

Abstract
Lecithin cholesterol acyl transferase (LCAT) deficiency is associated with low high-density lipoprotein (HDL) and the presence of an abnormal lipoprotein called lipoprotein X (Lp-X) that contributes to end-stage renal disease. We examined the possibility of using LCAT an as enzyme replacement therapy agent by testing the infusion of human recombinant (r)LCAT into several mouse models of LCAT deficiency. Infusion of plasma from human LCAT transgenic mice into LCAT-knockout (KO) mice rapidly increased HDL-cholesterol (C) and lowered cholesterol in fractions containing very-low-density lipoprotein (VLDL) and Lp-X. rLCAT was produced in a stably transfected human embryonic kidney 293f cell line and purified to homogeneity, with a specific activity of 1850 nmol/mg/h. Infusion of rLCAT intravenously, subcutaneously, or intramuscularly into human apoA-I transgenic mice showed a nearly identical effect in increasing HDL-C approximately 2-fold. When rLCAT was intravenously injected into LCAT-KO mice, it showed a similar effect as plasma from human LCAT transgenic mice in correcting the abnormal lipoprotein profile, but it had a considerably shorter half-life of approximately 1.23 ± 0.63 versus 8.29 ± 1.82 h for the plasma infusion. rLCAT intravenously injected in LCAT-KO mice crossed with human apolipoprotein (apo)A-I transgenic mice had a half-life of 7.39 ± 2.1 h and increased HDL-C more than 8-fold. rLCAT treatment of LCAT-KO mice was found to increase cholesterol efflux to HDL isolated from mice when added to cells transfected with either ATP-binding cassette (ABC) transporter A1 or ABCG1. In summary, rLCAT treatment rapidly restored the normal lipoprotein phenotype in LCAT-KO mice and increased cholesterol efflux, suggesting the possibility of using rLCAT as an enzyme replacement therapy agent for LCAT deficiency.
Introduction
Deficiency of lecithin cholesterol acyltransferase (LCAT; EC2.3.1.43), first described in 1962 (Glomset, 1962), can present with two different phenotypes, familial LCAT deficiency (FLD) and fish eye disease (FED) (Santamarina-Fojo et al., 2001; Rousset et al., 2009). Patients with FLD have almost an absence of LCAT activity. In contrast, patients with FED have partial LCAT activity, especially on LDL, and thus are relatively asymptomatic, with the exception of the deposition of cholesterol in their corneas. In addition to corneal cholesterol deposits, patients with FLD develop hepatosplenomegaly, normochromic normocytic anemia, and hypertension. Renal disease, however, is the main cause of morbidity in these patients. It usually presents as proteinuria that progresses to nephrotic syndrome, glomerulosclerosis, and eventually end-stage renal disease, as early as the third or fourth decade of life (Santamarina-Fojo et al., 2001). Low levels of HDL-C (<16 mg/dl for FLD; <27 mg/dl for FED) are typically observed in both disorders. In addition, patients with FLD often have a mild-to-moderate increase in triglycerides and produce an abnormal lipoprotein called Lp-X. This relatively large lipoprotein is devoid of cholesteryl esters (CE) and has excess phospholipids, thus it forms vesicular- or multilamellar-like structures. Lp-X is thought to contribute to the renal disease when it is filtered by the kidney and accumulates in mesangial cells of the glomerulus (Imbasciati et al., 1986).
LCAT is secreted into plasma by the liver and associates mostly with HDL, but also with LDL (Cheung et al., 1986). It catalyzes the conversion of cholesterol on the surface of lipoprotein particles to CE. LCAT cleaves fatty acids from phosphatidylcholine by a phospholipase A2-like activity and transfers the acyl group to the hydroxyl group on the A-ring of cholesterol (Rousset et al., 2009). Because CE is more hydrophobic than cholesterol, it partitions into the core of lipoproteins, which transforms nascent discoidally shaped HDL (pre-β-HDL) into spherically shaped HDL (α-HDL), with a neutral lipid core. When HDL does not undergo this maturation process, small pre-β-HDL is rapidly catabolized, leading overall to low HDL levels. LCAT is also believed to be a key enzyme in the reverse cholesterol transport (RCT) pathway, because esterification of cholesterol on HDL increases the concentration gradient for the movement of free cholesterol from cells onto HDL by the various cell transporters that efflux cholesterol (Zannis et al., 2006). HDL delivers its CE to the liver by the SR-BI receptor, and cholesterol is then excreted into the bile as cholesterol or as a bile salt, thus completing the RCT pathway (Zhang et al., 2005).
There is no specific treatment for FLD; patients are usually treated symptomatically and are candidates for corneal and renal transplantation, although the disease can reoccur in transplanted tissue (Panescu et al., 1997). There have been several case reports of lipid and lipoprotein abnormalities in patients with FLD being temporarily corrected, after transfusion of normal plasma containing LCAT (Murayama et al., 1984). Based on these studies, the half-life of human LCAT in plasma has been estimated to be 4 to 5 days (Stokke et al., 1974), which raises the possibility that FLD could be treated like several lysosomal enzyme storage diseases by enzyme replacement therapy (Brady, 2006). Because LCAT acts in the plasma compartment, it does not need to be targeted to a specific organ or cellular location like the lysosome to be effective. Other features that make LCAT an attractive target for enzyme replacement therapy are that it is a small single-subunit enzyme of 67 kDa and does not require any specific cofactors, with the exception of apoA-I, which is abundant in plasma. LCAT is also relatively stable and easy to produce in large quantities by recombinant protein expression systems (Lane et al., 2004). Furthermore, the concentration of LCAT in normal human plasma is a relatively low, approximately 5 to 6 μg/ml, and restoration of only 10 to 15% of activity is enough to prevent most of the manifestations of FLD, including kidney disease (Rousset et al., 2009).
In this study, we report on the production of human recombinant (r)LCAT and investigate its effect on lipid and lipoprotein metabolism in several mouse models, including LCAT-knockout (KO) mice. Intravenous infusion of rLCAT was found to rapidly raise HDL-C and correct the other lipid abnormalities in LCAT-KO mice, thus indicating that LCAT enzyme replacement therapy may be a useful approach for treating FLD.
Materials and Methods
Animals Procedures.
LCAT-KO and human LCAT-transgenic (Tg) mice were described previously (Vaisman et al., 1995; Lambert et al., 2001). LCAT-KO/apoA-I-Tg mice were produced by crossing human apoA-I-Tg mice [strain C57BL/6-Tg()1Rub/J, stock 001927; The Jackson Laboratory, Bar Harbor, ME] with LCAT-KO mice. All mice were fed ad libitum with a standard chow diet (NIH31 chow diet; Zeigler Brothers Inc., Gardners, PA). Intravenous treatments were done by injection into the retro-orbital sinus with a 27.5-gauge needle. Blood samples were collected from the periorbital sinus of the contralateral eye, with a heparinized capillary tube (50 or 250 μl) and placed into the tubes with K-EDTA as an anticoagulant (final concentration, 4 mM). Plasma was obtained after centrifugation for 10 min at 3000g at 4°C. LCAT activity in plasma was heat-inactivated by treatment for 20 min at 56°C, which inhibited more than 95% of activity. All animal procedures were approved by a National Institutes of Health Institutional Animal Care Committee (protocol H-0050R1).
Recombinant Human LCAT Production and Purification.
The plasmid pCMV6-XL4/LCAT, encoding human LCAT cDNA, was purchased from Origene (Rockville, MD) and ligated into pcDNA3.1/Hygro (Invitrogen, Carlsbad, CA). Stably transfected HEK293f cells were selected with 200 μg/ml hygromycin B and grown in Freestyle 293 serum-free medium (Invitrogen) in 10-liter shake flasks for 4 days. rLCAT was isolated from conditioned cell culture medium by precipitation with zinc chloride (Zaworski and Gill, 1988), followed by batch capture with phenyl-Sepharose (GE Healthcare, Little Chalfont, Buckinghamshire, UK) and batch elution, with 20 mM Tris and 0.5 M NaCl. Approximately 8 mg of rLCAT could be purified per liter of conditioned media.
LCAT Activity Assay.
LCAT activity was measured, using a proteoliposome as a substrate (Albers et al., 1986), with the following modifications (Vaisman and Remaley, 2010). Proteoliposomes were made by mixing of [14C]cholesterol (1.25 μCi/ml; PerkinElmer Life and Analytical Sciences, Boston, MA) and cholesterol (final concentration, 72 μM; Sigma-Aldrich, St. Louis, MO) and phosphatidylcholine (1.2 mM, α-l-lecithin; Calbiochem-EMD, La Jolla, CA) in chloroform and drying down under nitrogen. Lipids were solubilized in an assay buffer (110 mM Tris-HCl, 140 mM NaCl, and 1 mM EDTA, pH 7.4) containing an amphipathic peptide ETC-642 (PVLDLFRELLNELLEALKQKLK; Busseuil et al., 2008) and sodium cholic acid (final concentration, 62 mM; Sigma-Aldrich). After dialysis, proteoliposomes were stabilized by adding of equal volume of 2% BSA (w/v) with β-mercaptoethanol (10 mM) in the assay buffer and then incubated 20 min at 37°C. Proteoliposomes were then diluted 1.2-fold with assay buffer containing β-mercaptoethanol (35 mM) and stored in liquid nitrogen. To measure LCAT activity, 1 to 5 μl of sample was incubated with 60 μl of proteoliposomes for 30 min at 37°C. Reaction was stopped by adding of 1 ml of 100% cold (−20°C) ethanol, and samples were kept 20 min on dry ice and centrifuged for 10 min at 10,000g at 4°C. Supernatant was removed, evaporated in a Speed-Vac concentrator (Thermo Fisher Scientific, Waltham, MA), and resuspended in 30 μl of chloroform containing cholesterol (0.1 mg/ml) and cholesteryl oleate (0.1 mg/ml). After separation by thin layer chromatography (PE SIL G plates; Whatman, Clifton, NJ) with a mixture of diethyl ether (60 ml), petroleum ether (293 ml), and acetic acid (3 ml), radioactivity in the cholesterol and cholesteryl ester spots were measured by liquid scintillation counting. One unit of LCAT activity was defined as 1 nmol/ml/h of cholesteryl ester produced at 37°C.
Lipid and Lipoprotein Analysis.
Total cholesterol (Wako Chemicals, Richmond, VA), free cholesterol (Wako Chemicals), and triglycerides (Roche Diagnostics, Indianapolis, IN) were determined enzymatically, using a Victor3 plate reader (PerkinElmer Life and Analytical Sciences). Lipoproteins were fractionated by FPLC (Akta FPLC; GE Healthcare) on two Superose 6 columns in series. HDL (density = 1.21–1.063 g/ml) was isolated by density gradient ultracentrifugation (Schumaker et al., 1986). HDL-C was determined enzymatically from serum after precipitation of apoB-containing lipoproteins, with dextran sulfate/magnesium (Raichem, San Diego, CA). A semiautomated electrophoretic system (Hydrasys; Sebia, Norcross, GA), with agarose gels (Hydragel Lipoprotein[e] 15/30), was used for separation and detection of serum lipoproteins, after staining with Sudan black. Detection of apolipoproteins in gels was done by immunofixation, using a modified Sebia immunofixation electrophoresis method. Purified goat anti-human apoA-I (Meridian Life Science, Inc., Saco, ME) and anti-human apoB-48/100 (Meridian Life Science, Inc.) were used to stain the gels. Immunoprecipitates in the gel were detected by acid violet stain. Human serum from a 47-year-old FLD male, with the following lipid and lipoprotein test values, was obtained under an institutional review board-approved protocol/ total cholesterol, 175 mg/dl; triglycerides, 499 mg/dl; HDL-C, <5 mg/dl; apoA-I, 28 mg/dl; and apoB, <30 mg/dl.
Cholesterol Efflux Assay.
Baby hamster kidney (BHK) cells stably transfected with human ABCA1 (Oram et al., 2001), human ABCG1 (Vaughan and Oram, 2005), or human SR-BI, using the mifepristone inducible expression system (Invitrogen), were labeled with [3H]cholesterol (1 μCi/ml) for 18 h in DMEM with 10% fetal bovine serum. The cells were then washed and induced with mifepristone (10 nM) in DMEM plus 1 mg/ml BSA for 24 h. Efflux media were added for the time interval indicated, and released [3H]cholesterol was monitored by liquid scintillation counting. Residual counts in the cells were measured after solubilization in hexane/isopropanol (1:2). Efflux results are expressed as percentage of total radioactive counts effluxed into the media over time.
Statistical Analysis.
Unless otherwise indicated, all results are presented as the mean ± 1 S.D. of at least three replicates. Differences between groups were analyzed by unpaired t test and differences with P < 0.05 were considered to be significant.
Results
Effect of Infusion of Plasma from LCAT-Tg Mice into LCAT-KO Mice.
To test the feasibility of using rLCAT for enzyme replacement therapy, we first examined the effect of infusion of plasma from human LCAT-Tg mice into LCAT-KO mice. LCAT-Tg plasma contains 6600 units/ml LCAT activity, which is 220-fold higher than normal mouse plasma (Vaisman et al., 1995). Fresh plasma or plasma heat treated to inactivate LCAT was infused intravenously into LCAT-KO mice, and then plasma lipids were measured (Fig. 1). One hour after plasma infusion, total cholesterol nearly doubled for both the fresh and heat-inactivated plasma (Fig. 1A). After 24 h, total cholesterol in the heat-inactivated control group returned to baseline, whereas the mice treated with fresh plasma still had a 2-fold increase in total cholesterol. By 48 h, total cholesterol for both groups returned to near baseline. As expected, the level of CE at baseline (Fig. 1B) was very low, but it markedly increased after plasma infusion. Although this occurred in both treatment groups, there was a much larger increase of CE in mice treated with fresh plasma, particularly at 24 h, when CE was still more than 10-fold increased above baseline. Because LCAT-Tg mice have a substantial elevation of not only human LCAT but also more than a 2-fold increase in HDL-C and a 5-fold increase in HDL-CE (Vaisman et al., 1995), the exogenous lipids from the plasma infusion is the most likely source for the observed increase in total cholesterol and CE observed at 1 h, especially for the heat-inactivated plasma-treated group. In contrast, the difference in total cholesterol and CE observed between the fresh plasma and heat-inactivated plasma at later times when most of the exogenous lipids were catabolized and removed from the plasma compartment is probably due to the effect of the infused LCAT activity from the fresh plasma on endogenous lipoproteins.
Fig. 1.

Effect of plasma infusion from hLCAT-Tg mice on lipids in LCAT-KO mice. Heat-inactivated plasma (control; open bars) or fresh (LCAT; solid bars) plasma (220 μl) from hLCAT-Tg mice, containing 3300 units of LCAT, was injected intravenously into LCAT-KO mice (n = 5). Plasma from the treated mice was collected at the indicated times and measured for total cholesterol (A) or CE (B). Data represent mean ± S.D. (n = 5). **, p < 0.001; ***, p < 0.0001, LCAT versus control group. LCAT activity after plasma infusion in LCAT-KO mice was measured (C) at the indicated times after the plasma infusion. Results are expressed as the percentage of first time point (5 min) after treatment. Results are the mean ± 1 S.D. of five replicates.
The level of LCAT activity was monitored after injection of fresh plasma into LCAT-KO mice (Fig. 1C). Immediately after infusion of LCAT-Tg plasma, a greater than 10-fold increase above baseline in LCAT activity was observed. LCAT activity then followed a monoexponential decay, with an estimated half-life of 8.29 ± 1.82 h, which is close to the reported half-life of mouse HDL (Tape and Kisilevsky, 1990). Although the major change in CE occurred at 24 h (Fig. 1B), less than 20% of the original dose of infused LCAT was still present in the circulation after 24 h, which suggests that there is a delay in the catabolism of CE formed earlier by LCAT. No significant increase in LCAT activity was observed after infusion of heat-inactivated plasma (data not shown).
Changes in lipoprotein distribution and lipid composition of mice after plasma infusion was examined at the 24-h time point by FPLC analysis (Fig. 2), when it appeared that the infused LCAT had its greatest effect on plasma lipids (Fig. 1). For the heat-inactivated plasma group (Fig. 2B), the FPLC lipoprotein profile was similar to that of untreated LCAT-KO mice (Fig. 2A). The major cholesterol peak was found in fractions corresponding to where VLDL and Lp-X elute (elution volume, 12–19 ml), and only a relatively small fraction of cholesterol was esterified. A smaller cholesterol peak corresponding to LDL also was observed, similar to untreated LCAT-KO mice. In contrast, the major cholesterol peak after treatment of LCAT-KO mice with fresh plasma was in the HDL region (Fig. 2C). Cholesterol in VLDL fractions from mice treated with fresh plasma was decreased by approximately 40% compared with baseline (Fig. 2A). Unlike the heat-inactivated plasma treatment group, a large fraction, approximately 60% of cholesterol, was esterified in mice treated with fresh LCAT-Tg plasma.
Fig. 2.
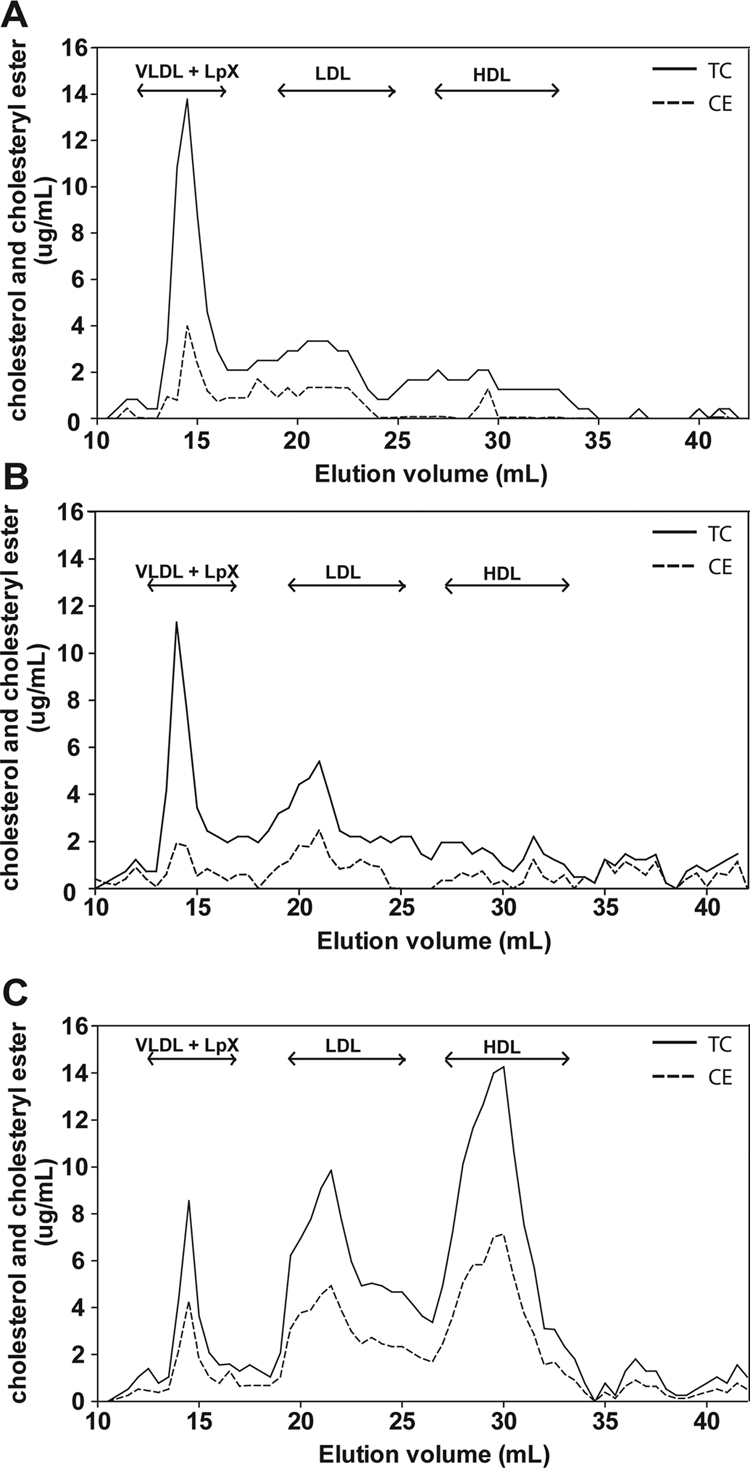
Lipoprotein profile of LCAT-KO mice infused hLCAT-Tg mice plasma. LCAT-KO mice were infused with plasma from LCAT-Tg mice, as described in Fig. 1. FPLC fractions from pooled plasma from untreated LCAT-KO mice (n = 5; A), heat-inactivated plasma-treated mice (n = 5; B), and fresh plasma-treated mice (n = 5; C) were analyzed for total cholesterol (solid line) and CE (dashed line).
Production and Characterization of rLCAT.
Human rLCAT produced in HEK cells was purified from conditioned serum-free medium as described under Materials and Methods. When analyzed on a 10% SDS-polyacrylamide gel electrophoresis gel and stained with Coomassie Blue-R250, a single band of approximately 70 kDa was observed, consistent with the reported size of the full-length glycosylated protein (Fig. 3; Supplemental Fig. 1). The band was found to stain with an anti-human LCAT antibody (Supplemental Fig. 1). The purified protein had a specific activity of 1850 nmol/mg/h, which is similar to previous reports of purified LCAT, using a proteoliposome substrate (Lane et al., 2004). Purified LCAT stored in an aqueous solution (PBS, pH 7.4), with 10% glycerol, was found to retain greater than 90% activity after storage at −70°C for at least 3 months.
Fig. 3.

In vitro effect of rLCAT on human FLD plasma. Serum (450 μl) from a normal subject (lanes 1–4) or an FLD subject (lanes 5–8) was incubated with rLCAT (185 units) (lanes 3, 4, 7, and 8) or the same volume of saline (lanes 1, 2, 5, and 6) at 37°C for 15 h. Then, 30 μg of purified human apoA-I was added to lanes 2, 4, 6, and 8. After rLCAT treatment, samples were analyzed by agarose gel electrophoresis and stained with Sudan black (A), immunofixed for apoA-I (B), or immunofixed for apoB (C). D, serum from an FLD subject (60 μl) was incubated with rLCAT (25 units) (LCAT; solid bars) or saline (control; open bars) at 37°C for 6 h. Samples were analyzed for the indicated lipids and lipoproteins. Results represent the mean ± 1 S.D. of triplicates. ***, p < 0.0001, experimental versus control group.
In Vitro Effect of rLCAT on Lipoproteins.
The in vitro effect of rLCAT on lipoproteins was examined after incubation of rLCAT with fasting human serum from an FLD subject and control subject (Fig. 3). rLCAT was incubated with serum for 15 h at 37°C and analyzed by agarose gel electrophoresis. As indicated, purified human apoA-I also was added to some samples. The addition of rLCAT to the control serum either in the presence or absence of exogenous apoA-I was found to shift LDL from its β-position and HDL from its α-position to faster migrating forms (Fig. 3A). Serum from a patient with FLD had no detectable HDL (α-lipoprotein), but it had a slow migrating band cathodal to the β-region and some residual lipoprotein trapped in the origin. After incubation of FLD serum with rLCAT, faint bands were detected in the α- and pre-α-region where HDL from normal serum also was found to migrate after rLCAT treatment. In addition, the major band cathodal to the β-region disappeared and the predominant band appeared with the same pre-β-position as the major band from normal serum treated with rLCAT. When the gels were immunofixed for apoA-I (Fig. 3B), the majority of apoA-I was found in the α-region for the control serum, and it shifted to a slightly faster migrating position after LCAT treatment. In FLD plasma, most of the apoA-I was present in the pre-β-region just in front of the major band observed with Sudan black staining (Fig. 3A). After treatment with LCAT, apoA-I shifted to the α- and pre-α-region similar in position observed for control serum after rLCAT treatment. When control serum was stained for apoB (Fig. 3C), the major Sudan black band in the β region was found to contain apoB and migrated slightly faster after treatment with LCAT. The slow-migrating band observed with Sudan black in FLD plasma also stained with apoB and shifted to the same position as the control serum after rLCAT treatment.
The effect of rLCAT incubation with serum from patients with FLD on lipid levels is presented in Fig. 3D. Total cholesterol, as expected, showed no change, but CE increased from approximately 20% of total cholesterol to approximately 75%, which is the typical percentage of CE in human serum (Rousset et al., 2009). HDL-C more than doubled, and there was a corresponding proportional decrease in cholesterol on non-HDL lipoproteins; thus, the in vitro incubation of rLCAT with serum from FLD subjects resulted in the net transfer of cholesterol to HDL from other lipoproteins.
Investigation of Different Routes of rLCAT Administration in Mice.
The effect of intravenous rLCAT administration on plasma lipids was tested in human apoA-I-Tg mice and compared with intramuscular and subcutaneous delivery of rLCAT in human apoA-I-Tg mice. We first selected mice overexpressing human apoA-I, because it has been shown previously that human LCAT has a preference for human apoA-I as an activator over mouse apoA-I (Francone et al., 1995). Compared with the saline-treated control group, rLCAT delivered intravenously, intramuscularly or subcutaneously showed a nearly identical effect on raising plasma CE over time (Fig. 4A). Likewise, all three routes of treatment showed a similar effect in raising HDL-C (Fig. 4B). HDL-C increased almost 2-fold by 24 h after rLCAT treatment, by all three routes, and thereafter began to decline to baseline. Even after 72 h, however, HDL-C was still elevated by approximately 25% in the rLCAT-treated mice.
Fig. 4.
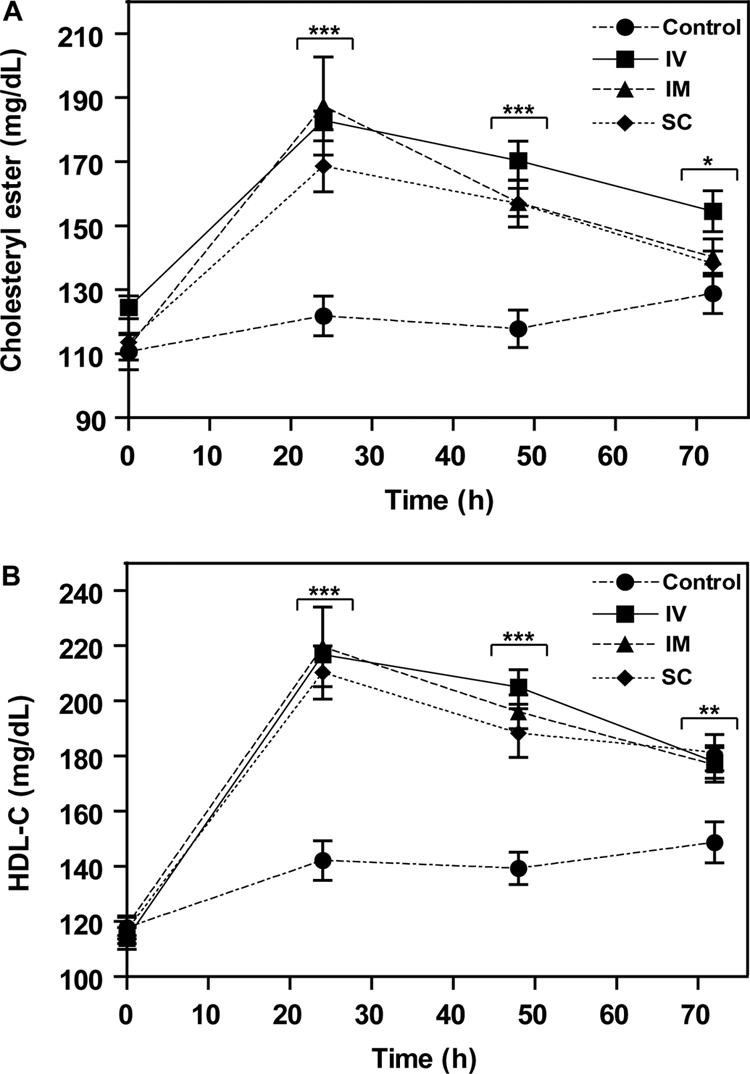
Effect of route of administration of rLCAT on mouse lipids. rLCAT (4200 units) was injected intravenously into the tail vein (squares), intramuscularly into hind limb (triangles) or subcutaneously (diamonds) into the back of hapoA-I-Tg mice, and then plasma was removed at the indicated times and analyzed for total cholesterol (A) and HDL-C (B). The control group was treated subcutaneously with saline (circles). Data represent mean ± S.D. (n = 10). *, p < 0.005; **, p < 0.001; and ***, p < 0.0001, experimental versus control group.
In Vivo Effect of rLCAT Infusion in LCAT-KO Mice.
rLCAT was infused intravenously into LCAT-KO mice, and plasma was analyzed for lipids after separation by FPLC (Fig. 5). Four hours after rLCAT injection, cholesterol on VLDL was reduced by more than 80% (Fig. 5A). This was associated with the appearance of a prominent HDL-C peak, which contained the majority of total cholesterol. At 24 h, cholesterol in the HDL fractions began to decrease, the size of the HDL also became smaller, whereas cholesterol in the LDL fractions increased and cholesterol on VLDL reappeared. At baseline, CE was only observed in the VLDL peak, but 4 h after infusion of rLCAT, almost all CE was observed in HDL (Fig. 5B). By 24 h, CE was still present on HDL but was reduced and on a smaller sized particle. CE also reappeared on VLDL at 24 h, and more CE was found on LDL compared with baseline.
Fig. 5.
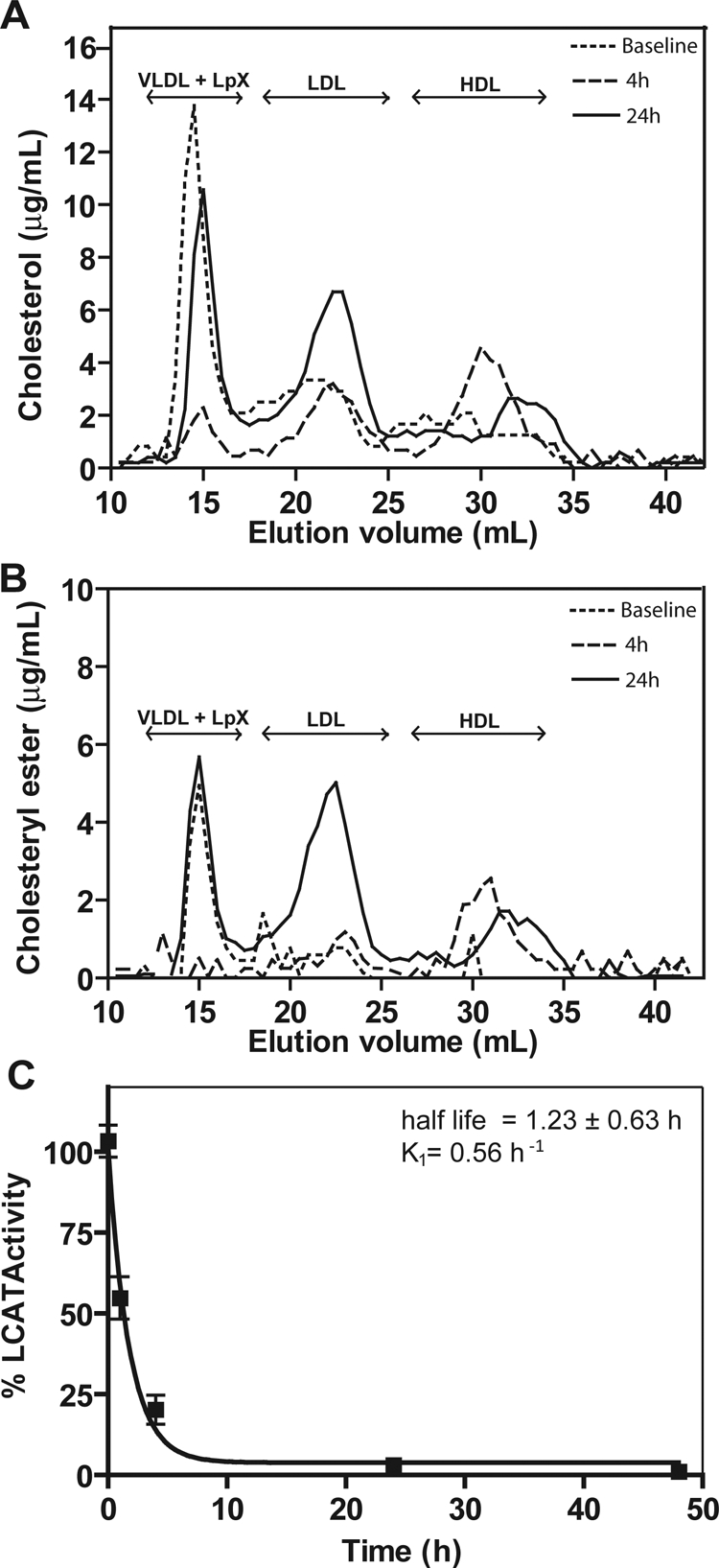
In vivo effect of rLCAT on lipoprotein profile of LCAT-KO mice. rLCAT (2900 units) was injected intravenously into LCAT-KO mice (n = 5). Control group of LCAT-KO mice (n = 5) was injected with PBS. Pooled plasma at baseline (dotted line), 4 h (large dashed line) and 24 h (solid line) after rLCAT treatment was fractionated by FPLC and analyzed for cholesterol (A) and CE (B). C, LCAT activity was measured after intravenous rLCAT injection for the indicated times. Results are expressed as percentage of first time point (5 min) after treatment. Results are the mean ± 1 S.D. of five replicates.
When the activity of rLCAT was monitored after infusion into LCAT-KO mice (Fig. 5C), it was found that rLCAT had a much shorter half-life of approximately 1.23 ± 0.63 h compared with that of hLCAT from plasma (Fig. 1C). By 24 h, there was nearly no residual LCAT activity detected, even though there were still changes in the lipoprotein distribution at this time (Fig. 5, A and B).
In Vivo Effect of rLCAT Infusion in LCAT-KO/apoA-I-Tg Mice.
The effect of intravenous infusion of rLCAT into LCAT-KO/apoA-I-Tg mice is presented in Fig. 6. In contrast to LCAT-KO mice (Fig. 2), at baseline LCAT-KO/apoA-I-Tg mice had a much smaller cholesterol peak on VLDL and had low but detectable level of cholesterol on a relatively large-sized HDL subfraction. It is interesting to note that they also had a prominent peak in fractions (elution volume, 32–35 ml) corresponding to a small-sized pre-β-HDL. Four hours after rLCAT injection, both the large- and small-sized HDL peaks disappeared, and an intermediate-sized HDL peak was formed. These changes largely persisted for 24 h, but the level of HDL-C continued to increase and by 24 h was approximately 8-fold higher compared with baseline (Fig. 6B). rLCAT had a half-life of 7.39 ± 2.1 h (Fig. 6C), which was similar to what was observed for hLCAT from LCAT-Tg plasma (Fig. 1C).
Fig. 6.
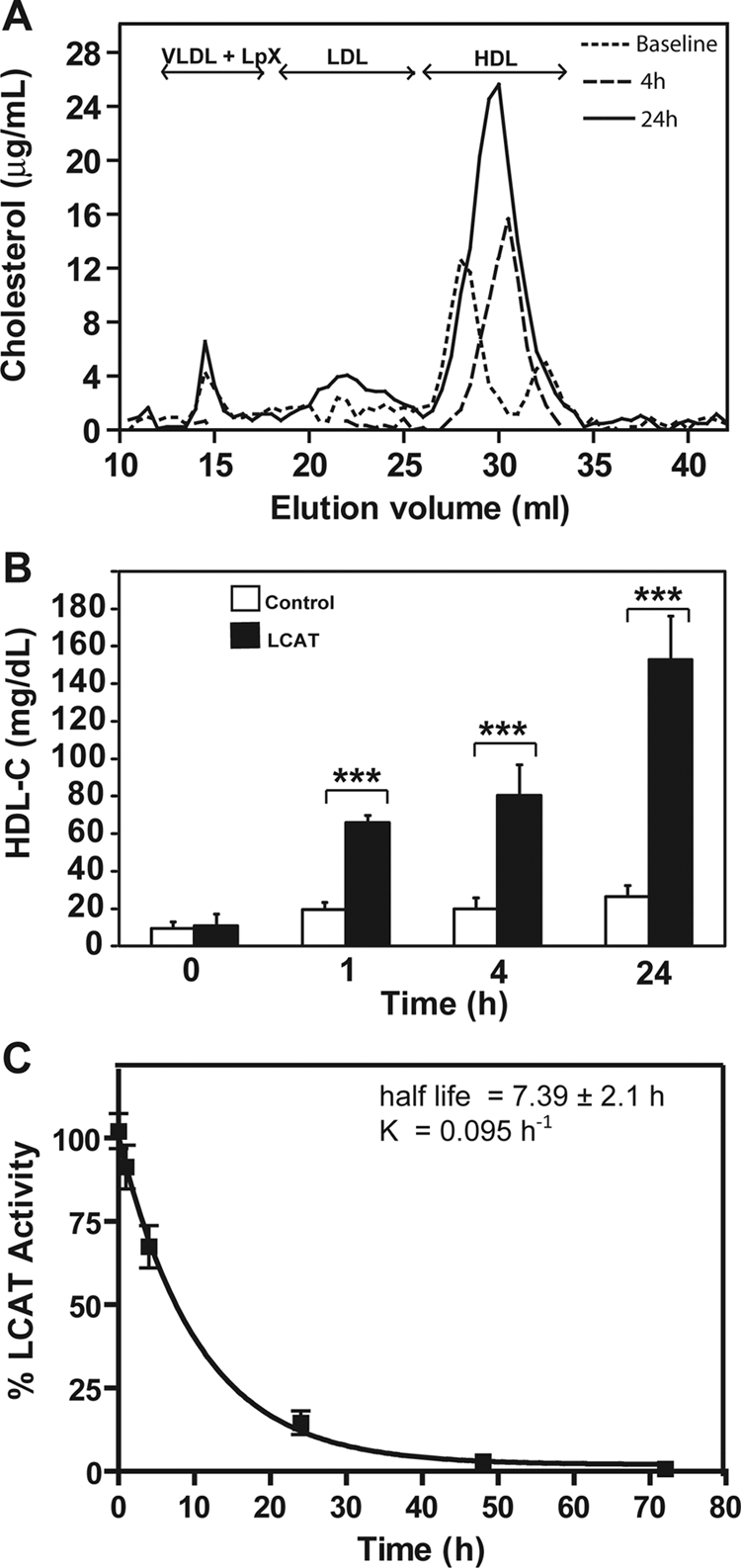
In vivo effect of rLCAT on lipoprotein profile of LCAT-KO × hapoA-I-Tg mice. rLCAT was injected (2900 units) intravenously into LCAT-KO/hapoA-I-Tg mice (n = 5). Pooled plasma at baseline (dotted line), 4 h (large dashed line), and 24 h (solid line) after rLCAT treatment was fractionated by FPLC and analyzed for total cholesterol (A). B, HDL-C was measured in plasma of control group (LCAT-KO mice, n = 5; injected with saline; open bars) and in LCAT group (injected with rLCAT; solid bars). C, LCAT activity was measured after intravenous rLCAT injection for the indicated times. Results are expressed as percentage of first time point (5 min) after treatment. Results are the mean ± 1 SD of five replicates. ***, p < 0.0001, experimental versus control group.
Effect of rLCAT on Cholesterol Efflux.
The functionality of HDL formed in plasma after rLCAT treatment was tested by assessing its ability to stimulate cholesterol efflux (Fig. 7). HDL was isolated by density gradient ultracentrifugation (density = 1.21–1.063 g/ml) from the plasma of LCAT-KO mice 4 h after infusion of either saline or rLCAT. The same volume of HDL from the isolated density fraction was then used from each group of mice to stimulate cholesterol efflux from BHK cells stably transfected with either ABCG1, SR-BI, or ABCA1. Increased cholesterol efflux was observed from the ABCA1- and ABCG1-transfected cell lines after rLCAT treatment, but no significant difference was observed in cholesterol efflux for the saline- versus rLCAT-treated group of mice from nontransfected BHK cells or SR-BI-transfected cells.
Fig. 7.
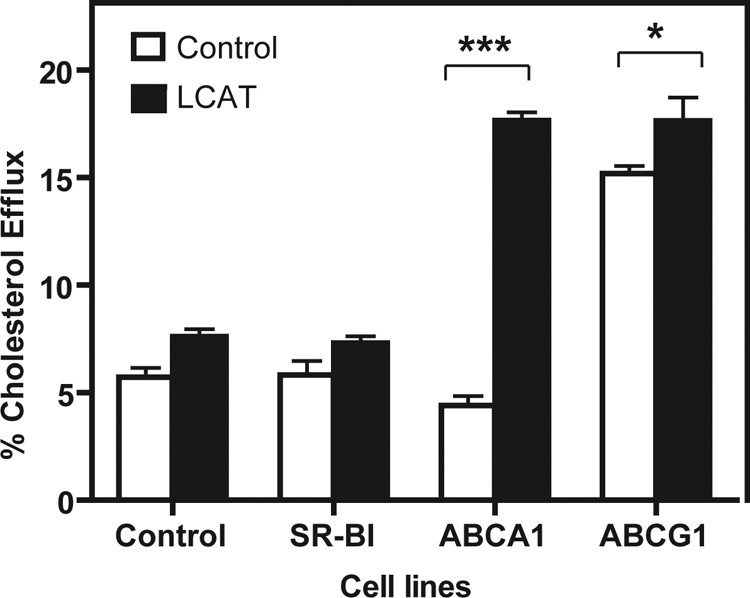
Effect of rLCAT on cholesterol efflux. rLCAT (2900 units; solid squares) or saline (open squares) was injected intravenously into LCAT-KO mice (n = 4), and plasma was collected 4 h after treatment. HDL was isolated by density gradient ultracentrifugation from 1.1 ml of plasma and contained 44 and 23 mg/dl total cholesterol from the rLCAT and saline-treated group, respectively. Volume of HDL corresponding to 10 μl of plasma was added to 1 ml of serum-free DMEM containing 1 mg/ml BSA and used to efflux cholesterol from the indicated transfected BHK cell lines or the control BHK cell line for 6 h. Results represent the mean percentage of total radioactive counts efflux ± 1 S.D. of triplicates. *, p < 0.005 and ***, p < 0.0001, LCAT versus control group (injected with saline).
Discussion
The results of this study support the feasibility of using rLCAT as an enzyme replacement therapy agent for FLD. Infusion of plasma from LCAT-Tg mice into LCAT-KO mice yielded results similar to previous reports of the rapid correction of the abnormal lipoproteins in FLD subjects after infusion with normal plasma (Norum and Gjone, 1968; Murayama et al., 1984). The half-life of rLCAT in mice, however, was significantly shorter than the 4- to 5-day half-life described in humans (Stokke et al., 1974). In LCAT-KO mice, which have very low HDL, the half-life of LCAT activity was only 1.23 ± 0.63 h (Fig. 5C). In contrast, hLCAT from LCAT-Tg plasma injected into LCAT-KO mice (Fig. 1) or rLCAT infused into LCAT-KO/apoA-I-Tg mice (Fig. 6) had a considerably longer half-life of 7 to 8 h. This is similar to the reported half-life of apoA-I in mice (Tape and Kisilevsky, 1990), which suggests that the half-life of rLCAT is shorter in mice than humans, because of the more rapid turnover of HDL in mice. Furthermore, the presence of endogenous HDL in mice infused with rLCAT, such as LCAT-KO/apoA-I-Tg mice (Fig. 6), may allow the association of LCAT onto HDL, which may stabilize it against rapid clearance. This suggests that for FLD subjects with low levels of HDL, a larger initial loading dose of rLCAT may be needed, but once some HDL is formed, less rLCAT may be needed to maintain a normal lipoprotein profile. That rLCAT produced in cells had a similar half-life in the presence of HDL (Fig. 6) as hLCAT endogenously produced in transgenic mice (Fig. 1) suggests that the two forms of LCAT have undergone a similar post-translational processing, such as glycosylation, which is well known to affect the half-life of proteins (Brady, 2006).
Based on the promising plasma infusion studies, we developed a system to produce rLCAT. Similar to a previous report (Lane et al., 2004), we were able to produce a stable cell line that synthesized relatively large amounts of rLCAT. More work is necessary to characterize the rLCAT product before human use, but preliminary results suggest that it should be possible to produce sufficient quantities of pure rLCAT to treat patients. Assuming a half-life of 4 to 5 days and that only 10 to 15% of LCAT activity is necessary to prevent renal disease (Santamarina-Fojo et al., 2001), we estimate that one treatment per week containing approximately 10 to 15 mg of pure LCAT should be sufficient to treat an average-sized adult with FLD. Based on Fig. 4, which shows a similar effect with three different routes of delivery, it may be possible to deliver rLCAT either subcutaneously or by intramuscular injection, which could potentially make possible the self-administration of rLCAT by patients with FLD.
Incubation of rLCAT with FLD plasma transformed the abnormal lipoproteins and changed their electrophoretic migration position to the same region as lipoproteins from normal plasma treated with rLCAT (Fig. 3). In FLD subjects, apoA-I was primarily in the pre-β-region, which did not stain with Sudan black, suggesting that it is relatively devoid of CE or triglycerides. This band probably represents pre-β-HDL, which is phospholipids-rich but neutral lipid-poor particle and is known to accumulate in FLD (Rousset et al., 2009). The esterification of cholesterol on pre-β-HDL by rLCAT resulted in the formation of normal-sized HDL. The change in the migration position also may be due to the production of lysophosphatidylcholine by rLCAT, which would increase on the electronegativity of HDL, therefore increasing its anodal migration. The main lipoprotein observed in FLD plasma was an apoB-containing particle that migrated at a position where both abnormal VLDL and Lp-X migrates (O K and Frohlich, 1995). After treatment with rLCAT, it shifted to a more anodal position to where LDL from normal plasma migrated after treatment with rLCAT. Treatment of LCAT-KO mice with rLCAT largely restored their lipoprotein profile to a more normal pattern (Fig. 5). In as short as 4 h, cholesterol in VLDL-sized particles was markedly reduced, and a large increase in cholesterol on HDL was observed and most of it was esterified. Based on the in vitro incubation of rLCAT with FLD plasma (Fig. 3), this may have occurred due to the conversion of pre-β-HDL in mice to larger α-HDL by the esterification of cholesterol. ApoA-I and other exchangeable apolipoproteins also have been observed on Lp-X (Santamarina-Fojo et al., 2001) as well as VLDL (Hamilton et al., 1991), so the esterification of cholesterol on these particles also could have resulted in the displacement of exchangeable apolipoproteins from these particles and the de novo formation of HDL. A similar process has been described to occur, during the postprandial lipolysis of VLDL and chylomicrons, which can generate de novo HDL (Sloop et al., 1983). That the ex vivo treatment of plasma with rLCAT caused the net transfer of cholesterol from non-HDL lipoproteins to HDL (Fig. 3C) is also consistent with this mechanism.
It is interesting to note that overexpression of hapoA-I in LCAT-KO mice reduced the level of cholesterol in the VLDL-sized fractions (Fig. 6). A peak corresponding to small, phospholipid-rich HDL also was observed (data not shown), which probably represents pre-β-HDL. Increased production of hapoA-I in the absence of sufficient LCAT would be expected to result in the production of more pre-β-HDL. Shortly after rLCAT treatment, almost of all the small-sized HDL was converted to an intermediate-sized HDL. In addition, a much greater increase in HDL-C was observed in these mice compared with LCAT-KO mice (Fig. 6). This probably occurs because there was more substrate, i.e., small-sized HDL for LCAT, and possibly because human apoA-I is a better activator for LCAT than mouse apoA-I (Francone et al., 1995).
As would be expected, if HDL formed by LCAT was functional, more cholesterol efflux was observed from ABCA1- and ABCG1-transfected cells to HDL isolated from mice treated with rLCAT (Fig. 7). Cholesterol efflux from ABCG1 is known to occur to lipid-rich forms of HDL, which were generated by the rLCAT treatment (Figs. 2 and 5). In contrast, ABCA1 effluxes cholesterol to lipid-poor HDL like pre-β-HDL (Zannis et al., 2006). The observed increase in cholesterol efflux from ABCA1-transfected cells was not anticipated. Several recent studies, however, have shown that lipid-rich HDL also can serve as an acceptor for cholesterol from ABCA1, possibly when apoA-I dissociates from HDL (Favari et al., 2009). As discussed, the displacement of exchangeable apolipoproteins from VLDL or Lp-X particles after treatment with rLCAT also could generate nascent-like HDL particles that could stimulate cholesterol efflux by ABCA1.
The increase in cholesterol efflux from cells after rLCAT treatment suggests that rLCAT also may be used for the treatment of atherosclerosis. LCAT has long been proposed to increase RCT, by trapping cholesterol effluxed from cells until it can be removed as CE by the liver (Glomset, 1968). Because cholesterol efflux is considered to be the first step in RCT, the cholesterol efflux data are consistent with a recent study showing that the flux of radiolabeled cholesterol from peritoneal macrophages to the stool was reduced by 45% in mice lacking LCAT (Tanigawa et al., 2009). LCAT expressed with adeno-associated virus did not however increase the amount of radiotracer cholesterol in the stool of apoA-I transgenic mice. It should be noted however that studies in mice that do not express CETP can lead to contradictory results compared with the effect of various genes on human lipoprotein metabolism (Briand et al., 2010). LCAT transgenic mice are not protected against atherosclerosis (Bérard et al., 1997), unless CETP also is expressed (Föger et al., 1999). In hamsters, which have CETP, overexpression of LCAT not only increased HDL but also increased biliary sterol excretion (Zhang et al., 2004). In rabbits, which express CETP, LCAT overexpression decreased the cholesterol content of the aorta (Van Craeyveld et al., 2009), and LCAT transgenic rabbits were protected from diet-induced atherosclerosis (Hoeg et al., 1996). A recent preliminary report showed that subcutaneous injection of rLCAT in rabbits stimulated whole-body RCT and reduced atherosclerosis (Zhou et al., 2009). Although the question whether FLD subjects have increased risk for atherosclerosis have yielded contradictory results (Hovingh et al., 2005; Calabresi et al., 2009), a recent study, using a more sensitive method (i.e., carotid 3.0 Tesla MRI; R. Duivenvoorden, unpublished data).
rLCAT treatment may be particularly useful when used in conjunction with HDL replacement therapy (Remaley et al., 2008). Under these conditions, LCAT levels may become rate-limiting, as evidenced by the increase in plasma pre-β-HDL (LCAT substrate) after administration of certain apoA-I mimetic peptides (Navab et al., 2004). If so, a combination of apoA-I mimetics along with rLCAT may show not only synergy in increasing HDL-C but also enhance the ability of HDL to remove cholesterol from atherosclerotic arteries. This is consistent with the result observed with the apoA-I Tg mice; despite having normal LCAT levels and increased apoA-I, they showed a much greater increase in HDL-C after rLCAT treatment than did just LCAT-KO mice (Figs. 5 and 6).
In summary, rLCAT treatment in LCAT-KO mice was observed to rapidly increase HDL-C and to decrease abnormal lipoproteins. These results demonstrate the potential of using rLCAT as an enzyme replacement therapy agent for patients with FLD.
Supplementary Material
Acknowledgments
We thank Rene Costello for excellent technical assistance.
This work was supported in part by the Intramural Research Program of the National Institutes of Health National Heart, Lung, and Blood Institute and the National Institutes of Health National Heart, Lung, and Blood Institute [Grant HL09265601].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.110.169540.
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
- LCAT
- lecithin cholesterol acyl transferase
- FLD
- familial lecithin cholesterol acyl transferase deficiency
- FED
- fish eye disease
- LDL
- low-density lipoprotein
- HDL-C
- high-density lipoprotein-cholesterol
- Lp-X
- lipoprotein X
- HDL
- high-density lipoprotein
- RCT
- reverse cholesterol transport
- apo
- apolipoprotein
- rLCAT
- recombinant human lecithin/cholesterol acyl transferase
- KO
- knockout
- Tg
- transgenic
- HEK
- human embryonic kidney
- BSA
- bovine serum albumin
- FPLC
- fast protein liquid chromatography
- BHK
- baby hamster kidney
- ABCA1
- ATP-binding cassette
- DMEM
- Dulbecco's modified Eagle's medium
- CE
- cholesteryl ester(s)
- CETP
- cholesteryl ester transport protein.
References
- Albers JJ, Chen C-H, Lacko AG. (1986) Isolation, characterization, and assay of lecithin-cholesterol acyltransferase. Methods Enzymol 129:763–783 [DOI] [PubMed] [Google Scholar]
- Bérard AM, Föger B, Remaley A, Shamburek R, Vaisman BL, Talley G, Paigen B, Hoyt RF, Jr, Marcovina S, Brewer HB, Jr, et al. (1997) High plasma HDL concentrations associated with enhanced atherosclerosis in transgenic mice overexpressing lecithin-cholesteryl acyltransferase. Nat Med 3:744–749 [DOI] [PubMed] [Google Scholar]
- Brady RO. (2006) Enzyme replacement for lysosomal diseases. Annu Rev Med 57:283–296 [DOI] [PubMed] [Google Scholar]
- Briand F, Tréguier M, André A, Grillot D, Issandou M, Ouguerram K, Sulpice T. (2010) Liver X receptor activation promotes macrophage-to-feces reverse cholesterol transport in a dyslipidemic hamster model. J Lipid Res 51:763–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busseuil D, Shi Y, Mecteau M, Brand G, Kernaleguen AE, Thorin E, Latour JG, Rhéaume E, Tardif JC. (2008) Regression of aortic valve stenosis by ApoA-I mimetic peptide infusions in rabbits. Br J Pharmacol 154:765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi L, Favari E, Moleri E, Adorni MP, Pedrelli M, Costa S, Jessup W, Gelissen IC, Kovanen PT, Bernini F, et al. (2009) Functional LCAT is not required for macrophage cholesterol efflux to human serum. Atherosclerosis 204:141–146 [DOI] [PubMed] [Google Scholar]
- Cheung MC, Wolf AC, Lum KD, Tollefson JH, Albers JJ. (1986) Distribution and localization of lecithin:cholesterol acyltransferase and cholesteryl ester transfer activity in A-I-containing lipoproteins. J Lipid Res 27:1135–1144 [PubMed] [Google Scholar]
- Favari E, Calabresi L, Adorni MP, Jessup W, Simonelli S, Franceschini G, Bernini F. (2009) Small discoidal pre-beta1 HDL particles are efficient acceptors of cell cholesterol via ABCA1 and ABCG1. Biochemistry 48:11067–11074 [DOI] [PubMed] [Google Scholar]
- Föger B, Chase M, Amar MJ, Vaisman BL, Shamburek RD, Paigen B, Fruchart-Najib J, Paiz JA, Koch CA, Hoyt RF, et al. (1999) Cholesteryl ester transfer protein corrects dysfunctional high density lipoproteins and reduces aortic atherosclerosis in lecithin cholesterol acyltransferase transgenic mice. J Biol Chem 274:36912–36920 [DOI] [PubMed] [Google Scholar]
- Francone OL, Gong EL, Ng DS, Fielding CJ, Rubin EM. (1995) Expression of human lecithin-cholesterol acyltransferase in transgenic mice. Effect of human apolipoprotein AI and human apolipoprotein all on plasma lipoprotein cholesterol metabolism. J Clin Invest 96:1440–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glomset JA. (1962) The mechanism of the plasma cholesterol esterification reaction: plasma fatty acid transferase. Biochim Biophys Acta 65:128–135 [DOI] [PubMed] [Google Scholar]
- Glomset JA. (1968) The plasma lecithins:cholesterol acyltransferase reaction. J Lipid Res 9:155–167 [PubMed] [Google Scholar]
- Hamilton RL, Moorehouse A, Havel RJ. (1991) Isolation and properties of nascent lipoproteins from highly purified rat hepatocytic Golgi fractions. J Lipid Res 32:529–543 [PubMed] [Google Scholar]
- Hoeg JM, Vaisman BL, Demosky SJ, Jr, Meyn SM, Talley GD, Hoyt RF, Jr, Feldman S, Bérard AM, Sakai N, Wood D, et al. (1996) Lecithin:cholesterol acyltransferase overexpression generates hyperalpha-lipoproteinemia and a nonatherogenic lipoprotein pattern in transgenic rabbits. J Biol Chem 271:4396–4402 [DOI] [PubMed] [Google Scholar]
- Hovingh GK, Hutten BA, Holleboom AG, Petersen W, Rol P, Stalenhoef A, Zwinderman AH, de Groot E, Kastelein JJ, Kuivenhoven JA. (2005) Compromised LCAT function is associated with increased atherosclerosis. Circulation 112:879–884 [DOI] [PubMed] [Google Scholar]
- Imbasciati E, Paties C, Scarpioni L, Mihatsch MJ. (1986) Renal lesions in familial lecithin-cholesterol acyltransferase deficiency. Ultrastructural heterogeneity of glomerular changes. Am J Nephrol 6:66–70 [DOI] [PubMed] [Google Scholar]
- Lambert G, Sakai N, Vaisman BL, Neufeld EB, Marteyn B, Chan CC, Paigen B, Lupia E, Thomas A, Striker LJ, Blanchette-Mackie J, Csako G, Brady JN, Costello R, Striker GE, et al. (2001) Analysis of glomerulosclerosis and atherosclerosis in lecithin cholesterol acyltransferase-deficient mice. J Biol Chem 276:15090–15098 [DOI] [PubMed] [Google Scholar]
- Lane SB, Tchedre KT, Nair MP, Thigpen AE, Lacko AG. (2004) Characterization of lecithin:cholesterol acyltransferase expressed in a human lung cell line. Protein Expr Purif 36:157–164 [DOI] [PubMed] [Google Scholar]
- Murayama N, Asano Y, Kato K, Sakamoto Y, Hosoda S, Yamada N, Kodama T, Murase T, Akanuma Y. (1984) Effects of plasma infusion on plasma lipids, apoproteins and plasma enzyme activities in familial lecithin: cholesterol acyltransferase deficiency. Eur J Clin Invest 14:122–129 [DOI] [PubMed] [Google Scholar]
- Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, Wagner AC, Frank JS, Datta G, Garber D, et al. (2004) Oral D-4F causes formation of pre-beta high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E-null mice. Circulation 109:3215–3220 [DOI] [PubMed] [Google Scholar]
- Norum KR, Gjone E. (1968) The effect of plasma transfusion on the plasma cholesterol esters in patients with familial plasma lecithin: cholesterol acyltransferase deficiency. Scand J Clin Lab Invest 22:339–342 [DOI] [PubMed] [Google Scholar]
- O K, Frohlich J. (1995) Role of lecithin:cholesterol acyltransferase and apolipoprotein A-I in cholesterol esterification in lipoprotein-X in vitro. J Lipid Res 36:2344–2354 [PubMed] [Google Scholar]
- Oram JF, Vaughan AM, Stocker R. (2001) ATP-binding cassette transporter A1 mediates cellular secretion of alpha-tocopherol. J Biol Chem 276:39898–39902 [DOI] [PubMed] [Google Scholar]
- Panescu V, Grignon Y, Hestin D, Rostoker G, Frimat L, Renoult E, Gamberoni J, Grignon G, Kessler M. (1997) Recurrence of lecithin cholesterol acyltransferase deficiency after kidney transplantation. Nephrol Dial Transplant 12:2430–2432 [DOI] [PubMed] [Google Scholar]
- Remaley AT, Amar M, Sviridov D. (2008) HDL-replacement therapy: mechanism of action, types of agents and potential clinical indications. Expert Rev Cardiovasc Ther 6:1203–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset X, Vaisman B, Amar M, Sethi AA, Remaley AT. (2009) Lecithin: cholesterol acyltransferase–from biochemistry to role in cardiovascular disease. Curr Opin Endocrinol Diabetes Obes 16:163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamarina-Fojo S, Hoeg JM, Assmann G, Bryan H., Brewer J. (2001) Lecithin cholesterol acyltransferase deficiency and fish eye disease, in Metabolic & Molecular Bases of Inherited Disease, Scriver CR, Beaudet AL, Sly WS. (eds.), pp 2817–2833, McGraw-Hill, New York [Google Scholar]
- Schumaker VN, Puppione DL, Segrest JP, Albers JJ. (1986) Sequential flotation ultracentrifugation, in Methods in Enzymology, Segrest JP, Alberspp JJ. pp 155–170, Academic Press, London, UK: [DOI] [PubMed] [Google Scholar]
- Sloop CH, Dory L, Hamilton R, Krause BR, Roheim PS. (1983) Characterization of dog peripheral lymph lipoproteins: the presence of a disc-shaped “nascent” high density lipoprotein. J Lipid Res 24:1429–1440 [PubMed] [Google Scholar]
- Stokke KT, Bjerve KS, Blomhoff JP, Oystese B, Flatmark A, Norum KR, Gjone E. (1974) Familial lecithin:cholesterol acyltransferase deficiency. Studies on lipid composition and morphology of tissues. Scand J Clin Lab Invest Suppl 137:93–100 [PubMed] [Google Scholar]
- Tanigawa H, Billheimer JT, Tohyama J, Fuki IV, Ng DS, Rothblat GH, Rader DJ. (2009) Lecithin: cholesterol acyltransferase expression has minimal effects on macrophage reverse cholesterol transport in vivo. Circulation 120:160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tape C, Kisilevsky R. (1990) Apolipoprotein A-I and apolipoprotein SAA half-lives during acute inflammation and amyloidogenesis. Biochim Biophys Acta 1043:295–300 [DOI] [PubMed] [Google Scholar]
- Vaisman B, Remaley AT. (2010) Measurement of lecithin:cholesterol acyltransferase activity with the use of a peptide-proteoliposome substrate, in Methods in Molecular Biology: Lipoprotein and Cardiovascular Disease (Freeman LA. ed), pp, Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- Vaisman BL, Klein HG, Rouis M, Bérard AM, Kindt MR, Talley GD, Meyn SM, Hoyt RF, Jr, Marcovina SM, Albers JJ. (1995) Overexpression of human lecithin cholesterol acyltransferase leads to hyperalphalipoproteinemia in transgenic mice. J Biol Chem 270:12269–12275 [DOI] [PubMed] [Google Scholar]
- Van Craeyveld E, Lievens J, Jacobs F, Feng Y, Snoeys J, De Geest B. (2009) Apolipoprotein A-I and lecithin:cholesterol acyltransferase transfer induce cholesterol unloading in complex atherosclerotic lesions. Gene Ther 16:757–765 [DOI] [PubMed] [Google Scholar]
- Vaughan AM, Oram JF. (2005) ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J Biol Chem 280:30150–30157 [DOI] [PubMed] [Google Scholar]
- Zannis VI, Chroni A, Krieger M. (2006) Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med 84:276–294 [DOI] [PubMed] [Google Scholar]
- Zaworski PG, Gill GS. (1988) Precipitation and recovery of proteins from culture supernatants using zinc. Anal Biochem 173:440–444 [DOI] [PubMed] [Google Scholar]
- Zhang AH, Gao S, Fan JL, Huang W, Zhao TQ, Liu G. (2004) Increased plasma HDL cholesterol levels and biliary cholesterol excretion in hamster by LCAT overexpression. FEBS Lett 570:25–29 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ. (2005) Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest 115:2870–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Sawyer J, Kelley K, Fordstrom P, Chan J, Tonn G, Carlson T, Retter M, Meininger D, Cheng J, et al. (2009) Lecithin cholesterol acyltransferase promotes reverse cholesterol transport and attenuates atherosclerosis progression in New Zealand White Rabbits (abstract 5920). Circulation 120:S1175b [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.