

Abstract
Mammalian target of rapamycin (mTOR) is an important mediator of phosphoinositol-3-kinase (PI3K) signaling. PI3K signaling regulates B cell development, homeostasis and immune responses. However, the function and molecular mechanism of mTOR mediated PI3K signaling in B cells has not been fully elucidated. Here we show that Sin1, an essential component of mTOR complex 2 (mTORC2), regulates B cell development. Sin1 deficiency results in increased IL-7 receptor (il7r) and RAG recombinase (rag1 and rag2) gene expression leading to enhanced pro-B cell survival and augmented V(D)J recombinase activity. We further show that Akt2 specifically mediates the Sin1-mTORC2 dependent suppression of il7r and rag gene expression in B cells by regulating FoxO1 phosphorylation. Finally, we demonstrate that the mTOR inhibitor rapamycin induces rag expression and promotes V(D)J recombination in B cells. Our study reveals that the Sin1/mTORC2-Akt2 signaling axis is a key regulator of FoxO1 transcriptional activity in B cells.
Introduction
Mammalian TOR is a constitutively expressed, evolutionarily conserved protein kinase which plays a central role in the regulation of cell growth, proliferation, apoptosis, and metabolism (Wullschleger et al., 2006). Mammalian TOR resides in two distinct protein complexes termed mammalian TOR complex 1 (mTORC1) and mammalian TORC2 (mTORC2) (Guertin and Sabatini, 2009; Wullschleger et al., 2006). mTORC1 contains mTOR, raptor, mLST8 (GβL) and PRAS40, and its function is acutely inhibited by rapamycin, a potent immunosuppressant with anti-tumor effect (Guertin and Sabatini, 2009; Wullschleger et al., 2006). Upon stimulation by nutrients, growth factors, hormones, and energy signals, mTORC1 phosphorylates the translational regulators S6K and 4EBP1 which leads to increased cellular protein synthesis and ribosome biogenesis (Gingras et al., 2004; Harris and Lawrence Jr., 2003; Wullschleger et al., 2006). mTORC2 contains Rictor, Sin1, and mLST8 in addition to mTOR, and regulates actin polymerization and cytoskeleton function (Guertin and Sabatini, 2009; Wullschleger et al., 2006). Mammalian TORC2 is resistant to acute rapamycin inhibition. However, chronic rapamycin exposure also inhibits mTORC2 in vitro and in vivo (Facchinetti et al., 2008; Sarbassov et al., 2006; Zeng et al., 2007). Recent studies show that mTORC2 regulates Akt/PKB in both a PI3K-dependent and PI3K-independent manner (Facchinetti et al., 2008; Jacinto et al., 2006; Sarbassov et al., 2005).
Akt/PKB is one of most studied members of the AGC kinase family, which also includes S6K, RSK, SGK, and PKC (Peterson and Schreiber, 1999; Woodgett, 2005). Like most members in this family, Akt is phosphorylated at two key residues that are located in the catalytic center (activation loop or T-loop) and the C-terminal hydrophobic motif (HM), respectively. Phosphorylation of Akt/PKB at the T-loop site (Thr308) is mediated by PDK1 and is essential for Akt catalytic activity (Alessi et al., 1997; Stephens et al., 1998). Phosphorylation of Akt at the HM site (Ser473) is independently mediated by mTORC2 (Jacinto et al., 2006; Sarbassov et al., 2005). Although Akt Ser473 phosphorylation is widely used as an indicator of Akt activation, the precise physiological function of this phosphorylation is still not fully understood. Phosphorylation at the Akt HM site may facilitate the PDK1 mediated phosphorylation of the T-loop site thereby enhancing Akt activity upon growth factor stimulation and PI3K activation (Alessi et al., 1996; Biondi, 2004; Scheid et al., 2002). Surprisingly however, genetic studies reveled that mTORC2 disruption, which completely abolishes Akt HM site phosphorylation, does not inhibit T-loop phosphorylation (Jacinto et al., 2006). Rather, Akt HM site phosphorylation regulates the substrate specificity of Akt (Jacinto et al., 2006). More recently, mTORC2 was shown to phosphorylate Akt at the turn motif (TM) residue Thr450, which controls Akt protein stability (Facchinetti et al., 2008).
B lymphocyte development is divided into distinct stages where immunoglobulin (Ig) variable (V), diversity (D) and joining (J) genes of the Ig heavy (IgH) chain and V and J genes of the Ig light (IgL) chain undergo somatic recombination, generally referred to as V(D)J recombination, to generate the B cell antigen receptor (BCR) (Schatz et al., 1989; Schlissel, 2003; Spicuglia et al., 2006). V(D)J recombination is mediated by the recombination activation genes rag1 and rag2 which associate and form the V(D)J recombinase (Leu and Schatz, 1995; Schatz et al., 1989). IgH gene recombination occurs first in progenitor B (pro-B) cells and, if successful, leads to the expression of the pre-BCR. Pre-BCR signals provide critical feedback about the functionality of the IgH chain allowing only those developing B cells with a functional pre-BCR to further differentiate into precursor B (pre-B) cells and begin IgL gene rearrangement (Herzog et al., 2009; Martensson et al., 2007). Pre-BCR signals promote cell survival and proliferation and suppress rag expression to prevent further IgH recombination (Geier and Schlissel, 2006; Schlissel, 2003). The pre-BCR dependent suppression of RAG expression contributes to allelic exclusion of IgH genes, terminates additional V(D)J recombination that could disrupt a productively rearranged IgH gene, and prevents aberrant V(D)J recombination which may result in genomic instability in proliferating pre-B cells. Subsequent IgL recombination leads to the expression of the BCR on immature B cells.
PI3K and Akt negatively regulate RAG expression and V(D)J recombination (Amin and Schlissel, 2008; Llorian et al., 2007; Verkoczy et al., 2007). The Forkhead family transcription factor FoxO1 is a direct regulator of rag genes downstream of PI3K and Akt (Amin and Schlissel, 2008; Dengler et al., 2008; Herzog et al., 2008; Herzog et al., 2009). Genetic or pharmacological inhibition of the PI3K pathway in B cells increases the expression of FoxO1 target genes and results in abnormal B cell function (Donahue and Fruman, 2004; Llorian et al., 2007; Suzuki et al., 1999; Verkoczy et al., 2007). These studies suggest that Akt may mediate PI3K signaling to control FoxO1 activity in B cells, however it is unclear how PI3K signals are integrated through Akt to regulate FoxO1 and it is not known if Akt is the sole mediator of PI3K dependent signals which regulate FoxO1. Additionally, the Akt isoform that regulates FoxO1 phosphorylation and function in B cells has not been identified and the molecular mechanism through which pre-BCR/BCR signals activate Akt and suppress FoxO1 activity is unknown.
In this study we reveal the function of mTORC2 in B cells and elucidate mechanisms of mTORC2 regulation of B cell development. We show that genetic ablation of Sin1 in mice disrupts mTORC2 and abolishes Akt phosphorylation at Ser473 and Thr450 but not at Thr308 in developing B cells. Developing Sin1-/- B cells show increased IL-7 receptor expression, enhanced response to IL-7, augmented RAG expression, and elevated V(D)J recombinase activity. We demonstrate that the mTORC2 dependent Akt HM phosphorylation is specifically required for the suppression of rag gene expression and FoxO1 phosphorylation is dependent on both Sin1 and Akt2 in B cells. Finally, we show that the mTOR inhibitor rapamycin increases rag1 expression and promotes V(D)J recombination in B cells. These data reveal that the Sin1/mTORC2-Akt2 signaling axis regulates IL-7 responsiveness, RAG expression, and V(D)J recombination in developing B cells.
Results
Sin1 regulates B cell development
To investigate the function of Sin1 in vivo, we generated Sin1 KO mice (Jacinto et al., 2006). The Sin1-/- embryos die during gestation between embryonic day (E) 10.5 and 15.5 due to severe defects in cardiovascular development (this will be described in another study). We successfully reconstituted lethally irradiated CD45.1 congenic mice with wild type and Sin1-/- fetal liver hematopoietic cells from E11.5-E12.5 embryos, demonstrating that Sin1-/- hematopoietic stem cells are capable of reconstitute the hematopoietic system of adult mice (data not shown).
We examined bone marrow of chimeric mice and found that the proportion of immature IgM+ bone marrow B cells was reduced in Sin1-/- chimeric mice when compared to Sin1+/+ chimeras (Fig. 1a). Specifically, in a representative pair of Sin1+/+ and Sin1-/- chimeric mice, 70% (12% CD19+IgM+ / 17% CD19+) of the bone marrow B cells were IgM+ while only 43% (3% CD19+IgM+ / 7% CD19+) of Sin1-/- bone marrow B cells were IgM+. We also analyzed a chimeric mouse that contained a 1:1 ratio of host (wild type) to Sin1-/- donor cells allowing us to directly compare developing Sin1-/- B cells and wild type B cells within the same animal. The proportion and number of Sin1-/- IgM+ B cells was reduced by approximately three fold when compared to the wild type IgM+ B cells (Fig. 1b-1c). We also observed a four fold increase in the total number of Sin1-/- IgM- bone marrow B cells when compared to the wild type IgM- B cells. The CD19+IgM- population contains pro-B cells, which are the most immature bone marrow B cell population that also express the pro-B cell surface marker CD45R (B220+) and high levels of CD43 (CD43hi). Analysis of IgM- bone marrow B cells from the Sin1-/- chimeric mice revealed a two-fold increase in the proportion of Sin1-/- B220+CD43hiIgM- pro-B cells relative to wild type cells (Fig. 1d). Together, these data show that the developmental defect is intrinsic to the Sin1-/- B cells and Sin1 deficiency results in expansion of pro-B cells.
Figure 1. Sin1 regulates B cell development.
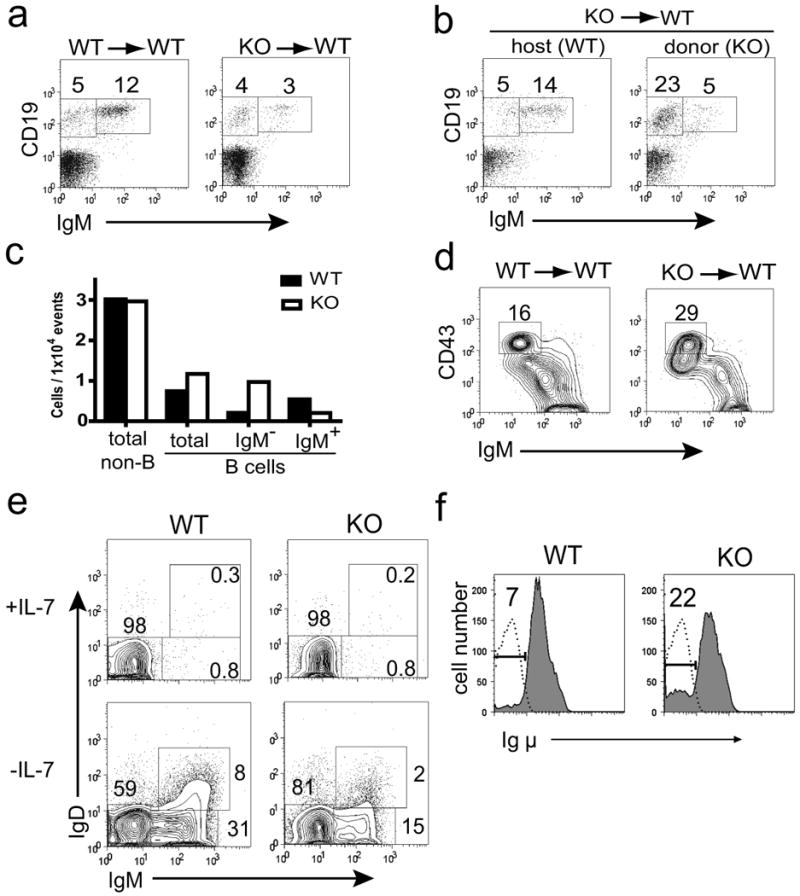
a) Bone marrow from Sin1+/+ or Sin1-/- chimeric mice was analyzed by flow cytometry and the percentage of CD19+ IgM- and CD19+ IgM+ B cells is indicated. The data are representative of Sin1+/+ (n=3) and Sin1-/- (n=4) fetal liver chimeric mice.
b) Bone marrow from a chimeric mouse containing a 1:1 ratio of wild type host and Sin1-/- donor cells was analyzed by flow cytometry. The plots shown are pre-gated on CD45.1+ host cells or CD45.1- Sin1-/- donor cells and the percentage of CD19+IgM- and CD19+IgM+ B cells is indicated.
c) Bar graph illustrating the total number of Sin1+/+ host or Sin1-/- donor bone marrow cells from the chimeric mouse shown in (b).
d) The proportion of B220+CD43hiIgM- bone marrow pro-B cells from wild type or Sin1-/- fetal liver chimeric mice. The plots shown are pre-gated on CD45.2+ donor cells and are representative of Sin1+/+ (n=3) and Sin1-/- (n=4) fetal liver chimeric mice.
e) Sin1+/+ or Sin1-/- pro-B cells were cultured in vitro on OP9 cells with or without IL-7 for 7 days and surface IgM and IgD expression was analyzed. The plots are representative of 4 independent experiments.
f) Sin1+/+ or Sin1-/- pro-B cells were cultured on OP9 cells without IL-7 for 7 days, fixed, permeablized and stained for Igμ chain expression (shaded area). The proportion of Igμ- pro-B cells is indicated. Rag1-/- pro-B cells are a negative control for Igμ staining (dotted line). Representative plots are shown from 3 independent experiments.
Sin1 deficiency perturbs B cell development in vitro
We established primary pro-B cell lines from the fetal livers of paired E12.5 Sin1-/- and Sin1+/+ littermate embryos using OP9 stromal cells supplemented with IL-7 (Vieira and Cumano, 2004). We generated 4 independent pairs of wild type and Sin1-/- pro-B cell lines and observed no defect in the ability in Sin1-/- fetal liver hematopoietic cells to give rise to pro-B cells when compared to the wild type fetal liver hematopoietic cells (Fig. S1a-1c).
To determine the differentiation potential of Sin1+/+ or Sin1-/- pro-B cells, we differentiated those cells in vitro on OP9 cells in the absence of IL-7 for 7 days. We found that Sin1-/- pro-B cells differentiated in vitro gave rise to a smaller proportion of IgM+ cells than the wild type cells (Fig. 1e). We also observed 3 fold more Igμ- pro-B cells in the Sin1-/- culture than in the Sin1+/+ B cell culture (Fig. 1f). Together, these results show that Sin1 is required for the proper development of IgM+ B cells and suggest that Sin1 deficiency may enhance the survival and/or proliferation of pro-B cells when IL-7 is limiting.
Sin1-/- pro-B cells exhibit increased IL-7Rα expression and enhanced IL-7 dependent survival
IL-7 provides the primary pro-B cell survival signal. Since we observed an increased proportion of Sin1-/- pro-B cells in the bone marrow and in the OP9 co-culture differentiation assay, we speculated that Sin1 deficiency might enhance pro-B cell responsiveness to IL-7. We examined IL-7 receptor (IL7R) expression in Sin1+/+ or Sin1-/- pro-B cells and found that il7r mRNA levels were increased approximately 2 fold in Sin1-/- pro-B cells relative to Sin1+/+ pro-B cells (Fig. 2a). The expression of membrane bound IL-7R was also increased on Sin1-/- pro-B cells when compared to the Sin1+/+ pro-B cells (Fig. 2b). IL-7R was not expressed on Sin1+/+ or Sin1-/- IgM+ immature B cells indicating that Sin1 only regulates il7r expression in pro-B cells (Fig. S2).
Figure 2. Sin1-/- pro-B cells exhibit increased il7r expression and enhanced IL-7 dependent survival.
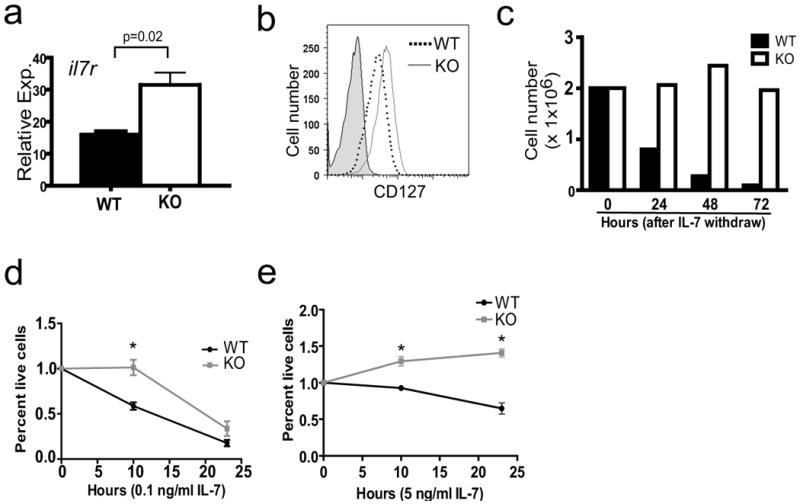
a) IL-7 receptor (il7r) mRNA levels in Sin1+/+ (WT) or Sin1-/- (KO) pro-B cells were measured by qPCR and normalized to GAPDH expression. Samples were run in triplicate and data are representative of two independent experiments.
b) Expression of IL-7R (CD127) on Sin1+/+ or Sin1-/- pro-B cells was measured by flow cytometry. Shaded area is the isotype control staining. The plots are representative of 3 independent experiments.
c) Sin1+/+ or Sin1-/- pro-B cells were cultured on OP9 cells without exogenous IL-7. The total number of live cells recovered at each time point was measured. Dead cells were excluded from the analysis by trypan blue staining. Data are representative of two independent experiments.
d) Sin1+/+ or Sin1-/- pro-B cells were cultured without OP9 cells in medium supplemented with 0.1 ng/ml IL-7. The number of live cells at 10 and 24 hours after plating was determined by trypan blue dye exclusion. Each data point represents triplicate wells from one of two independent experiments. (* = p<0.01)
e) Sin1+/+ or Sin1-/- pro-B cells were cultured without OP9 cells in medium supplemented with 5 ng/ml IL-7. The number of live cells at various time points after plating was determined by trypan blue dye exclusion. Each data point represents triplicate wells from one of two independent experiments. (* = p<0.01)
Error bars indicate standard deviation.
These data suggested that the increased IL-7R expression on Sin1-/- pro-B cells may render these cells more sensitive to IL-7 than Sin1+/+ pro-B cells. Indeed, we found that the number of Sin1-/- B cells recovered at each time point after IL-7 withdraw was substantially greater than that of Sin1+/+ cells (Fig. 2c). Most notably, the number of viable Sin1+/+ B cells decreased by approximately 90 percent 72 hours after IL-7 withdraw while the number of viable Sin1-/- B cells at 72 hours was similar to the number of cells initially plated at time zero. These data suggest that Sin1-/- pro-B cells exhibit enhanced survival to the IL-7 produced by the OP9 cells.
To further confirm that Sin1-/- pro-B cells exhibit enhanced IL-7 responsiveness, we measured IL-7 dependant survival of Sin1+/+ and Sin1-/- pro-B cells in the absence of OP9 cells. Sin1+/+ or Sin1-/- pro-B cells were washed and cultured in medium containing a low concentration of IL-7 (0.1 ng/ml). The number of live cells was measured at 10 hours or 24 hours after plating. We observed significantly more live Sin1-/- pro-B cells than wild type pro-B cells after 10 hours in culture (Fig. 2d). There was little difference between Sin1+/+ and Sin1-/- cell viability by 24 hours indicating that IL-7 is absolutely required for the survival of both wild type and Sin1-/- pro-B cells. We also cultured pro-B cells in medium supplemented with a high concentration of IL-7 (5 ng/ml) and found that Sin1-/- pro-B cells showed enhanced survival over Sin1+/+ pro-B cells under these conditions (Fig. 2e). Together, these data show that Sin1 deficient pro-B cells have increased IL-7 receptor expression and exhibit enhanced IL-7 dependent survival.
Sin1-/- B cells lack functional mTORC2 and exhibit defective Akt phosphorylation
To investigate the mechanism of Sin1 function in B cells we established Abelson murine leukemia virus (Ab-MuLV) transformed pre-B cells from Sin1+/+, Sin1+/- and Sin1-/- pro-B cells. Sin1-/- Ab-MuLV pre-B cells expressed B lineage surface markers and showed no defect in growth, proliferation, or survival when compared to Sin1+/+ or Sin1+/- pre-B cells (data not shown). To determine if Sin1 is essential for mTORC2 integrity in B cells, the endogenous mTORC2 was immunoprecipitated from Sin1+/- or Sin1-/- Ab-MuLV pre-B cells using an anti-Rictor antibody. As expected, mTOR and Sin1 co-immunoprecipitated with Rictor in the Sin1+/- but not Sin1-/- pre-B cells (Fig. 3a). In addition, Rictor and mTOR co-immunoprecipitated with Sin1 in the Sin1+/- cells (data not shown). These data show that Sin1 is required for the mTORC2 integrity in B cells. Consistent with these results, phosphorylation of Akt at the mTORC2 target sites Ser473 and Thr450 was abolished in the Sin1-/- pro-B cells and Ab-MuLV pre-B cells but not in the Sin1 sufficient B cells (Fig. 3b). The Akt T-loop (Thr308) phosphorylation was approximately 1.5 fold more in Sin1-/- pro-B cells and about 10 fold more in Sin1-/- Ab-MuLV pre-B cells than that in Sin1+/+ pro-B and pre-B cells, respectively (Fig. 3b). Furthermore, Akt expression in both primary and transformed Sin1-/- B cells was decreased as well, consistent with our previous studies showing that the TM phosphorylation regulates Akt protein stability (Facchinetti et al., 2008).
Figure 3. Sin1 is required for mTORC2 dependent Akt hydrophobic motif and Akt turn motif phosphorylation in B cells.
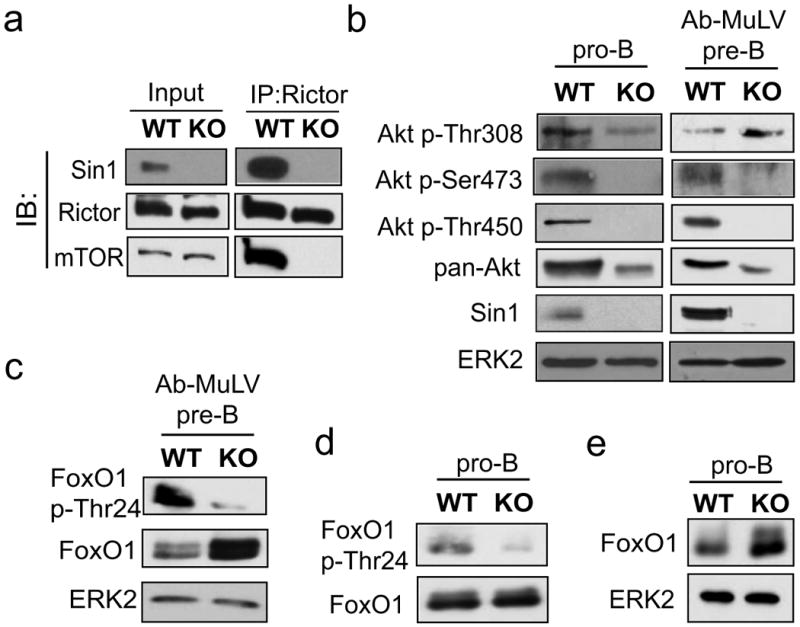
a) The mTORC2 complex integrity in Sin1+/- (WT) or Sin1-/- (KO) Ab-MuLV pre-B cells was determined by immunoprecipitation (IP) of Rictor followed by immunoblotting (IB) of Sin1, Rictor and mTOR.
b) Akt phosphorylation and expression was measured in primary Sin1+/+ or Sin1-/- pro-B cells cultured on OP9 cells with IL-7 and Ab-MuLV transformed Sin1+/- or Sin1-/- pre-B cells. Samples were normalized to total cellular protein and blotted as indicated.
c) FoxO1 phosphorylation and expression was measured in Sin1+/- or Sin1-/- Ab-MuLV pre-B cells. Samples were normalized to total cellular protein and blotted for FoxO1 or FoxO1 p-Thr24.
d) FoxO1 phosphorylation was measured in Sin1+/+ or Sin1-/- pro-B cells cultured with serum and IL-7. The samples were normalized to total FoxO1 protein then blotted for FoxO1 p-Thr24.
e) FoxO1 expression was measured in Sin1+/+ or Sin1-/- pro-B cells. The samples were normalized to total cellular protein.
Sin1 is required for FoxO1 phosphorylation in B cells
The FoxO transcription factors are evolutionarily conserved targets of Akt and disruption of mTORC2 results in the selective impairment of FoxO1/3a phosphorylation in embryonic fibroblasts (Jacinto et al., 2006). Therefore, we examined FoxO1 phosphorylation in control and Sin1-/- Ab-MuLV pre-B cells and found that FoxO1 phosphorylation at Thr24 was impaired (Fig. 3c). The Akt dependent phosphorylation of FoxO proteins is known to promote the ubiquitination and subsequent proteasome-dependent degradation of FoxO proteins (Plas and Thompson, 2003). Consistently, more FoxO1 protein was detected in Sin1-/- Ab-MuLV pre-B cells than the control cells (Fig. 3c). FoxO1 Thr24 phosphorylation was also impaired and FoxO1 protein levels were increased in Sin1-/- pro-B cells when compared to Sin1+/+ pro-B cells (Fig. 3d and Fig. 3e). Together, these data show that Sin1 deletion impairs FoxO1 phosphorylation in B developing cells.
Sin1 suppresses RAG expression in developing B cells
Our observation that FoxO1 phosphorylation is impaired and FoxO1 expression is increased in Sin1-/- B cells provides a possible mechanism to explain the elevated expression of il7r, which was recently identified as a FoxO1 target gene (Kerdiles et al., 2009). The genes rag1 and rag2 are also regulated by FoxO1 in B cells (Amin and Schlissel, 2008; Herzog et al., 2008; Llorian et al., 2007; Verkoczy et al., 2007). We measured rag1 transcript levels in Sin1+/- and Sin1-/- Ab-MuLV pre-B cells and found that rag1 expression was significantly elevated in Sin1-/- pre-B cells compared to the Sin1+/- cells (Fig. 4a). Rag expression is suppressed by the v-Abl kinase and can be rapidly induced by inhibiting v-Abl activity with Imatinib (Muljo and Schlissel, 2003). We found that Imatinib treatment substantially increased rag1 expression in both control and Sin1-/- pre-B cells. However, rag1 expression was increased an additional three fold in Imatinib treated Sin1-/- pre-B cells when compared to the Sin1+/- pre-B cells (Fig. 4a). In addition, RAG1 protein was readily detectable in Sin1-/- but not Sin1+/- Ab-MuLV pre-B cells cultured in the absence of Imatinib (Fig. 4b).
Figure 4. Increased RAG expression and V(D)J recombinase activity in developing Sin1-/- B cells.
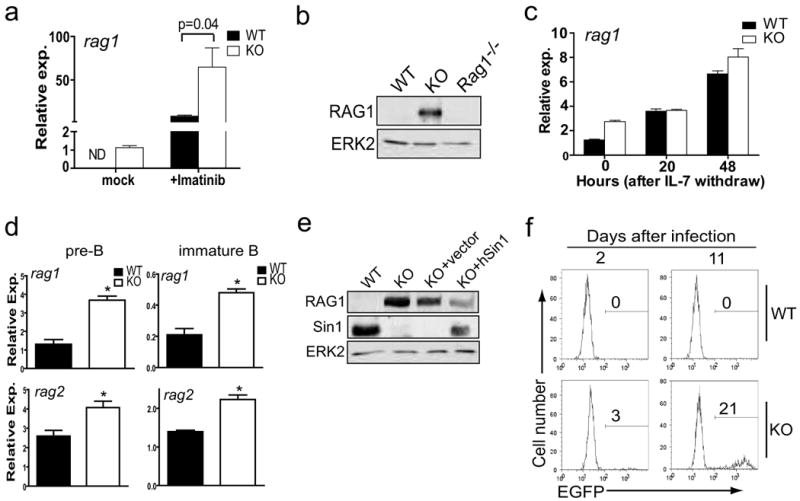
a) Rag1 mRNA expression was measured by qPCR in Sin1+/- (WT) or Sin1-/- (KO) Ab-MuLV pre-B cells cultured for 5 hours in the presence or absence of Imatinib. Rag1 mRNA was normalized to GAPDH mRNA. ND: not detected.
b) RAG1 protein in Sin1+/- (WT), Sin1-/- (KO) or rag1-/- Ab-MuLV pre-B cells was measured by immunoblotting. The ERK2 protein is used as a loading control.
c) Sin1+/+ or Sin1-/- pro-B cells were cultured on OP9 cells without exogenous IL-7 for 0, 20, or 48 hours. Rag1 expression was measured by qPCR. Rag1 expression was normalized to GAPDH mRNA. Samples were run in triplicate and are representative of two independent experiments.
d) Sin1+/+ or Sin1-/- pro-B cells were differentiated in vitro on OP9 cells for 7 days, and rag1 or rag2 expression in pre-B and immature B cells obtained from these cultures was measured by qPCR as described in panel a). Samples were run in triplicate and are representative of two independent experiments. (* p<0.01)
e) Sin1-/- Ab-MuLV pre-B cells were infected with a retrovirus expressing human Sin1 or a control empty retrovirus, and the RAG1 expression was measured by immunoblotting. Data are representative of two independent experiments.
f) Sin1+/- or Sin1-/- Ab-MuLV pre-B cells were infected with a retrovirus containing an EGFP RAG recombinase reporter. The infected Ab-MuLV pre-B cells were then grown under normal culture conditions and the EGFP+ cells were determined by flow cytometry 2 days and 11 days later. The plots shown are gated on infected cells (hCD4+ cells). The data are representative of three independent experiments.
Error bars indicate standard deviation.
Next, we examined rag1 expression in Sin1+/+ or Sin1-/- pro-B cells cultured on OP9 cells with IL-7 and found that rag1 expression was approximately 2 fold more in Sin1-/- pro-B cells than in Sin1+/+ pro-B cells (Fig. 4c, at the 0 hr point). IL-7 attenuates rag1 expression in pro-B cells (Melamed et al., 1997). Therefore we measured rag1 expression in Sin1+/+ or Sin1-/- pro-B cells 20 hours after IL-7 withdrawal and found that rag1 expression increased two fold in Sin1+/+ pro-B cells while rag1 expression showed little increase in Sin1-/- pro-B cells (Fig 4c). At 48 hours after IL-7 withdrawal, Sin1+/+ pro-B cells showed a 5 fold increase in rag1 expression while Sin1-/- pro-B cells showed a 3 fold increase in rag1 expression relative to the 0 hour time point. These data suggest that IL-7 signals suppress rag expression through a mechanism which is not dependent on Sin1.
We also examined rag1 expression in Sin1+/+ or Sin1-/- pre-B cells and immature B cells differentiated in vitro on OP9 cells. We found that rag1 expression was 3 fold higher in Sin1-/- pre-B cells than Sin1+/+ pre-B cells and 2 fold higher in Sin1-/- immature B cells than Sin1+/+ immature B cells (Fig. 4d). Expression of rag2 was also higher in both Sin1-/- pre-B and immature B cells than that in Sin1+/+ B cells (Fig. 4d). To determine if Sin1 deficiency influences IgL recombination we examined the Igκ and Igλ chain expression on immature IgM+ B cells. Analysis of IgL chain expression on immature IgM+ Sin1+/+ or Sin1-/- B cells revealed a 2 fold increase in the proportion of Igλ light chain expressing Sin1-/- B cells when compared to Sin1+/+ B cells (Fig. S3). Together, these results show that Sin1 regulates rag expression in B cells and suggest that Sin1 influences IgL recombination in developing B cells.
Finally, we reconstituted the Sin1-/- Ab-MuLV pre-B cells with human Sin1 cDNA and examined RAG1 protein levels in these cells. Restoration of Sin1 expression in Sin1-/- Ab-MuLV pre-B cells decreased RAG1 protein expression while a control virus lacking human Sin1 expression did not decrease RAG1 expression (Fig. 4e). These data confirm that Sin1 is a negative regulator of rag expression in developing B cells.
Sin1 suppresses V(D)J recombinase activity in developing B cells
To determine if the elevated rag gene expression and RAG1 protein observed in the Sin1-/- pre-B cells correlates with V(D)J recombinase activity, we infected wild type or Sin1-/- Ab-MuLV pre-B cells with a retrovirus containing an EGFP-based V(D)J recombinase reporter. The reporter contains an anti-sense orientated EGFP cDNA flanked by 12- and 23-recombination signal sequences. RAG mediated recombination flips the EGFP cDNA sequence to the sense orientation permanently marking the cell with EGFP expression. Sin1+/- or Sin1-/- pre-B cells infected with the reporter virus were assayed 2 days and 11 days post-infection by flow cytometry. The infected cells were first identified by human CD4 expression from an IRES-hCD4 cassette in the retroviral vector and then analyzed for EGFP expression. At day 2, no Sin1+/- pre-B cells expressed EGFP while 3% of the Sin1-/- hCD4+ pre-B cells expressed EGFP. Eleven days after infection, 21% of the hCD4+ Sin1-/- pre-B cells expressed EGFP while none of the Sin1+/- hCD4+ pre-B cells expressed EGFP (Fig. 4f). These data show that Sin1 deficiency results in increased V(D)J recombinase activity in developing B cells.
Akt2 regulates FoxO1 phosphorylation and suppresses il7r and rag expression in B cells
Our data shows that Sin1/mTORC2 regulates the expression of the FoxO1 target genes il7r, rag1 and rag2. Studies from our group and other laboratories suggest that Sin1/mTORC2 mediates PI3K signals to activate Akt which in turn suppresses FoxO1 activity. However, genetic evidence supporting this model is currently lacking. If this model is correct, we predict that the Sin1 and Akt deficient B cells will share a common phenotype showing defective FoxO1 regulation and augmented FoxO1 target gene expression. To test this model, we established primary pro-B cell lines from Akt1-/-, Akt2-/- and Akt1-/-/Akt2-/- mice. We first examined rag expression in these cells by quantitative RT-PCR, and found that Akt1 deficiency had no effect on rag1 expression in pro-B cells (Fig. 5a). In contrast, the rag1 expression was elevated approximately 3 fold in Akt2-/- pro-B cells relative to the wild type cells (Fig. 5a). Consistent with the increased rag1 mRNA expression, RAG1 protein levels were also significantly increased in Sin1-/-, Akt2-/-, and Akt1-/-/Akt2-/- pro-B cells but not wild type or Akt1-/- pro-B cells (Fig. 5b). We also examined IL-7R expression in Akt deficient pro-B cells and found that il7r expression was increased in Akt2-/- and Ak1-/-/Akt2-/- but not wild type or Akt1-/- pro-B cells (Fig. 5c). These results indicate that Akt2 is the principle mediator of Sin1/mTORC2 signaling which suppresses il7r and rag expression in developing B cells.
Figure 5. Akt2 regulates Rag and IL-7R expression in pro-B cells.
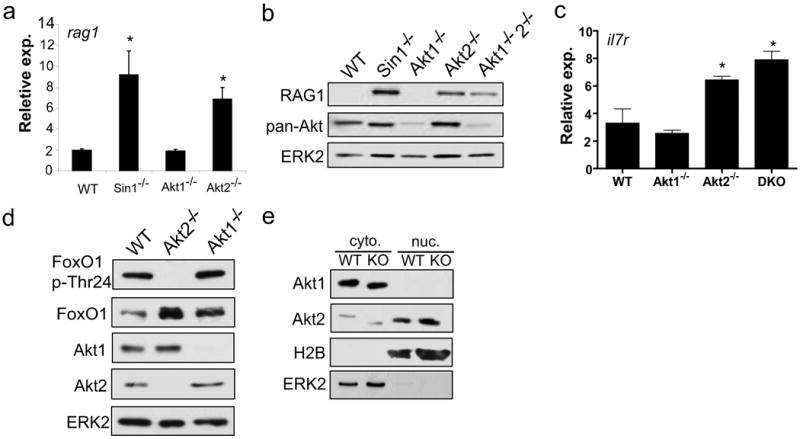
a) Wild type (WT), Sin1-/- and Akt deficient pro-B cells were cultured on OP9 cells with IL-7. Rag1 mRNA levels were measured by qPCR and normalized to GAPDH mRNA. Samples were run in triplicate and are representative of two independent experiments. (* = p<0.01)
b) RAG1 protein was measured in primary pro-B cells cultured on OP9 cells with IL-7. Total Akt expression was measured with a pan-Akt antibody. Samples were normalized to total cellular protein.
c) Il7r mRNA expression in wild type, Akt1-/-, Akt2-/- or Akt1-/- Akt2-/- (DKO) pro-B cells cultured on OP9 cells with IL-7 was measured by quantitative RT-PCR and normalized to GAPDH mRNA. Samples were run in triplicate and are representative of two independent experiments. (* = p<0.01)
d) FoxO1 phosphorylation was measured in wild type, Akt2-/-, or Akt1-/- pro-B cells. Samples were normalized to total cellular protein.
e) Cytoplasmic and nuclear Akt1 and Akt2 was determined in Sin1+/+ (WT) or Sin1-/- (KO) pro-B cells cultured on OP9 cells with IL-7. ERK2 and H2B proteins were used to verify the quality of the cytoplasmic and nuclear fractions. Three times more nuclear proteins (based on the total cell numbers) than cytosolic proteins were loaded in this assay to enhance the nuclear signals.
Standard deviation is indicated by all error bars.
To explore the mechanism though which Akt2 regulates rag1 and il7r in B cells, we analyzed FoxO1 phosphorylation in Akt1-/- and Akt2-/- pro-B cells and found that the deletion of Akt2 but not Akt1 blocked FoxO1 Thr24 phosphorylation (Fig. 5d). These results prompted us to examine how Akt2 might specifically regulate FoxO1 phosphorylation in B cells. We speculated that the differential sub-cellular localization of Akt1 and Akt2 proteins within pro-B cells may contribute to the specific regulation of FoxO1 by Akt2. Therefore, we fractionated cytosol and nuclear proteins from Sin1+/+ and Sin1-/- pro-B cells and determined the distribution of Akt1 and Akt2 proteins in these sub-cellular fractions. We found that Akt1 was localized exclusively in the cytosol while Akt2 was localized in both cytosol and nucleus of pro-B cells (Fig. 5e). These studies also revealed that the differential sub-cellular localization of Akt1 and Akt2 proteins in pro-B cells is not dependent on Sin1. Together, these data show that Akt2 specifically regulates FoxO1 phosphorylation in pro-B cells and suggests that the selective localization of Akt2 to the B cell nucleus contributes to the specific regulation of FoxO1 by Akt2.
Rapamycin induces rag expression in B cells by blocking mTORC2 dependent Akt HM phosphorylation
Prolonged rapamycin treatment is known to disrupt mTORC2 and inhibit Akt Ser473 phosphorylation in some cell types (Facchinetti et al., 2008; Sarbassov et al., 2006). Therefore we asked if rapamycin treatment may disrupt mTORC2, inhibit Akt HM phosphorylation and induce rag gene expression in B cells. We treated wild type Ab-MuLV pre-B cells with rapamycin for 5 hours or 25 hours and measured rag expression by quantitative RT-PCR. The acute rapamycin treatment (5 hours) did not alter rag expression, but prolonged rapamycin treatment (25 hours) substantially increased rag1 and rag2 mRNA levels (Fig. 6a). In addition, rapamycin treatment also markedly increased the RAG1 protein level in Ab-MuLV pre-B cells (Fig. 6b). We also observed that FoxO1 protein levels were increased after rapamycin treatment (Fig. 6b). Furthermore, we observed that phosphorylation of Akt Ser473 was inhibited after 25 hours but not 5 hours of rapamycin treatment (Fig. 6c). The overall Akt protein level was also reduced after 25 hours of rapamycin treatment. As expected, phosphorylation of the mTORC1 target 4E-BP1 was inhibited by rapamycin at 5- and 25-hour points (Fig. 6c). These data show that rapamycin induces rag gene expression in a manner that correlates with the loss of Akt Ser473 phosphorylation in B cells.
Figure 6. Rapamycin induces RAG gene expression and inhibits Akt Ser473 phosphorylation in B cells.
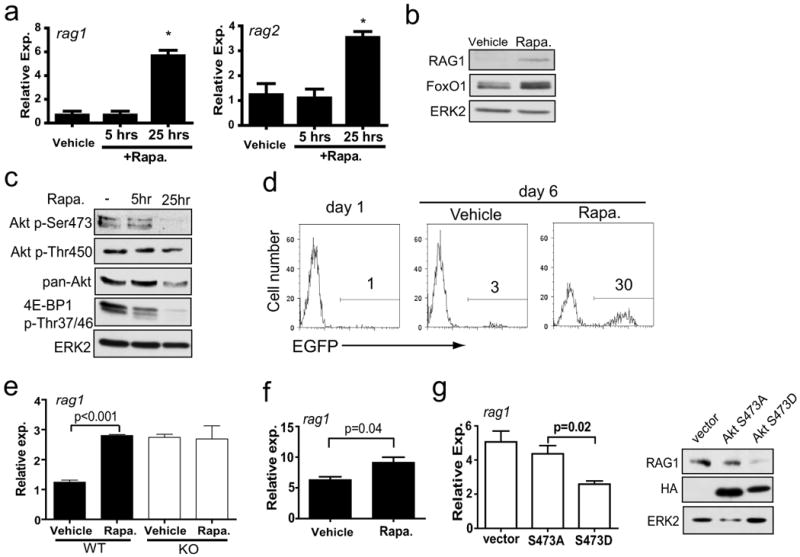
a) Rag mRNA levels were measured by qPCR in Sin1+/- Ab-MuLV pre-B cells treated with rapamycin for the indicated periods of time. Rag1 and rag2 expression was normalized to GAPDH mRNA. Samples were run in triplicate and are representative of two independent experiments. (* = p<0.01)
b) Sin1+/- Ab-MuLV pre-B cells were cultured in the presence or absence of rapamycin for 25 hours and RAG1 or FoxO1 protein was measured. ERK2 is used as a loading control.
c) Sin1+/- Ab-MuLV pre-B cells were vehicle or rapamycin treated for 5 or 25 hours. Cell lysates were normalized to total protein and immunoblotted as indicated.
d) Sin1+/- Ab-MuLV pre-B cells were infected with a retrovirus containing an EGFP RAG recombinase reporter. The infected cells were continuously cultured in the presence (Rapa.) or absence (vehicle) of rapamycin for 6 days. The EGFP+ cells were determined by flow cytometry at 1 day and 6 days after infection. The plots are gated on infected cells (hCD4+ cells) and are representative of three independent experiments.
e) Sin1+/+ (WT) or Sin1-/- (KO) pro-B cells cultured on OP9 cells with IL-7 were treated with rapamycin or vehicle only for 25 hours and rag1 expression was measured by qPCR. Samples were normalized to GAPDH expression. Samples were run in triplicate and the data are representative of two independent experiments.
f) Resting Sin1+/- splenic B cells were enriched by negative selection and cultured in vitro in the presence (Rapa) or absence (Vehicle) of rapamycin for 25 hours. Rag1 expression was measured by qPCR and normalized to GAPDH expression. Samples were run in triplicate and the data are representative of two independent experiments.
g) Sin1-/- Ab-MuLV pre-B cells were infected with retrovirus expressing human Akt with a null HM mutation (Ser473Ala) or a phospho-mimetic HM mutation (Ser473Asp). Rag1 mRNA expression was measured by qPCR and normalized to GAPDH. RAG1 protein levels were measured by immunoblotting and the HA blotting verifies expression of the virally expressed Akt. Quantitative RT-PCR samples were run in triplicate and the data are representative of two independent experiments.
Standard deviation is indicated by all error bars.
The increased rag expression in Sin1+/- pre-B cells following rapamycin treatment suggested that rapamycin may also increase V(D)J recombinase activity in B cells. We infected Sin1+/- Ab-MuLV pre-B cells with the EGFP RAG recombinase reporter and cultured these cells in the presence or absence of rapamycin. We observed that about 30% of the rapamycin treated, infected pre-B cells expressed EGFP while only 3% of the vehicle treated, infected cells expressed EGFP after 6 days (Fig. 6d). These data show that rapamycin mimics the effect of Sin1 deficiency (Fig. 4f) and promotes V(D)J recombinase activity in B cells.
Next we asked if rapamycin induces rag expression in non-transformed B cells. We cultured Sin1+/+ or Sin1-/- pro-B cells on OP9 cells with IL-7 in the presence or absence of rapamycin for 25 hours then measured rag1 expression by quantitative RT-PCR. Rapamycin induced rag1 expression in the Sin1+/+ pro-B cells by approximately 3 fold while rapamycin had no effect on rag1 expression in Sin1-/- pro-B cells (Fig. 6e). Additionally, we also observed that rapamycin induced il7r mRNA levels by 2 fold in Sin1+/+ pro-B cells (Fig. S4). To further explore the effect of rapamycin on rag expression in B cells, we purified total splenic B cells from a Sin1+/- mouse and cultured these cells in vitro for 24 hours with or without rapamycin. We observed that rag1 expression was increased in the rapamycin treated Sin1+/- splenic B cells when compared to vehicle treated B cells (Fig. 6f). These data show that rapamycin treatment induces il7r expression in pro-B cells and rag expression in pro-B and splenic B cells.
Since Akt HM phosphorylation is dependent on mTORC2, we asked if Akt HM phosphorylation specifically mediates the Sin1/mTORC2 dependent inhibition of rag expression in B cells. We infected Sin1-/- Ab-MuLV pre-B cells with retrovirus expressing a human Akt cDNA with either a Ser473 to Ala null mutation or a phosphomimetic Ser473 to Asp mutation. Infected cells were sorted based on virally expressed GFP and rag1 expression was measured by quantitative RT-PCR. Ectopic expression of the Ser473Ala Akt mutant failed to inhibit rag1 expression in Sin1-/- pre-B cells. In contrast, expression of the Ser473Asp Akt mutant markedly reduced rag1 expression in Sin1-/- pre-B cells (Fig. 6g). Consistently, expression of the Ser473Asp Akt mutant but not the Ser473Ala Akt mutant suppressed RAG1 protein levels in Sin1-/- pre-B cells (Fig. 6g). In addition, Ser473Asp Akt mutant also induced more FoxO1 phosphorylation than the Ser473Ala Akt mutant (data not shown). These data demonstrate that Akt HM site function is necessary and sufficient to complement Sin1 deficiency and suppress rag expression in developing B cells.
Discussion
In this study we show that Sin1, an essential component of mTORC2, plays a critical role in B cell development. As illustrated in the model in Figure 7, Sin1/mTORC2 mediates PI3K dependent signals (i.e. pre-BCR or BCR) to phosphorylate the hydrophobic motif (Ser473) of Akt2. PI3K dependent PDK1 phosphorylates the T-loop (Thr308) of Akt2 resulting in full Akt2 activation. Ser473 phosphorylation directs Akt2 activity towards its substrate FoxO1 Thr24 resulting in phosphorylation of FoxO1 thus suppressing the expression of FoxO1 target genes il7r, rag1, and rag2 in developing B cells. Our study demonstrates the specific role of Sin1/mTORC2 and Akt2 as key regulators of il7r and rag gene expression in B cells.
Figure 7.
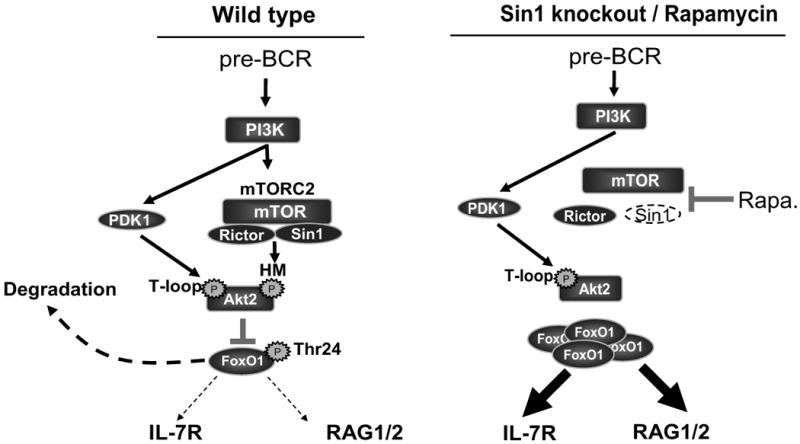
A model illustrating the regulation of il7r and rag expression by the Sin1/mTORC2-Akt2 signaling axis in developing B cells. PDK1 and Sin1/mTORC2 mediate the PI3K signals to phosphorylate Akt2 at Thr308 and Ser473, respectively. Activated Akt2 then phosphorylates FoxO1, resulting in FoxO1 degradation and down regulation of il7r and rag gene expression. In the absence of Sin1 or during chronic rapamycin treatment, mTORC2 is disrupted and Akt2 is not fully activated resulting in FoxO1 hypophosphorylation and accumulation; this leads to the augmented il7r and rag gene expression.
Interestingly, Sin1 is not required for pro-B cell proliferation and survival (Fig. S1a & S1b). Rather, Sin1-/- pro-B cells exhibit enhanced survival when cultured in the presence of IL-7. Our data indicates that this enhanced response to IL-7 is due to increased IL-7R expression on Sin1-/- pro-B cells. Our data also indicates that Sin1 regulates pro-B cell to pre-B cell differentiation since we observe an accumulation of pro-B and a reduction of IgM+ immature B cells in the bone marrow of Sin1-/- chimeric mice and in Sin1-/- B cells differentiated in vitro on OP9 cells. Therefore, we propose that Sin1 mediates PI3K dependent pre-BCR signaling to suppress rag and il7r expression, which inhibits further IgH recombination.
It is of note that, mice lacking key structural components of pre-BCR such as Igμ, surrogate light chain genes λ5 and VpreB, and mice deficient in key pre-BCR signaling mediators such as Igα or Igβ all exhibit a B cell developmental block at the pro-B to pre-B transition (Herzog et al., 2009). The Sin1/mTORC2-Akt2-FoxO1 axis may also operate in a similar manner in transducing BCR signals in immature B cells following IgL recombination since the downstream mediators of pre-BCR and BCR signaling are conserved (Herzog et al., 2009). Therefore we predict that Sin1 deficiency may also perturb BCR signaling in immature and mature B cells. PI3K dependent BCR signaling suppresses rag expression and inhibits IgL receptor editing in immature B cells (Verkoczy et al., 2007). We observe that rag gene expression is elevated in Sin1-/- immature B cells and that Igλ chain usage is increased in Sin1-/- B cells (Fig. 4d and Fig. S3), suggesting that Sin1 mediates BCR dependent PI3K signaling as well.
Sin1 deletion does not appear to impair Ig recombination. Rather we observe increased rag expression and V(D)J recombinase activity in Sin1-/- B cells. Furthermore, the induction of Igκ germline transcription, which is an indicator of locus activation, is not impaired in Sin1-/- pre-B cells indicating that Sin1 is not required for the IgL locus accessibility (data not shown). In fact, the augmented V(D)J recombinase activity in developing Sin1-/- B cells may even promote IgL recombination since we observe an increase in the percentage of immature Sin1-/- B cells that express Igλ (Fig. S3 and Fig. 4d).
Sin1 is an evolutionally conserved adapter molecule and is essential for the integrity of mTORC2 in a diverse array of organisms and cell types (Jacinto et al., 2006; Yang et al., 2006). Sin1-deficiency completely blocks the phosphorylation of Akt at Ser473 and Thr450 in both primary pro-B and transformed pre-B cells without impairing Akt Thr308 phosphorylation. These results are consistent with our previous studies in non-immune cells (Facchinetti et al., 2008; Jacinto et al., 2006). Our results also demonstrate that the mTORC2 is the sole kinase (PDK2) for Akt HM site phosphorylation in B cells.
The expression of rag1, rag2 and il7r is stringently regulated in developing B cells through a mechanism, which involves multiple signaling pathways. Our study reveals Sin1 as a key negative regulator of these genes in B cells whose activity is most likely mediated by FoxO1 since rag1, rag2 and il7r are direct targets of FoxO1 regulation (Amin and Schlissel, 2008; Dengler et al., 2008; Herzog et al., 2008). We present evidence showing that disruption of Sin1 or Akt2 impairs FoxO1 phosphorylation, which correlates with increased rag1/2 and il7r expression. Furthermore, we demonstrate the functional importance of Akt HM phosphorylation in regulating FoxO1 activity by showing that ectopic expression of the phosphomimetic Ser473Asp Akt mutant but not the phosphorylation null Ser473Ala Akt mutant inhibits rag expression in Sin1-/- pre-B cells. Interestingly, we observed that the relative amount of nuclear localized FoxO1 protein is similar in Sin1+/+ and Sin1-/- pro-B cells (data not shown). These data suggest that the relative abundance of nuclear FoxO1 protein is not tightly correlated with FoxO1 transcriptional activity and that regulation of FoxO1 localization in B cells is Sin1/mTORC2 independent.
Although Sin1/mTORC2 regulates both Akt1 and Akt2 HM and TM site phosphorylation, Akt1 and Akt2 appear to be differentially utilized by developing B cells with respect to the regulation of rag1, rag2 and il7r. We were particularly surprised to find that Akt1 does not regulate FoxO1 phosphorylation or rag1, rag2 and il7r expression since Akt1 is the most abundantly expressed Akt isoform in developing B cells (Fig. 5b). We show that Akt2, but not Akt1, is selectively localized to the nucleus of B cells. Based on these data we propose that nuclear localized Akt2 is responsible for phosphorylating FoxO1. Exactly how this selective, isoform specific, cellular distribution of Akt proteins is achieved and regulated in B cells remains to be elucidated. However, we observe that Akt2 is nuclear localized in Sin1-/- pro-B cells suggesting that Akt HM and TM phosphorylation are not required for Akt2 import into the nucleus. These results indicate that Akt2 HM phosphorylation may be required to specifically facilitate interaction of Akt2 with FoxO1 or promote the Akt2 dependent phosphorylation of FoxO1 at Thr24.
V(D)J recombination is normally suppressed in proliferating B cells by coupling RAG2 protein degradation with mitosis (Li et al., 1996). Cell cycle dependent regulation of V(D)J recombination helps to ensure that aberrant recombination products are not generated during the cell cycle which may result in mutations that promote tumor formation. Our data reveals that Sin1/mTORC2 signaling provides an additional level of protection against abnormal V(D)J recombinase activity by suppressing expression of rag1 and rag2 in proliferating B cells. Surprisingly, we observed substantial V(D)J recombinase activity in Sin1-/- Ab-MuLV pre-B leukemia cells that were actively proliferating (Fig. 4f). These findings raise the possibility that mTOR inhibitors, which disrupt mTORC2 function and induce rag expression, may promote genome instability in B cells by promoting aberrant V(D)J recombinase activity. This is a very important point in light of our finding that rapamycin increases rag expression in mature B cells. Our data argues that the rag locus is not irreversibly silenced upon B cell maturation and suggests that mTORC2 dependent signaling actively suppresses rag expression in mature B cells. This has significant implications with regard to the mechanisms that lead to genome instability and B cell tumors. Perturbations of mTORC2 signaling that induce rag expression in immature or mature B cells may increase the likelihood of co-expression of rag with activation-induced cytidine deaminase (AID), a circumstance that has been strongly implicated in the generation of chromosomal translocations (Tsai et al., 2008; Wang et al., 2008; Wang et al., 2009). Future studies will elucidate the role of mTORC2 in promoting B cell genome stability and determine if pharmacologic mTOR inhibition increases the likelihood of generating B cell tumors.
Methods
A detailed explanation of the experimental methods can be found in the Supplemental Methods.
Mice
Sin1 knockout mice were described previously (Jacinto et al., 2006). Akt1 and Akt2 knockout mice were described previously (Di Lorenzo et al., 2009). CD45.1+ congenic (B6.SJL-Ptprca) mice were purchased from The Jackson Laboratory and used as recipients for the fetal liver hematopoietic cell transfers. Mice receiving fetal liver cell transplants were irradiated with 700-900 cGy. All mice were housed in the animal facilities at Yale University and all animal procedures were approved by the Yale Institutional Animal Care and Use Committee.
B cell cultures
Pro-B cells were derived from paired Sin1+/+ and Sin1-/- littermate E12.5 embryos. Akt2-/- and Akt1-/-/Akt2-/- pro-B cells were derived from E13.5 embryos and Akt1-/- pro-B cells were derived from the bone marrow of a 6 week old Akt1-/- mouse. All pro-B cells were cultured on OP9 stromal cells in medium supplemented with recombinant mIL-7 (PeproTech). Abelson murine leukemia virus transformed pre-B cells were generated by infecting cultured pro-B cells with viral supernatant (kindly provided by Dr. Yuan Zhuang, Duke University).
Inhibitors
Imatinib (10 mM, LC Laboratories) stocks were prepared in sterile water and used at a final concentration of 10 uM. Rapamycin (LC Laboratories) was prepared as a 10 uM stock in ethanol and used at a final concentration of 10 nM in all studies unless otherwise indicated.
Flow Cytometry
Single cell suspensions were stained in cold FACS buffer (1× PBS pH7.4 + 2% FBS) with the appropriate fluorophore or biotin conjugated antibodies for 15 min on ice. For biotin conjugated antibodies, cells were washed with FACS buffer and incubated with the appropriate streptavidin conjugated fluorophores for 15 min on ice. Anti-IgM μ chain specific F(ab′)2 fragment (Jackson ImmunoResearch) was used for intracellular IgH staining. All cells were washed and resuspended in FACS buffer for analysis with a FACSCalibur (BD).
Quantitative RT-PCR
Cells were lysed in TRIzol (Invitrogen), total RNA was purified by isopropanol precipitation. Total RNA was treated with RNAse free DNAse I (Sigma) and reverse transcribed with Super-Script II reverse transcriptase (Invitrogen) using random primers. Quantitative RT-PCR was performed with an iQ5 multicolor RT-PCR detection system (Bio-Rad) using the Power SYBR green PCR master mix kit (Applied Biosystems).
V(D)J recombinase activity reporter
Ab-MuLV pre-B cells were infected with the retroviral vector pMX-RSS-GFP/IRES-hCD4, which contains a RAG recombinase activity reporter cassette and an IRES-hCD4 expressing cassette (Liang et al., 2002). The infected cells were gated for human CD4 expression and analyzed for the EGFP-positive cells at various times following infection.
Immunoblotting and antibodies
Cells were washed 2 × with ice cold 1×PBS and lysed in cold RIPA buffer with freshly added protease and phosphatase. Total cell lysates were resolved by SDS-PAGE and blotted with the following antibodies: anti-Akt p-Thr308, anti-Akt p-Ser473 (587F11), anti-panAkt (11E7), anti-Akt2 (D6G4), anti-PKCα/βII p-Thr641/641, anti-Foxo1/3a p-Thr23/32, anti-Foxo1 (C29H4), anti-4E-BP1 p-Thr37/36 from Cell Signaling, anti-Akt1 (E45W), anti-H2B (EP819Y) (Epitomics), anti-Sin1 (K87) (Jacinto et al., 2006), anti-Rictor (Bethyl Inc.), anti-mTOR (N5D11) (IBL), anti-RAG1 (35.2) (Leu and Schatz, 1995), ERK2 (381A10) (Invitrogen). Densitometry analysis was performed with a BioRad Molecular Imager Gel Doc XR system and Quantity One software (BioRad). Immunoprecipitation of mTORC2 was performed as previously described (Facchinetti et al., 2008).
Supplementary Material
Acknowledgments
We thank Drs. Mark Shlomchik and Adriano Flora for critically reading the manuscript and providing helpful suggestions, Dr. Yuan Zhuang for the gift of Abelson murine leukemia virus. This work is supported in part by grant AI 063348 (NIH) (to B.S.), an America Heart Association grant 0765060Y (AHA) (to V.F.). A.S.L. is a recipient of Brown-Cox Fellowship from Yale University and a Leukemia & Lymphoma Society fellow. D.G.S. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contributions: A.S.L. designed and performed experiments, analyzed data and wrote the paper, D.L and V.F. performed experiments, A.D.L., W.C.S., and D.G.S. provided key experimental reagents, D.G.S. assisted in experiment design, B.S. designed experiments, analyzed data and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Amin RH, Schlissel MS. Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat Immunol. 2008;9:613–622. doi: 10.1038/ni.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondi RM. Phosphoinositide-dependent protein kinase 1, a sensor of protein conformation. Trends Biochem Sci. 2004;29:136–142. doi: 10.1016/j.tibs.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH, DePinho RA, Rickert RC. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol. 2008;9:1388–1398. doi: 10.1038/ni.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo A, Fernandez-Hernando C, Cirino G, Sessa WC. Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc Natl Acad Sci U S A. 2009;106:14552–14557. doi: 10.1073/pnas.0904073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue AC, Fruman DA. PI3K signaling controls cell fate at many points in B lymphocyte development and activation. Semin Cell Dev Biol. 2004;15:183–197. doi: 10.1016/j.semcdb.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. Embo J. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geier JK, Schlissel MS. Pre-BCR signals and the control of Ig gene rearrangements. Semin Immunol. 2006;18:31–39. doi: 10.1016/j.smim.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. mTOR signaling to translation. Curr Top Microbiol Immunol. 2004;279:169–197. doi: 10.1007/978-3-642-18930-2_11. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- Harris TE, Lawrence JC., Jr TOR signaling. Sci STKE. 2003;15:1–17. doi: 10.1126/stke.2122003re15. [DOI] [PubMed] [Google Scholar]
- Herzog S, Hug E, Meixlsperger S, Paik JH, DePinho RA, Reth M, Jumaa H. SLP-65 regulates immunoglobulin light chain gene recombination through the PI(3)K-PKB-Foxo pathway. Nat Immunol. 2008;9:623–631. doi: 10.1038/ni.1616. [DOI] [PubMed] [Google Scholar]
- Herzog S, Reth M, Jumaa H. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat Rev Immunol. 2009;9:195–205. doi: 10.1038/nri2491. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, Hedrick SM. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–184. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu TM, Schatz DG. rag-1 and rag-2 are components of a high-molecular-weight complex, and association of rag-2 with this complex is rag-1 dependent. Mol Cell Biol. 1995;15:5657–5670. doi: 10.1128/mcb.15.10.5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Dordai DI, Lee J, Desiderio S. A conserved degradation signal regulates RAG-2 accumulation during cell division and links V(D)J recombination to the cell cycle. Immunity. 1996;5:575–589. doi: 10.1016/s1074-7613(00)80272-1. [DOI] [PubMed] [Google Scholar]
- Liang HE, Hsu LY, Cado D, Cowell LG, Kelsoe G, Schlissel MS. The “dispensable” portion of RAG2 is necessary for efficient V-to-DJ rearrangement during B and T cell development. Immunity. 2002;17:639–651. doi: 10.1016/s1074-7613(02)00448-x. [DOI] [PubMed] [Google Scholar]
- Llorian M, Stamataki Z, Hill S, Turner M, Martensson IL. The PI3K p110delta is required for down-regulation of RAG expression in immature B cells. J Immunol. 2007;178:1981–1985. doi: 10.4049/jimmunol.178.4.1981. [DOI] [PubMed] [Google Scholar]
- Martensson IL, Keenan RA, Licence S. The pre-B-cell receptor. Curr Opin Immunol. 2007;19:137–142. doi: 10.1016/j.coi.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Melamed D, Kench JA, Grabstein K, Rolink A, Nemazee D. A functional B cell receptor transgene allows efficient IL-7-independent maturation of B cell precursors. J Immunol. 1997;159:1233–1239. [PubMed] [Google Scholar]
- Muljo SA, Schlissel MS. A small molecule Abl kinase inhibitor induces differentiation of Abelson virus-transformed pre-B cell lines. Nature Immunology. 2003;4:31–37. doi: 10.1038/ni870. [DOI] [PubMed] [Google Scholar]
- Peterson RT, Schreiber SL. Kinase phosphorylation: Keeping it all in the family. Curr Biol. 1999;9:R521–524. doi: 10.1016/s0960-9822(99)80326-1. [DOI] [PubMed] [Google Scholar]
- Plas DR, Thompson CB. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem. 2003;278:12361–12366. doi: 10.1074/jbc.M213069200. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Marignani PA, Woodgett JR. Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol Cell Biol. 2002;22:6247–6260. doi: 10.1128/MCB.22.17.6247-6260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlissel MS. Regulating antigen-receptor gene assembly. Nat Rev Immunol. 2003;3:890–899. doi: 10.1038/nri1225. [DOI] [PubMed] [Google Scholar]
- Spicuglia S, Franchini DM, Ferrier P. Regulation of V(D)J recombination. Curr Opin Immunol. 2006;18:158–163. doi: 10.1016/j.coi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Terauchi Y, Fujiwara M, Aizawa S, Yazaki Y, Kadowaki T, Koyasu S. Xid-like immunodeficiency in mice with disruption of the p85alpha subunit of phosphoinositide 3-kinase. Science. 1999;283:390–392. doi: 10.1126/science.283.5400.390. [DOI] [PubMed] [Google Scholar]
- Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135:1130–1142. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkoczy L, Duong B, Skog P, Ait-Azzouzene D, Puri K, Vela JL, Nemazee D. Basal B cell receptor-directed phosphatidylinositol 3-kinase signaling turns off RAGs and promotes B cell-positive selection. J Immunol. 2007;178:6332–6341. doi: 10.4049/jimmunol.178.10.6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira P, Cumano A. Differentiation of B lymphocytes from hematopoietic stem cells. Methods in Molecular Biology. 2004;271:67–76. doi: 10.1385/1-59259-796-3:067. [DOI] [PubMed] [Google Scholar]
- Wang JH, Alt FW, Gostissa M, Datta A, Murphy M, Alimzhanov MB, Coakley KM, Rajewsky K, Manis JP, Yan CT. Oncogenic transformation in the absence of Xrcc4 targets peripheral B cells that have undergone editing and switching. J Exp Med. 2008;205:3079–3090. doi: 10.1084/jem.20082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Gostissa M, Yan CT, Goff P, Hickernell T, Hansen E, Difilippantonio S, Wesemann DR, Zarrin AA, Rajewsky K, et al. Mechanisms promoting translocations in editing and switching peripheral B cells. Nature. 2009;460:231–236. doi: 10.1038/nature08159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR. Recent advances in the protein kinase B signaling pathway. Current Opinion in Cell Biology. 2005;17:150. doi: 10.1016/j.ceb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR Signaling in Growth and Metabolism. Cell. 2006;124:471. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z, Sarbassov DD, Samudio IJ, Yee KWL, Munsell MF, Ellen Jackson C, Giles FJ, Sabatini DM, Andreeff M, Konopleva M. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109:3509–3512. doi: 10.1182/blood-2006-06-030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.