = grade 5. The number of mice with a particular histopathological grade is shown in each column. Mice with grades ≥2 are considered to have significant kidney injury. The ratio of mice with grades ≥2 compared to the total number of mice is presented as percentages in the right column. Histopathology grades were rank-ordered prior to statistical analysis.
| Histopathology Grade |
Percent of Mice with Grades of 2 or Greater | ||||||
|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | ||
| Wild-type | |||||||
| Control | 8 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cisplatin (18 mg/kg) | 1 | 11 | 2 | 0 | 0 | 0 | 14* |
| Cisplatin (25 mg/kg) | 1 | 6 | 2 | 2 | 0 | 2 | 46* |
| Nrf2-null | |||||||
| Control | 8 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cisplatin (18 mg/kg) | 0 | 3 | 1 | 2 | 5 | 1 | 75*† |
| Cisplatin (25 mg/kg) | 0 | 1 | 0 | 0 | 1 | 7 | 89*† |
Statistically significant differences (p < 0.05) compared with genotype control mice.
Statistically significant difference (p < 0.05) from cisplatin-treated wild-type mice.
Platinum-DNA Adducts and TUNEL Staining.
To determine whether Nrf2-null mice were exposed to greater kidney concentrations of cisplatin, we quantified the mRNA expression of the uptake organic cation transporter 2 (Oct2) and the binding of platinum to GG residues of DNA. Oct2/OCT2 is the primary transporter for renal uptake of cisplatin in mice and humans (Filipski et al., 2009). Messenger RNA expression of renal Oct2 was similar between genotypes (data not shown).
After administration of cisplatin to rodents, cisplatin rapidly accumulates in the kidneys, and the majority of the administered dose is eliminated within the first 12 to 24 h (Siddik et al., 1987). To assess kidney exposure to cisplatin, we quantified platinum (guanine-guanine, GG)-DNA adducts. Formation of platinum (GG)-DNA adducts in the proximal tubule epithelium and other cortical cells was similar in wild-type and Nrf2-null mice at 1 and 4 h after cisplatin administration (Fig. 3). In addition, mRNA expression of DNA repair enzymes was similarly regulated between genotypes on day 4 in response to cisplatin (data not shown). Four days after cisplatin, renal Xrcc1 (also known as X-ray repair complementing defective repair in Chinese hamster cells) mRNA was unchanged in wild-type and Nrf2-null mice, whereas 8-oxoguanine DNA-glycosylase 1 and apurinic/apyrimidinic endonuclease 1 mRNA were elevated similarly (1.5–2-fold) in both genotypes (data not shown). Collectively, differences in the binding of cisplatin to DNA and subsequent repair of adducts are not likely mechanisms for greater susceptibility of Nrf2-null mice to cisplatin nephrotoxicity.
Fig. 3.
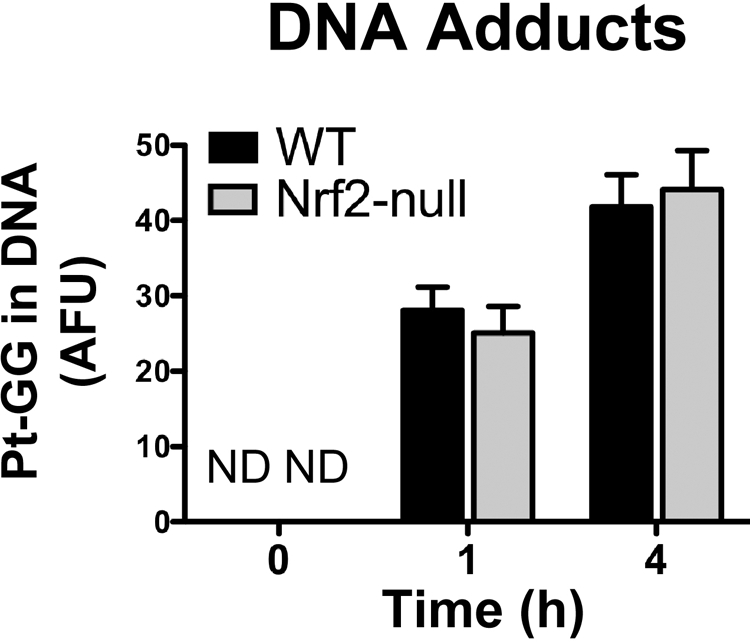
Platinum-DNA adducts in kidneys of wild-type and Nrf2-null mice after cisplatin treatment. Platinum-(GG) DNA adducts were quantified after immunofluorescent staining in frozen kidney sections (5 μm) from vehicle and cisplatin (18 mg/kg)-treated wild-type and Nrf2-null mice at 1 and 4 h according to Liedert et al. (2006). Adduct counts are expressed in arbitrary fluorescence units.
Because histological analysis suggested differences in proximal tubule cell apoptosis between genotypes (Fig. 2), TUNEL staining was performed (data not shown). TUNEL-positive nuclei were infrequently observed in vehicle-treated wild-type and Nrf2-null mice on day 4. Cisplatin treatment increased the number of TUNEL-positive nuclei in both genotypes. Compared with wild types, there were 2-fold more TUNEL-positive nuclei observed in Nrf2-null mice at the 18 mg/kg dose but not at 25 mg/kg (data not shown).
Compensatory Proliferation.
Messenger RNA analysis and immunohistochemical staining were used to assess compensatory proliferation (Fig. 4). Proliferation was quantified by PCNA staining of nuclei (brown) in wild-type and Nrf2-null mice (Fig. 4, A and B). Minimal PCNA-positive nuclei were observed in the kidneys of vehicle-treated mice (images not shown). Cisplatin increased the number of PCNA-positive nuclei in the kidneys of both genotypes in a dose-dependent manner at 4 days (Figs. 4, A and B); however, no differences in PCNA staining were observed between genotypes. The mRNA expression of cell cycle- and DNA synthesis-related genes was also quantified in kidneys from wild-type and Nrf2-null mice 4 days after cisplatin (Fig. 4C). There were no differences in PCNA, c-Myc, Ki67, or Topo2a mRNA between vehicle-treated wild-type and Nrf2-null mice. Cisplatin increased the mRNA expression of PCNA (2–3-fold) and c-Myc (5–8-fold) to similar extents in both genotypes. In contrast, Ki67 and Topo2a mRNA levels were increased to a greater extent in the kidneys of Nrf2-null mice (3.4–5.2- and 12.7–15-fold higher than genotype controls, respectively) than in wild-type mice.
Fig. 4.

Proliferation mRNA expression and immunohistochemical staining in kidneys of wild-type and Nrf2-null mice after cisplatin. A, PCNA staining (brown) in cisplatin (18 mg/kg)-treated wild-type and Nrf2-null kidney sections. Sections were counterstained with hematoxylin. Images were acquired at 40× magnification. B, PCNA staining was quantified in paraffin-embedded kidney sections (5 μm) from vehicle and cisplatin (18 and 25 mg/kg)-treated wild-type and Nrf2-null mice on day 4. PCNA-positive nuclei were quantified by counting three high-powered fields at 40× magnification. C, messenger RNA expression of PCNA, c-Myc, Ki67, and Topo2a was quantified using total kidney RNA from control and cisplatin (18 or 25 mg/kg)-treated wild-type and Nrf2-null mice on day 4. Data (n = 4–9) are presented as means ± S.E. Messenger RNA data are normalized to wild-type control mice. Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. * represents statistically significant differences (p < 0.05) compared with genotype control mice. † represents a statistically significant difference (p < 0.05) from cisplatin-treated wild-type mice.
Inflammation and NFκB Activation.
Inflammation is an important determinant of cisplatin-induced nephrotoxicity in rodents (Pabla and Dong, 2008). Few neutrophils were observed in kidneys of vehicle-treated wild-type and Nrf2-null mice. After cisplatin treatment, neutrophils were more numerous in kidneys of both genotypes, with higher numbers observed in Nrf2-null mice (Fig. 5A). NFκB is an important transcription factor involved in the acute phase inflammatory response (Guijarro and Egido, 2001). DNA binding of the p65 subunit of NFκB was increased on day 4 in response to cisplatin, particularly in Nrf2-null mice (Fig. 5B). In agreement with neutrophil accumulation and p65 binding, the mRNA expression of acute phase cytokines tumor necrosis factor α and interleukins (IL) 6 and 1β were induced to a greater extent in Nrf2-null mice (Fig. 5C). Additionally, the prostaglandin synthesis gene cyclooxygenase 2, the profibrogenic extracellular matrix gene Col1a1 (collagen 1a1), and the proinflammatory chemokine Ccl2 [chemokine (C-C motif) ligand 2] were preferentially higher in a dose-dependent manner in the kidneys of Nrf2-null mice (Fig. 5C). The mRNA expression of chemokine (C-X-C motif) ligands 1 and 10 were up-regulated in response to cisplatin, with little difference between genotypes, whereas Cxcl2 [chemokine (C-X-C motif) ligand 2] mRNA was elevated to a greater extent in Nrf2-null mice (Supplemental Fig. 1).
Fig. 5.

Neutrophil infiltration, p65 NFκB binding, and inflammatory mediator mRNA expression in kidneys of wild-type and Nrf2-null mice after cisplatin treatment. A, the number of neutrophils in three nonoverlapping high-powered fields were quantified in hematoxylin and eosin-stained kidney sections from vehicle or cisplatin (18 or 25 mg/kg)-treated wild-type and Nrf2-null mice on day 4. B, binding of kidney nuclear extracts from vehicle and cisplatin (18 or 25 mg/kg)-treated mice to p65 NFκB DNA-response element using an ELISA-based format. Data are presented as optical density (OD) at 450 nm. C, messenger RNA expression of tumor necrosis factor α, IL-6, IL-1β, cyclooxygenase 2, Col1a1, and Ccl2 was quantified using total kidney RNA from control and cisplatin (18 or 25 mg/kg)-treated wild-type and Nrf2-null mice on day 4. Data (n = 3–9) are presented as means ± S.E. Messenger RNA data were normalized to wild-type control mice. Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. * represents statistically significant differences (p < 0.05) compared with genotype control mice. † represents a statistically significant difference (p < 0.05) from cisplatin-treated wild-type mice.
Activation of Nrf2 Signaling.
Activation of Nrf2-mediated gene transcription is a mechanism for cell recovery after toxic insult (Aleksunes and Manautou, 2007). In response to cisplatin, Nrf2 mRNA expression in kidneys was increased 2-fold (Fig. 6A). Likewise, translocation of Nrf2 protein to the nucleus was enhanced 4-fold, and DNA binding to the ARE was enriched 2- to 3-fold in wild-type mice on day 4 after cisplatin administration (Fig. 6, B and C).
Fig. 6.

Nrf2 mRNA, DNA binding, and nuclear translocation in kidneys of wild-type and Nrf2-null mice after cisplatin treatment. A, messenger RNA expression of Nrf2 was quantified using total kidney RNA from control and cisplatin (18 or 25 mg/kg)-treated wild-type and Nrf2-null mice on day 4. B, kidney expression of Nrf2 protein was quantified by Western blot in nuclear extracts (50 μg of protein/lane) from cisplatin (18 mg/kg)-treated wild-type and Nrf2-null mice on day 4. Histone H3 was used as a loading control. The Western blot data are presented as individual blots and mean relative protein expression. C, binding of kidney nuclear extracts from vehicle and cisplatin (18 or 25 mg/kg)-treated mice to ARE using an ELISA-based format. Data are presented as optical density (OD) at 450 nm. Data (n = 3–5) are presented as means ± S.E. Messenger RNA and Western blot data are normalized to wild-type control mice. Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. * represents statistically significant differences (p < 0.05) compared with genotype control mice. † represents a statistically significant difference (p < 0.05) from cisplatin-treated wild-type mice. ND, not detected.
Renal Expression of Nrf2 Target Genes and Proteins.
Functional activation of Nrf2 signaling in response to cisplatin was evident by induction of a number of Nrf2 target genes and proteins (Fig. 7). Messenger RNA expression of Nqo1, Ho-1, and Gclc was up-regulated 3.5-, 5.5-, and 2.2-fold, respectively, in the kidneys of cisplatin-treated wild-type mice on day 4 (Fig. 7A). Little (Ho-1) or no change (Nqo1, Gclc) was observed in cisplatin-treated Nrf2-null mice. Parallel increases in protein expression of Nqo1, Ho-1, and Gclc occurred in kidneys of wild-type mice but not in Nrf2-null mice (Fig. 7B).
Fig. 7.

Renal mRNA and protein expression of Nrf2 targets in kidneys of wild-type and Nrf2-null mice after cisplatin. A, messenger RNA expression of Nrf2 targets (Nqo1, Ho-1, and Gclc) was quantified using total kidney RNA from control and cisplatin (18 mg/kg)-treated wild-type and Nrf2-null mice on day 4. B, kidney expression of Nrf2 target proteins (Nqo1, Ho-1, and Gclc) was quantified by Western blot using cytosol (Nqo1, Gclc) and membrane (Ho-1) preparations (50 μg of protein/lane) from cisplatin (18 mg/kg)-treated wild-type and Nrf2-null mice on day 4. β-Actin was used as a loading control. The Western blot data are presented as individual blots and mean relative protein expression. Data (n = 3–6) are normalized to wild-type controls and presented as means ± S.E. Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. * represents statistically significant differences (p < 0.05) compared with genotype control mice. † represents a statistically significant difference (p < 0.05) from cisplatin-treated wild-type mice.
Messenger RNA expression of additional detoxification and cytoprotective enzymes was quantified. Similar to Nqo1, thioredoxin reductase-1 mRNA was up-regulated 2.6- to 3.6-fold in a Nrf2-dependent manner 4 days after cisplatin treatment (Supplemental Fig. 2). Expression of epoxide hydrolase-1, glutaredoxin-1, metallothionein-1, and thioredoxin reductase-3 mRNA was elevated to a similar extent in cisplatin-treated wild-type and Nrf2-null mice (Supplemental Fig. 2).
Renal Expression of Efflux Transporter Genes and Proteins.
Mrp2, Mrp4, and Mdr1b are apical transporters on the brush-border membrane that efflux chemicals into urine. In addition to renal excretion of drugs, these transporters efflux signaling molecules that are involved in cellular injury and recovery, including glutathione, leukotrienes, prostaglandins, and cyclic nucleotides. Previous reports demonstrate renal induction of Mrp and Mdr1 genes in cisplatin-treated mice (Aleksunes et al., 2008) and rats (Thompson et al., 2004). Cisplatin increased mRNA expression on day 4 of Mrp2 (2-fold), Mrp4 (2.7-fold), and Mdr1b (6-fold) in wild-type but not in Nrf2-null mice (Fig. 8A). Expression of Mrp1, Mrp3, and Mdr1a mRNA was unchanged by cisplatin treatment (data not shown). Nrf2-dependent induction of Mrp2 (5.2-fold) and Mrp4 (3.7-fold) proteins was also observed in cisplatin-treated wild-type mice (Fig. 8B). Protein levels of Mdr1b (also named P-glycoprotein) were increased approximately 5-fold in both genotypes after cisplatin treatment. However, it should be noted that the antibody (C219) used to detect Mdr1b protein is not specific for only this isoform.
Fig. 8.

Renal mRNA and protein expression of efflux Mrp and Mdr transporters in kidneys of wild-type and Nrf2-null mice after cisplatin treatment. A, messenger RNA expression of Mrp2, Mrp4, and Mdr1b transporters was quantified using total kidney RNA from control and cisplatin (18 mg/kg)-treated wild-type and Nrf2-null mice on day 4. B, kidney expression of Mrp2, Mrp4, Mdr1b proteins was quantified by Western blot (50 μg of protein/lane) from cisplatin (18 mg/kg)-treated wild-type and Nrf2-null mice on day 4. β-Actin was used as a loading control. The Western blot data are presented as individual blots and mean relative protein expression. Data (n = 3–6) are normalized to wild-type controls and presented as means ± S.E. Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. * represents statistically significant differences (p < 0.05) compared with genotype control mice. † represents a statistically significant difference (p < 0.05) from cisplatin-treated wild-type mice.
Immunofluorescent detection confirmed apical staining of Mrp2 and Mrp4 proteins (Fig. 9, green) in proximal tubules of vehicle- and cisplatin-treated mice. There were no differences in staining intensity for either protein in kidneys of vehicle-treated wild-type and Nrf2-null mice. Increased Mrp2 and Mrp4 protein staining upon cisplatin treatment was observed in the kidneys of wild-type but not Nrf2-null mice.
Fig. 9.

Immunofluorescent staining of Mrp2 and Mrp4 in kidney sections of wild-type and Nrf2-null mice after cisplatin. Indirect immunofluorescence against brush-border membrane transporters Mrp2 and Mrp4 (green) was conducted on kidney cryosections (5 μm) obtained on day 4 from control and cisplatin (18 mg/kg)-treated wild-type and Nrf2-null mice. Representative cortex regions are shown. Magnification 20×.
To ascertain whether Mrp transporter mRNA induction in cisplatin-treated wild-type mice was due to binding of Nrf2 to regulatory response elements, an ELISA-based format was used to assess Nrf2 binding to the proximal ARE of Mrp2 at −185 bp. Using nuclear extracts from Nrf2-transfected COS-7 cells, we confirmed that Nrf2 binds to an ARE (−185 bp) of the mouse Mrp2 gene (Supplemental Fig. 3). Binding of Nrf2 to the biotinylated Mrp2 ARE was competed by unlabeled wild-type oligonucleotides but not mutant ARE oligonucleotides. Compared with vehicle controls, DNA binding of nuclear extracts from wild-type mice treated with cisplatin was increased 20 to 35% in a dose-dependent manner (Fig. 10). Mrp2 ARE DNA binding was unchanged in Nrf2-null mice.
Fig. 10.

Binding of Nrf2 to Mrp2 promoter ARE in kidneys of wild-type and Nrf2-null mice after cisplatin treatment. Binding of kidney nuclear extracts from vehicle and cisplatin-treated mice to the −185 bp of ARE of the mouse Mrp2 gene using an ELISA-based format. Data are presented as optical density (OD) at 450 nm. Data (n = 3–4) are normalized to wild-type controls and presented as means ± S.E. Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. * represents statistically significant differences (p < 0.05) compared with genotype control mice. † represents a statistically significant difference (p < 0.05) from cisplatin-treated wild-type mice.
Effect of CDDO-Im on Renal Nrf2 Signaling and Protection against Cisplatin-Induced Nephrotoxicity in Wild-Type and Nrf2-Null Mice.
Recently, the triterpenoid CDDO-Im was shown to activate Nrf2 signaling and protect against acetaminophen-induced liver injury (Reisman et al., 2009) as well as lipopolysaccharide-induced inflammation and mortality (Thimmulappa et al., 2006). The potential of CDDO-Im to prevent cisplatin-induced nephrotoxicity was assessed in wild-type and Nrf2-null mice. Two daily doses of CDDO-Im were administered to wild-type and Nrf2-null mice followed by a single dose of cisplatin (20 mg/kg) a day later. An intermediary dose of cisplatin was selected to achieve sufficient renal injury in wild-type mice to test CDDO-Im efficacy but not enough to cause irreparable toxicity in Nrf2-null mice. Blood urea nitrogen levels were assessed 4 days after cisplatin treatment and demonstrated dose-dependent lowering by CDDO-Im in wild-type mice (30–53%) and to a lesser extent in Nrf2-null mice (15–32%) (Fig. 11A). Histopathological evaluation of wild-type mice revealed that CDDO-Im pretreatment before cisplatin dosing reduced the severity of proximal tubule degeneration and necrosis as well as the incidence of significant injury (Fig. 11B; Table 2). Similar severity of renal injury was observed in cisplatin-treated Nrf2-null mice irrespective of vehicle- or CDDO-Im pretreatment, suggesting that CDDO-Im conferred protection against cisplatin nephrotoxicity via Nrf2.
Fig. 11.

Effect of CDDO-Im on cisplatin-induced nephrotoxicity in wild-type and Nrf2-null mice. A, wild-type and Nrf2-null mice were administered CDDO-Im (3 or 10 mg/kg per day p.o.) for 2 days, challenged with cisplatin (20 mg/kg i.p.), and evaluated 4 days later for changes in blood urea nitrogen. Data (n = 3–7) are presented as means ± S.E. Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. * represents statistically significant differences (p < 0.05) compared with genotype control mice. † represents a statistically significant difference (p < 0.05) from cisplatin-treated wild-type mice. B, samples were fixed in zinc formalin prior to routine processing and paraffin embedding. Sections (5 μm) of kidneys were stained with hematoxylin and eosin and examined by light microscopy for the presence and severity of proximal tubule degeneration, apoptosis, and necrosis as well as renal cast formation and neutrophil infiltration.
TABLE 2.
Histopathological analysis of kidneys from wild-type and Nrf2-null mice after CDDO-IM pretreatment and cisplatin challenge
Wild-type and Nrf2-null mice were pretreated with CDDO-Im (oral gavage, 3, 10 mg/kg per day for 2 days) and challenged with cisplatin (20 mg/kg i.p.). Kidneys were removed 4 days after cisplatin and fixed in formalin prior to paraffin embedding and staining with hematoxylin and eosin. Kidney slices were evaluated for the severity of degeneration and necrosis in proximal tubule segments. Histopathology scoring of renal proximal tubule degeneration and necrosis by a veterinary pathologist: no injury = grade 0; minimal injury (less than 10% of cells with degeneration or necrosis) = grade 1; mild injury involving 10–25% of cells = grade 2; moderate injury involving 25–40% of cells = grade 3; marked injury involving 40–50% of cells = grade 4; severe injury involving greater than 50% of cells = grade 5. The number of mice with a particular histopathological grade is shown in each column. Mice with grades ≥2 are considered to have significant kidney injury. The ratio of mice with grades ≥2 compared to the total number of mice is presented as percentages in the right column. Histopathology grades were rank-ordered prior to statistical analysis. * represents statistically significant differences (p < 0.05) compared with genotype control mice. † represents a statistically significant difference (p < 0.05) from treatment-matched wild-type mice.
| Histopathology Grade |
Percent of Mice with Grades of 2 or Greater | ||||||
|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | ||
| Wild-type | |||||||
| Control | 3 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cisplatin | 0 | 3 | 2 | 0 | 0 | 1 | 50* |
| Cisplatin/CDDO-Im (3 mg/kg) | 0 | 7 | 0 | 0 | 0 | 0 | 0 |
| Cisplatin/CDDO-Im (10 mg/kg) | 1 | 5 | 0 | 0 | 0 | 1 | 14 |
| Nrf2-null | |||||||
| Control | 3 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cisplatin | 0 | 2 | 0 | 1 | 2 | 1 | 67* |
| Cisplatin/CDDO-Im (3 mg/kg) | 0 | 1 | 1 | 1 | 0 | 4 | 86*† |
| Cisplatin/CDDO-Im (10 mg/kg) | 0 | 1 | 3 | 0 | 0 | 3 | 86*† |
In an attempt to determine whether CDDO-Im activates Nrf2 at the time of cisplatin administration, mice were dosed with CDDO-Im (3 and 10 mg/kg per day for 2 days), and kidneys were analyzed 24 h after the second dose. CDDO-Im increased the mRNA expression of Nqo1 dose-dependently and enriched DNA binding of Nrf2 to a prototypical ARE in wild-type mice (Fig. 12, A and B).
Fig. 12.

Effect of CDDO-Im on renal Nrf2 DNA binding and target gene expression in wild-type and Nrf2-null mice. A, messenger RNA expression of Nqo1 was quantified using total kidney RNA from control and CDDO-Im (3 or 10 mg/kg per day for 2 days p.o.)-treated wild-type and Nrf2-null mice 24 h after the last dose. B, binding of kidney nuclear extracts from vehicle and CDDO-Im-treated mice to the ARE using an ELISA-based format. Data are presented as optical density (OD) at 450 nm. Data (n = 3–5) are normalized to wild-type controls and presented as means ± S.E. Black bars represent wild-type mice, and gray bars represent Nrf2-null mice. * represents statistically significant differences (p < 0.05) compared with genotype control mice. † represents a statistically significant difference (p < 0.05) from cisplatin-treated wild-type mice.
Discussion
The present study investigated the mechanisms underlying the heightened sensitivity of Nrf2-null mice to cisplatin nephrotoxicity. Our findings are an in-depth extension of a recent report also demonstrating that renal function and survival are reduced in cisplatin-treated Nrf2-null mice (Liu et al., 2009). We have shown enhanced susceptibility of Nrf2-null mice by multiple endpoints (histopathology, urea nitrogen, urinary flow rate, Kim-1 expression, TUNEL). Differences in the initiation of toxicity and pharmacokinetics are unlikely because the two genotypes have similar expression of the uptake transporter (Oct2), an equal extent of DNA adduct formation, and comparable blood urea nitrogen elevations on day 3.
Subsequent experiments focused on inflammation, DNA repair, compensatory proliferation, and the adaptive gene response, which can influence the progression and recovery from renal damage. Compensatory proliferation was similar between the two genotypes 4 days after cisplatin, and in fact, up-regulation of some cell cycle and DNA synthesis genes (Ki67 and Topo2a) was higher in Nrf2-null mice, reflective of more damage. Instead, there were dramatic differences in the extent of inflammation between the two genotypes. In the kidneys of Nrf2-null mice, there were more neutrophils, greater induction of acute phase cytokines, fibrogenic and inflammatory mediators, and enriched DNA binding of NFκB, a key transcription factor in the regulation of inflammatory genes. Likewise, expression of detoxification, heat shock, and efflux transport genes and proteins was up-regulated in the kidneys of wild-type but not Nrf2-null mice. Therefore, an exaggerated inflammatory response and impaired adaptive gene regulation (as well as basal expression) likely sensitized Nrf2-null mice to cisplatin-induced nephrotoxicity. Lastly, CDDO-Im protected wild-type mice from cisplatin injury, suggesting the therapeutic utility of triterpenoid Nrf2 activators for renal disease. Collectively, this study capitalized on both genetic (Nrf2-null mice) and pharmacological (CDDO-Im) approaches to evaluate the roles of Nrf2 in cisplatin toxicity.
In addition to enhanced nuclear accumulation of Nrf2 protein, mRNA levels of Nrf2 were increased in kidneys from cisplatin-treated wild-type mice, likely reflecting autoregulation of Nrf2 via an ARE in its upstream promoter region (Kwak et al., 2002). Activation of Nrf2 in response to renal injury is not specific to cisplatin exposure and has been documented in vivo and in vitro after ischemia reperfusion (Leonard et al., 2006) or treatment with cadmium chloride (Chen and Shaikh, 2009), cephaloridine (Rokushima et al., 2008), and ochratoxin (Boesch-Saadatmandi et al., 2009). Likewise, Nrf2-null mice are more susceptible to ferric nitrilotriacetate nephrotoxicity (Kanki et al., 2008; Tanaka et al., 2008), ischemia-reperfusion renal injury (Liu et al., 2009), and diabetic nephropathy (Yoh et al., 2008). Although Nrf2 is important in limiting renal damage, other mechanisms appear to be activated in cisplatin-treated Nrf2-null mice to allow the kidney to repair, albeit at a delayed rate. Up-regulation of alternate protective genes (including epoxide hydrolase-1, glutaredoxin-1, metallothionein-1, and thioredoxin reductase-2) occurred in both genotypes and may have contributed to the ultimate recovery of Nrf2-null mice from cisplatin toxicity.
Efflux drug transporters can be important in toxicology by excreting the insulting toxicant (such as cisplatin). Mrp2 has been shown to transport cisplatin-glutathione conjugates (Ishikawa and Ali-Osman, 1993; Cui et al., 1999) and protect against platinum-DNA formation in cancer cells (Liedert et al., 2003; Materna et al., 2005). Thus, up-regulation of this transporter may enhance renal elimination of a subsequent exposure to cisplatin. In contrast, Mdr overexpression is not thought to be involved in cisplatin transport or cellular resistance (Hamaguchi et al., 1993). Up-regulation of Mrps occurs not only after cisplatin (Thompson et al., 2004; Aleksunes et al., 2008) but also after ferric nitrilotriacetate (Tanaka et al., 2008) and cephaloridine toxicity (Rokushima et al., 2008), suggesting that this event is one component of a general adaptive response of the kidney regardless of the toxicant. In these cases, up-regulation of efflux transporters in diverse models of nephrotoxicity suggests that transporters may contribute to the repair of proximal tubules by effluxing byproducts of toxicity (such as oxidized glutathione, which is a substrate of Mrp2) (Keppler et al., 1997) or paracrine signaling of endogenous cellular mediators (such as cyclic nucleotides, leukotrienes, and prostaglandins) (Toyoda et al., 2008). For example, prostaglandin production increases in rat kidneys within 3 days after cisplatin treatment and appears to be responsible for changes in renal concentrating ability (Moel et al., 1987). It is possible that expression of transporters, such as Mrp2 and Mrp4, that efflux prostaglandins (Reid et al., 2003; de Waart et al., 2006) may be enhanced to regulate the intracellular and extracellular levels of prostaglandins during injury. Moreover, increases in Mrp2 and Mrp4 may be part of the global Nrf2 transcriptional response in response to tissue damage. Nrf2-mediated Mrp2 and Mrp4 induction occurs in the liver and in vitro (Vollrath et al., 2006; Maher et al., 2007). However, the present study extends this phenomenon to the kidneys and provides evidence that Nrf2-dependent regulation of Mrp2 during nephrotoxicity probably involves direct transcription factor binding.
Suppression of inflammation alleviates cisplatin toxicity (Pabla and Dong, 2008). Renal inflammation is exaggerated in Nrf2-null mice after cisplatin, probably due to enhanced p65 NFκB binding. Because inflammation and proliferative responses typically follow each other, it was unexpected that there was little difference in proliferation-related pathways between genotypes. Evaluation of PCNA staining on day 4 revealed a similar extent of stained nuclei in kidneys of wild-type and Nrf2-null mice after cisplatin; however, subsequent time points were not evaluated. It is likely that the delayed recovery of Nrf2-null mice to cisplatin toxicity is due to enhanced inflammation, blunted defensive gene transactivation, and impaired cellular repair.
CDDO-Im is a potent activator of Nrf2 and up-regulates target genes in multiple tissues (Liby et al., 2005; Yates et al., 2007). Pretreatment with CDDO-Im protected wild-type mice against cisplatin toxicity, with limited protection of Nrf2-null mice. CDDO-Im not only activates Nrf2 but can suppress NFκB by directly inhibiting IκB kinase β, thus explaining some protection of Nrf2-null mice (Yore et al., 2006). CDDO-Im-mediated protection is probably due to suppression of inflammation as well as coordinated up-regulation of detoxification and transport genes (Liby et al., 2005; Yates et al., 2007). Moreover, CDDO-Im may represent a novel effective drug for protecting the kidneys via Nrf2 signaling. In preliminary studies, we treated mice with other known Nrf2 activators (including oltipraz, butylated hydroxyanisole, ethoxyquin, and sulforaphane) at doses that enhanced Nrf2-mediated transcription in liver but not in kidneys (data not shown). It is currently unknown whether the discrepancy in tissue-specific Nrf2 activation represents differences in the pharmacokinetics or pharmacodynamics of CDDO-Im in mice compared with other known Nrf2 activators. Additional studies should be designed to better delineate how CDDO-Im activates Nrf2 in the kidneys.
In conclusion, the absence of Nrf2 exacerbates cisplatin-induced nephrotoxicity in mice, and pharmacological Nrf2 activation may be a novel therapeutic strategy to suppress renal injury. Moreover, these findings mechanistically reflect the stages of toxicity and repair that are modulated by Nrf2 signaling and demonstrate that coordinated regulation of detoxification enzymes and transporters and suppression of inflammation by Nrf2 are key events in proximal tubule cell recovery.
Supplementary Material
Acknowledgments
We thank K. Brian Lee for assistance with imaging, Dr. Sarah Campion for assistance with pilot studies, Dr. Scott Reisman for assistance with mRNA analysis, and graduate students and fellows of the Klaassen laboratory for contributions to this project.
This work was supported by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grants DK080774, DK081461]; the National Institutes of Health National Institute of Environmental Health Sciences [Grants ES009649, ES009716, ES007079, ES013714]; and the National Institutes of Health National Center for Research Resources [Grant RR021940].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.110.170084.
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
- Nrf2
- nuclear factor erythroid 2-related factor 2
- CDDO-Im
- 2-cyano-3,12-dioxooleana-1,9-dien-28-oic imidazolide
- ARE
- antioxidant-response element
- Mrp(s)
- multidrug resistance-associated protein(s)
- Ho-1
- heme oxygenase-1
- Nqo1
- NADPH:quinone oxidoreductase 1
- PCNA
- proliferating cell nuclear antigen
- Mdr1b
- multidrug resistance protein 1b
- Kim-1
- kidney injury molecule-1
- Topo2a
- topoisomerase 2a
- Gclc
- glutamate cysteine ligase, catalytic subunit
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick-end labeling
- NFκB
- nuclear factor κB
- Oct2
- organic cation transporter 2
- IL
- interleukin
- ELISA
- enzyme-linked immunosorbent assay
- bp
- base pair.
References
- Aleksunes LM, Augustine LM, Scheffer GL, Cherrington NJ, Manautou JE. (2008) Renal xenobiotic transporters are differentially expressed in mice following cisplatin treatment. Toxicology 250:82–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksunes LM, Manautou JE. (2007) Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol Pathol 35:459–473 [DOI] [PubMed] [Google Scholar]
- Aleksunes LM, Yeager RL, Klaassen CD. (2009) Application of multivariate statistical procedures to identify transcription factors that correlate with MRP2, 3, and 4 mRNA in adult human livers. Xenobiotica 39:514–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesch-Saadatmandi C, Wagner AE, Graeser AC, Hundhausen C, Wolffram S, Rimbach G. (2009) Ochratoxin A impairs Nrf2-dependent gene expression in porcine kidney tubulus cells. J Anim Physiol Anim Nutr (Berl) 93:547–554 [DOI] [PubMed] [Google Scholar]
- Chen J, Shaikh ZA. (2009) Activation of Nrf2 by cadmium and its role in protection against cadmium-induced apoptosis in rat kidney cells. Toxicol Appl Pharmacol 241:81–89 [DOI] [PubMed] [Google Scholar]
- Cui Y, König J, Buchholz JK, Spring H, Leier I, Keppler D. (1999) Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol Pharmacol 55:929–937 [PubMed] [Google Scholar]
- de Waart DR, Paulusma CC, Kunne C, Oude Elferink RP. (2006) Multidrug resistance associated protein 2 mediates transport of prostaglandin E2. Liver Int 26:362–368 [DOI] [PubMed] [Google Scholar]
- Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, Sparreboom A. (2009) Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther 86:396–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friling RS, Bensimon A, Tichauer Y, Daniel V. (1990) Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc Natl Acad Sci USA 87:6258–6262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guijarro C, Egido J. (2001) Transcription factor-kappa B (NF-kappa B) and renal disease. Kidney Int 59:415–424 [DOI] [PubMed] [Google Scholar]
- Hamaguchi K, Godwin AK, Yakushiji M, O'Dwyer PJ, Ozols RF, Hamilton TC. (1993) Cross-resistance to diverse drugs is associated with primary cisplatin resistance in ovarian cancer cell lines. Cancer Res 53:5225–5232 [PubMed] [Google Scholar]
- Hartley DP, Klaassen CD. (2000) Detection of chemical-induced differential expression of rat hepatic cytochrome P450 mRNA transcripts using branched DNA signal amplification technology. Drug Metab Dispos 28:608–616 [PubMed] [Google Scholar]
- Hirayama A, Yoh K, Nagase S, Ueda A, Itoh K, Morito N, Hirayama K, Takahashi S, Yamamoto M, Koyama A. (2003) EPR imaging of reducing activity in Nrf2 transcriptional factor-deficient mice. Free Radic Biol Med 34:1236–1242 [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Ali-Osman F. (1993) Glutathione-associated cis-diamminedichloroplatinum(II) metabolism and ATP-dependent efflux from leukemia cells. Molecular characterization of glutathione-platinum complex and its biological significance. J Biol Chem 268:20116–20125 [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13:76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki K, Umemura T, Kitamura Y, Ishii Y, Kuroiwa Y, Kodama Y, Itoh K, Yamamoto M, Nishikawa A, Hirose M. (2008) A possible role of nrf2 in prevention of renal oxidative damage by ferric nitrilotriacetate. Toxicol Pathol 36:353–361 [DOI] [PubMed] [Google Scholar]
- Keppler D, Leier I, Jedlitschky G. (1997) Transport of glutathione conjugates and glucuronides by the multidrug resistance proteins MRP1 and MRP2. Biol Chem 378:787–791 [PubMed] [Google Scholar]
- Kwak MK, Itoh K, Yamamoto M, Kensler TW. (2002) Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol 22:2883–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard MO, Kieran NE, Howell K, Burne MJ, Varadarajan R, Dhakshinamoorthy S, Porter AG, O'Farrelly C, Rabb H, Taylor CT. (2006) Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury. FASEB J 20:2624–2626 [DOI] [PubMed] [Google Scholar]
- Li J, Stein TD, Johnson JA. (2004) Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol Genomics 18:261–272 [DOI] [PubMed] [Google Scholar]
- Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, et al. (2005) The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res 65:4789–4798 [DOI] [PubMed] [Google Scholar]
- Liedert B, Materna V, Schadendorf D, Thomale J, Lage H. (2003) Overexpression of cMOAT (MRP2/ABCC2) is associated with decreased formation of platinum-DNA adducts and decreased G2-arrest in melanoma cells resistant to cisplatin. J Invest Dermatol 121:172–176 [DOI] [PubMed] [Google Scholar]
- Liedert B, Pluim D, Schellens J, Thomale J. (2006) Adduct-specific monoclonal antibodies for the measurement of cisplatin-induced DNA lesions in individual cell nuclei. Nucleic Acids Res 34:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Grigoryev DN, Crow MT, Haas M, Yamamoto M, Reddy SP, Rabb H. (2009) Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int 76:277–285 [DOI] [PubMed] [Google Scholar]
- Maher JM, Dieter MZ, Aleksunes LM, Slitt AL, Guo G, Tanaka Y, Scheffer GL, Chan JY, Manautou JE, Chen Y, et al. (2007) Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology 46:1597–1610 [DOI] [PubMed] [Google Scholar]
- Manautou JE, Silva VM, Hennig GE, Whiteley HE. (1998) Repeated dosing with the peroxisome proliferator clofibrate decreases the toxicity of model hepatotoxic agents in male mice. Toxicology 127:1–10 [DOI] [PubMed] [Google Scholar]
- Materna V, Liedert B, Thomale J, Lage H. (2005) Protection of platinum-DNA adduct formation and reversal of cisplatin resistance by anti-MRP2 hammerhead ribozymes in human cancer cells. Int J Cancer 115:393–402 [DOI] [PubMed] [Google Scholar]
- Mistry P, Lee C, McBrien DC. (1989) Intracellular metabolites of cisplatin in the rat kidney. Cancer Chemother Pharmacol 24:73–79 [DOI] [PubMed] [Google Scholar]
- Moel DI, Safirstein RL, Cohn RA, Penning J. (1987) The role of prostaglandins in early polyuria induced by cisplatin in the rat. Nephron 46:91–95 [DOI] [PubMed] [Google Scholar]
- Pabla N, Dong Z. (2008) Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73:994–1007 [DOI] [PubMed] [Google Scholar]
- Reid G, Wielinga P, Zelcer N, van der Heijden I, Kuil A, de Haas M, Wijnholds J, Borst P. (2003) The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci USA 100:9244–9249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisman SA, Buckley DB, Tanaka Y, Klaassen CD. (2009) CDDO-Im protects from acetaminophen hepatotoxicity through induction of Nrf2-dependent genes. Toxicol Appl Pharmacol 236:109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokushima M, Fujisawa K, Furukawa N, Itoh F, Yanagimoto T, Fukushima R, Araki A, Okada M, Torii M, Kato I, et al. (2008) Transcriptomic analysis of nephrotoxicity induced by cephaloridine, a representative cephalosporin antibiotic. Chem Res Toxicol 21:1186–1196 [DOI] [PubMed] [Google Scholar]
- Rushmore TH, Morton MR, Pickett CB. (1991) The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem 266:11632–11639 [PubMed] [Google Scholar]
- Shord SS, Thompson DM, Krempl GA, Hanigan MH. (2006) Effect of concurrent medications on cisplatin-induced nephrotoxicity in patients with head and neck cancer. Anticancer Drugs 17:207–215 [DOI] [PubMed] [Google Scholar]
- Siddik ZH, Newell DR, Boxall FE, Harrap KR. (1987) The comparative pharmacokinetics of carboplatin and cisplatin in mice and rats. Biochem Pharmacol 36:1925–1932 [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Aleksunes LM, Goedken MJ, Chen C, Reisman SA, Manautou JE, Klaassen CD. (2008) Coordinated induction of Nrf2 target genes protects against iron nitrilotriacetate (FeNTA)-induced nephrotoxicity. Toxicol Appl Pharmacol 231:364–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW, Biswal S. (2006) Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-imidazolide. Biochem Biophys Res Commun 351:883–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KL, Afshari CA, Amin RP, Bertram TA, Car B, Cunningham M, Kind C, Kramer JA, Lawton M, Mirsky M, et al. (2004) Identification of platform-independent gene expression markers of cisplatin nephrotoxicity. Environ Health Perspect 112:488–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda Y, Hagiya Y, Adachi T, Hoshijima K, Kuo MT, Ishikawa T. (2008) MRP class of human ATP binding cassette (ABC) transporters: historical background and new research directions. Xenobiotica 38:833–862 [DOI] [PubMed] [Google Scholar]
- Vollrath V, Wielandt AM, Iruretagoyena M, Chianale J. (2006) Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem J 395:599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T, Gribble GW, Johnson DA, Johnson JA, Burton NC, et al. (2007) Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther 6:154–162 [DOI] [PubMed] [Google Scholar]
- Yoh K, Hirayama A, Ishizaki K, Yamada A, Takeuchi M, Yamagishi S, Morito N, Nakano T, Ojima M, Shimohata H, et al. (2008) Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells 13:1159–1170 [DOI] [PubMed] [Google Scholar]
- Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, Morito N, Koyama A, Yamamoto M, Takahashi S. (2001) Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int 60:1343–1353 [DOI] [PubMed] [Google Scholar]
- Yore MM, Liby KT, Honda T, Gribble GW, Sporn MB. (2006) The synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole blocks nuclear factor-kappaB activation through direct inhibition of IkappaB kinase beta. Mol Cancer Ther 5:3232–3239 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.