

Abstract
Psoriasis is a common incurable inflammatory skin disease affecting 2–3% of the European population. Psoriatic skin contains large numbers of immune cells which produce many cytokines, chemokines and inflammatory molecules. The epidermis divides much faster than normal and has a defective outer layer or barrier which under normal circumstances protects from infection and dehydration. Psoriatic skin is characterized by a distinct set of inflammation and epidermal proliferation and differentiation markers, and it has not been clear if the genetic basis of psoriasis is due to defects of the immune system or the skin. One genetic determinant lies within the major histocompatibility complex class 1 region. Genome-wide association studies have revealed genetic susceptibility factors that play a role in the formation of immune cells found in psoriasis lesions. Others affect epidermal proliferation and the formation of the skin’s barrier. Hence, genetic components of both the immune system and the epidermis predispose to disease.
Psoriasis: a common inflammatory skin disease with a genetic component
Psoriasis affects 2–3% of the European population, is rarer in individuals of Asian descent (0.1% or less), and exceedingly rare in Africa [1]. Initial outbreaks typically affect individuals in their twenties, but can occur at any age [2]. Psoriasis patients have a natural history of outbreaks (flares) followed by temporary remissions. Environmental factors can trigger or exacerbate flares. These factors include HIV infection [3], use of drugs such as lithium, beta-blockers, or anti-malarials, and the withdrawal of corticosteroids [4]. As many as 10–30% of psoriasis patients develop an inflammatory arthritis termed psoriatic arthritis which is progressive and leads to destruction of the joints if it is not treated aggressively [5]. Although psoriasis often appears sporadically, with patients having no family history of disease, it is a complex disease which includes a familial component. Having a family member with the disease will increase ones risk of being affected. Siblings of an individual with psoriasis are at a 4–6 fold increased risk of developing psoriasis compared to the general population [1].
Psoriasis and psoriatic arthritis are serious, poorly understood, diseases. There are no cures and they require sophisticated medical care and treatments. Moreover, having psoriasis increases the risk of heart disease and stroke [6, 7]. Recent developments in genetic analysis have been instrumental in highlighting important biological pathways in disease susceptibility. In the case of psoriasis genetic studies such as these are providing a far better understanding of the fundamental biological pathways leading to disease. It is expected that this in turn will lead to the development of better and specific forms of treatment. Here we review recent genetic findings concerning risk factors for psoriasis susceptibility, how they relate to the altered biology of the diseased skin and the promise of additional findings from future genetic studies.
Histopathology and molecular biology of psoriasis
There are several different forms of psoriasis (Figure 1). The most common form of psoriasis termed plaque psoriasis or psoriasis vulgaris (PV), accounts for approximately 90% of all psoriasis cases [8]. Individuals with PV also will develop lesions in response to physical trauma (termed the Koebner response [9]). This is obtained by repeatedly applying and removing a piece of tape from the individual’s skin, a procedure referred to as tape stripping, which effectively disrupts the skin barrier and leads to inflammation. Less common forms of psoriasis include seborrheic psoriasis, nail psoriasis, inverse psoriasis, guttate psoriasis and pustular psoriasis.
Figure 1.
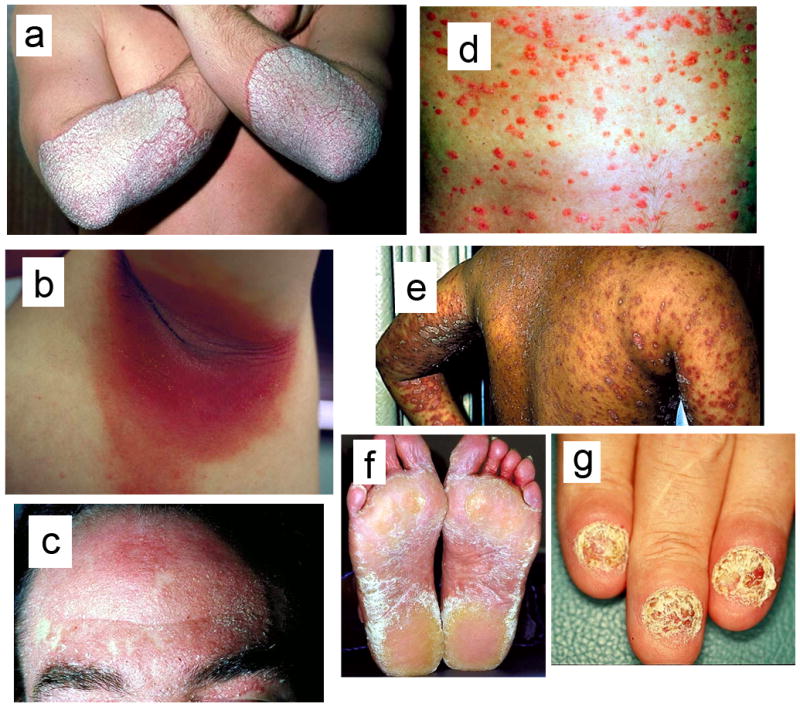
Different forms of psoriasis. (a) Plaque psoriasis is characterized by patches of inflamed skin with silvery scales. These lesions mostly target the elbows, knees and trunk. (b) Flexural (inverse) psoriasis is characterized by painful smooth, red and inflamed lesions in natural skin folds such as the inside of elbows and knees. (c) Seborrheic psoriasis targets areas such as the scalp or eyebrows with that have an oily appearance. (d) Guttate psoriasis lesions are punctate inflamed dots appearing in children or adolescents following infection with Streptococci bacteria. Guttate psoriasis in contrast to other forms is not chronic and typically resolves on its own. (e) Pustular psoriasis is more common in adults than in children. The lesions are numerous skin eruptions that are filled with sterile pus. (f) Non-pustular palmar-plantar psoriasis. (g) Nail psoriasis can lead to pitting and clouding of the nails, sometimes with total loss of the nail bed.
Psoriatic skin is characterized by inflammation, hyperproliferation, abnormal differentiation and changes in the transcriptome [10, 11]. Psoriasis was initially described as a “Th1” disease due to proinflammatory cytokines found in the skin that are produced by T helper type 1 (Th1) cells such as interleukin-1 (IL1), [JS1] tumor necrosis factor alpha (TNFA) and gamma interferon (INFG). However cytokines such as IL-17, IL-20 and IL-22, that are produced by Th17 cells have been found in psoriatic lesions as well [12]. The cytokine mixture produced by the T helper cells in psoriasis act on dermal and epidermal cells, altering the gene expression and maturation of keratinocytes and other cells. Changes in psoriasis are distinct from those in atopic dermatitis (AD) which has a Th2 cytokine signature [13–15]. The number of regulatory T cells is reduced in individuals with psoriasis as well [16] which prevents the exaggerated immune response found in psoriatic skin from being easily modulated.
Keratinocytes of psoriatic skin reach the surface of the skin from the basal layer in as few as 6–8 days compared to approximately 40 days in normal skin [17]. In psoriasis as well as upon injury (tape stripping), sunburn, irritation, infection and disease (such as AD), keratins 6, 16 and 17 are induced [18] (Figure 2) which is similar to that seen in a wound-healing response [19]. The hyper-proliferative basal layer forms finger like projections known as papillae into the dermis. Overall, the epidermis also demonstrates hyperkeratosis and parakeratosis, and blood vessels in the dermis of a psoriatic lesion are increased in number, lumen diameter and tortuosity.
Figure 2.

Diagram of normal and psoriatic involved epidermis found in lesions (not to scale). Normal epidermis contains approximately 10 cell layers made up of the basal layer, spinous layer, granular layer, and the cornified layer or stratum corneum (SC). The SC is constantly being sloughed off and replenished via proliferation in the basal layer. Keratin proteins including Keratins 5/14, 1/10 and 6/16/17 are generated in large amounts and represent the bulk of the keratinocyte weight [86]. S100 proteins such as S100A7 and S100A11 are present in the basal and spinous layers in normal epidermis. They appear in the nucleus and cytoplasm in basal cells but are associated with the plasma membrane in spinous cells in normal and psoriatic tissue[87]. S100A7 is also termed psoriasin and has antimicrobial functions [88]. Terminal differentiation proteins are made in the granular layer where nuclei break down, keratins condense, and cornified envelope (CE) proteins are cross-linked by transglutaminase. Mature keratinocytes in the cornified layer are known as corneocytes, lack nuclei and are flattened and condensed. The space between cells is filled neutral lipids that have been secreted from lamellar granules (LG) into the intercellular spaces of the upper granular layer to form intracellular lipid lamellae. The resultant organization of lipid and corneocytes protects the organism from infection and dehydration and has been compared to bricks and mortar [89]. Defensins are stored in lamellar granules and extruded with lipids into the intracellular space of normal skin [90]. At the same time desmosomes are transformed into corneodesmosomes by insertion of the protein corneodesmosin into the adhesive portion of these structures.
In psoriasis lesions the granular layer is often absent, and corneocytes retain their nuclei (parakeratosis). The SC is thicker and disorganized. Components of the CE are also prematurely synthesized in the spinous layer. In psoriasis lesions neutral lipids are not secreted in a normal fashion into the extracellular space. This leads to a defective water/vapor barrier and the shedding of strateum corneum fragments in large sheets called scales or flakes in psoriasis plaques. In psoriasis antimicrobial peptides such as S100A7 and beta-defensin are highly upregulated. S100A7 is Connexin 26 (Cx26) is a gap junction protein that is also highly upregulated in psoriasis [48]. Transgenic over-expression of Cx26 in mouse epidermis keeps wounded epidermis in a hyper-proliferative state and blocks the transition to remodeling [81]. Its upregulation also leads to infiltration of immune cells.
In psoriatic skin some genes that are normally expressed only in the basal layer, such as integrins, are expressed into the thickened spinous layer [20]. Granular layer products, such as psoriasis associated fatty acid binding protein (FABP5), filaggrin (FLG), corneodesmosin (CDSN), and proteins involved in the formation of the cornified envelope (CE) such as epidermal transglutaminase (TGM3), involucrin (IVL), and loricrin (LOR), can also be prematurely expressed in the spinous layer [21, 22]. The granular layer is sometimes reduced in psoriatic skin [23]. If the granular layer is completely absent, the overlying stratum corneum (SC) is often parakeratotic. Keratinocytes also can retain their nuclei in the stratum corneum, whereas in normal skin the nucleus is not normally present. There are other changes that reflect the altered maturation of keratinocytes of psoriasis lesions that are also shown in Figure 2.
Keratinocytes in psoriasis lesions produce an array of proteins that attract leukocytes. These include the S100 proteins (A7, A8, A9, A12) that are encoded by EDC genes, beta-defensins, ICAM-1, CD40, IL-8 and IP-10 and HLA-DR [24]. Some of the proteins upregulated in psoriatic keratinocytes such as S100A8 and S100A9 are also synthesized by neutrophils or myeloid dendritic cells that are recruited into psoriatic epidermis. In this way, once inflammation is initiated by keratinocyte products, leukocyte recruitment or inflammation is amplified [24]. Keratinocytes also produce endothelial cell mitogens such as VEGF and PDGF, leading to angiogenesis [24].
Pre-GWAS psoriasis genetics
Because of the inflammatory nature of psoriatic skin, it was hypothesized that its immune cells were reacting to an as-yet unidentified antigen. Hence, the earliest genetic studies on psoriasis were case/control studies performed with classical MHC alleles. This revealed association with HLA class 1 alleles with the strongest association being with the HLA-C allele, Cw6 [25]. In these early studies, which were performed in Northern European populations, the frequency of HLA-Cw6 was ~46% in cases with psoriasis vulgaris and 7.4% in controls. The frequency of HLA-Cw6 was even higher in patients with guttate psoriasis (~73%). However, harboring HLA-Cw6 was not sufficient to develop disease, and the penetrance of this allele was estimated to be only 10% [26]. Later studies examined the association with polymorphisms in specific candidate genes that were thought to play a role in psoriasis pathogenesis. This was followed with family based approaches such as genetic linkage analyses. Approximately 10 genome-wide linkage scans, primarily with polymorphic microsatellites, have been conducted in psoriasis. This has lead to the identification of over 20 possible linked regions [27]. One of these linked regions (PSORS2) has been identified several times and segregates as an autosomal dominant mapping to chromosome 17q25 in psoriasis families from the U.S. and Taiwan [28, 29]. Further analysis of a second psoriasis locus (PSORS5 on chromosome 3q21) lead to the identification of a gene coding for a member of the solute carrier family 12 proteins (SLC12A8) [30]. However, its role in psoriasis susceptibility is still not clear. Many linkage analysis regions did not achieve genome-wide significance for linkage and could not be replicated. Moreover, many candidate gene associations have not been replicated either. With the advent of genome-wide association studies, however, several psoriasis risk factors that are common alleles in the general population have been identified. Findings that have been confirmed have provided important insights into the genetics of psoriasis and are discussed below.
Psoriasis genetic associations
Inflammatory genes
Several large genome-wide association studies for psoriasis in both the European and Asian populations have been performed to date [31–35] replicating association with several risk genes (Table 1). Interestingly the most highly significant associations in both populations are with SNPs from the MHC class I region that contains the human leukocyte antigens (HLA) HLAA, HLAB and HLAC. The psoriasis associated SNPs are physically close to the gene encoding HLAC [33–35] and the associated SNP alleles are highly correlated with the MHC HLA-Cw*-0602 allele which corresponds to the HLA-Cw6 serological protein. Hence, GWAS findings are consistent with the very early psoriasis associations performed with classical MHC alleles.
Table 1.
Validated psoriasis associated genes
| Gene | Functional category | Studies |
|---|---|---|
| HLAC | Inflammatory | [33];[34];[35] |
| IL12B | Inflammatory | [34, 39];[35] |
| IL23A | Inflammatory | [34] |
| IL23R | Inflammatory | [34] |
| IL2/Il21 | Inflammatory | [33] |
| TNFAIP3 | Inflammatory | [34] |
| TNIP1 | Inflammatory | [34] |
| SLC12A8 | Epidermal | [85] |
| ZNF313 | Inflammatory | [42] |
| HBD | Epidermal/Antimicrobial | [43] |
| LCE | Epidermal/Cornified Envelope | [35] |
There is some evidence that HLA-Cw*-0602 is the actual risk variant (Box 1) and not just linked to the risk variant [36]. Two additional independent MHC loci also confer risk of psoriasis in both European and Chinese populations [37]. One is within chromosome 6 open reading frame 10 (c6orf10), and the second locus is 30 kb centromeric of human leukocyte antigen B (HLAB) and 16 kb telomeric of MHC class I polypeptide-related sequence A (MICA). In the case of HLAB, higher risk of developing psoriasis is conferred by the haplotype of HLA-B*57, whereas the HLA-B*40 haplotype confers protection. The gene c6orf10 does not have a well-documented function, so its role in psoriasis pathogenesis is unclear. It is known that expression of this gene in keratinocytes is induced by exposure to TNF-α.
Box 1. Inflammatory reactions in psoriatic skin.
Early studies on T cells in the skin revealed the presence of oligoclonal T-cells which could indicate the presence of a common antigen as a major target of the lesional psoriatic immune response [83]. HLA-Cw*-0602 protein product binds peptide motifs that are shared between the M proteins of Streptococci and the keratins K16 and K17 in the skin. This provides a mechanism for guttate psoriasis where Streptococcal infection leads to T cell activation. Within antigen presenting cells (APCs) variants in both HLAB and HLAC can present antigens and specific alleles could process different peptide signatures more effectively, providing a stronger signal for the activation of T helper cells. APCs in the skin such as resident dendritic cells (DCs) could also present keratin fragments similar to M protein, activating cytotoxic T cells in the skin in the absence of true infection [84].
Psoriasis is also associated with two independent variants within and upstream of interleukin 12B (IL12B) and with the interleukin 23 receptor (IL23R) in European and Chinese populations [31, 34, 38]. The association with IL12B is of interest, because it replicates a much earlier candidate gene association with psoriasis seen in the Japanese population [39]. Psoriasis is also associated with interleukin 23, alpha subunit p19 (IL23A) in European populations [34]. IL12B and IL23A heterodimerize to form IL23. This binds the IL-23 receptor (a heterodimer of IL23R and IL12RB1) on naïve CD4+ T cells to induce the development of Th17 cells [40]. These cells play an important role in driving disease in psoriatic skin. Figure 3 compares the major immune cells in normal versus psoriatic skin and how the genetic risk factors contribute to their formation and act on keratinocytes to trigger inflammation or proliferation.
Figure 3.

Comparison of immune cells in normal versus psoriatic skin illustrating how genetic risk factors could trigger and perpetuate epidermal inflammation and proliferation. (a) In normal skin there are a number of resident cells of the immune system. These include specialized dendritic cells (DCs) called Langerhan’s cells, as well as skin homing T cells and neutrophils. (b) In psoriasis lesions DCs and keratinocytes can act as antigen presenting cells (APCs) to lymphocytes via their major histocompatibility complex (MHC) proteins (see Box 1). Such an initiating trigger could lead to enhanced production of IL12 and IL23 by DCs in genetically susceptible individuals. In psoriasis lesions, a subset of DCs express high levels of tumour necrosis factor (TNF) and the enzyme inducible nitric oxide synthase (iNOS). These are termed TIP-DCs (TNF and iNOS-producing DCs). It is thought that these DCs produce the cytokines IL-23 and IL-20, which have the potential to activate T cells and keratinocytes, respectively [24]. IL23 triggers differentiation of Th17 cells following binding to the IL23R on naïve T cells. Production of Th17 cells would be enhanced by the presence of the IL23R genetic risk factor. TNF, gamma interferon, Th1 and Th17 cytokines induce keratinocytes in psoriasis lesions to synthesize numerous proteins that can attract the array of leukocytes found in lesional epidermis. This includes beta-defensins, and the S100A7 protein also known as psoriasin. Neutrophils accumulate in small aggregates in the cornified epidermis in the form of Munro abscesses. IL-22 cytokines act on keratinocytes to increase proliferation [46].
The nuclear factor kappa B (NFκB) pathway plays a central role in numerous cellular processes in psoriasis, including the stress response and keratinocyte proliferation and differentiation [41]. Proteins modulating this pathway such as TNF-α-induced protein 3 (TNFAIP3) and TNFAIP3-interacting protein 1 (TNIP1) [34] are also encoded by genes associated with psoriasis and are likely to enhance NFκB activation. Variants of zinc finger protein 313 (ZNF313) are associated with psoriasis as well [42]. ZNF313 is a paralogue of TRAC1, an ubiquitin ligase regulating T-cell activation, and it is thought that ZNF313 has a similar role.
A β-defensin cluster on human chromosome 8p21 has highly variable copy number (2–12 copies) and a recent analysis of this copy number variation has revealed significant association between increased β-defensin copy number and psoriasis susceptibility [43]. This is significant given that defensins, along with other innate immunity genes are secreted by keratinocytes of psoriatic skin as a result of the cytokine environment. This is enhanced by NFκB signaling and genetic risk factors such as TNFAIP3 and TNIP1 might affect the levels of β-defensin in the skin. β-defensins are expressed at a remarkably high level in psoriatic skin and attract many different immune cell types such as T cells, dendritic cells (DCs) and neutrophils [44–46]. They also trigger keratinocyte proliferation and stimulate cytokine secretion by keratinocytes, thereby playing an important role in perpetuating the psoriatic response.
Barrier development genes
There is now evidence that compromised skin barrier function also plays a role in psoriasis susceptibility. The epidermal differentiation complex (EDC) lies on human chromosome 1q21, spans 2 Mb and encodes at least 45 genes that play a role in the generation or maintenance of the epidermis [47]. Many genes of the EDC are upregulated in psoriatic lesions suggesting underlying alterations in co-ordinate regulation of genes of this complex [48]. Genes within the EDC include those encoding the CE precursors (loricrin, LOR; involucrin, IVL; the small proline rich proteins, SPRRs; and late cornified envelope proteins, LCEs); those encoding granular layer keratin bundling proteins (filaggrin, FLG; trichohyalin, TCHH; filaggrin 2, FLG2; hornerin, HRNR; and cornulin; CRNN); and those encoding proteins involved in signaling and cell cycle progression such as S100A7 and other S100 calcium binding proteins (S100s) [49]). The LCE genes are arrayed into an approximately 340 kb cluster of paralogous family members within the EDC. Within this region are three major LCE families of genes; LCE1, LCE2 and LCE3. Under normal circumstances, LCE1 and LCE2 family members are expressed in the skin, and are incorporated into the CE very late in its development [50]. However, in psoriasis, and following tape stripping, LCE3 family members are induced [51]. Moreover, a deletion polymorphism of ~30 kb that removes LCE3B and LCE3C is associated with psoriasis in both Europeans and in Chinese [35, 51]. It has been suggested that the absence of intact LCE3C and LCE3B genes could lead to an inappropriate repair response following barrier disruption, which is insufficiently backed up by other LCE genes [51]. However, the LCE3B/3C deletion also removes an enhancer important for keratinocyte differentiation, and loss of this regulatory element could also be responsible for the psoriasis association [52]. Although altered epithelial differentiation and barrier formation are hallmarks of both psoriasis and AD, there is no evidence however that this deletion is associated with AD. Furthermore, mutations in FLG, lying near the centromeric end of the EDC have been linked to AD [53], but FLG is not associated with psoriasis [54] providing further evidence that these diseases have different barrier defects [55].
Psoriasis-like mouse models with barrier dysfunction
Genetic perturbations within mouse skin frequently result in a psoriasis-like phenotype that includes inflammation and hyperproliferation and are consistent with the observation that keratinocyte hyperplasia, vascular hyperplasia and cell-mediated immunity in the skin are interrelated. There are many such models (reviewed elsewhere [56]) and some of these are discussed here.
Transgenic mice in which growth factors selective for keratinocytes (amphiregulin, Areg; [57]) or vascular endothelial cells (vascular endothelial growth factor, Vegf; [58]) or leukocytes (Il23; [59]) are overexpressed in the skin (via keratin promoters) leads to keratinocyte hyperplasia, increased vascularity, and skin infiltration by mononuclear leukocytes, including T cells which is similar to changes seen in psoriasis. Similar skin responses have been produced by altering transcription factors: Overexpression of Stat3 (signal transducer and activator of transcription 3) in epidermal keratinocytes [60]; knock-out of Irf2 (interferon-regulatory factor 2) a transcription factor that serves as a negative regulator of interferon signaling [61]; epidermal specific knock-out of Gata3 [62], a transcription factor that binds GATA DNA sequences; knock-out of Klf4 (Kruppel-like factor 4; [63]); and epidermal specific deletion of Ap1 (activator-protein 1; [64]) all result in similar epidermal phenotypes. The Vegf and Stat3 transgenic models develop lesions at skin wounds, as is seen in human psoriasis. Directly perturbing components of the epidermal matrix, such as the transgenic overexpression of Itgb1 (integrin beta-1), also produces thickened skin, inflammatory infiltrates and increased vascularity [65].
In each of these transgenic models, dysregulated immunity or an immune-stimulated pathway leads to background alterations in the epithelium and blood vessels that are similar to changes in psoriasis vulgaris. T cells can directly trigger reactive skin changes, as in mice transplanted with CD45RBhiCD4+ T cells [66] or in rats bearing a human HLA-B27 transgene [67]. However, keratinocyte hyperplasia does not always result in vascular involvement and immune activation, as is the case in transgenic IL-20 (Il20; [68]) and transgenic Kgf (keratinocyte growth factor) mice [69]. Interestingly, the transplantation of non-involved skin from human psoriasis patients onto AGR129 mice eventually leads to plaque formation [70] providing further evidence for the importance for the gamma interferon pathway in psoriasis [71].
A mouse strain with a targeted epidermal deletion of Cdsn has also been created which leads to detachment of the upper layers of the skin (the stratum granulosum (SG) and between the SC and the SG). When grafted onto immunodeficient mice, Cdsn-deficient skin undergoes rapid hair loss together with epidermal abnormalities resembling psoriasis [72]. This is significant because it indicates that disruption of the skin barrier will trigger keratinocyte hyper-proliferation.
In summary, even though the cutaneous alterations in these models do not exactly reproduce the human disease the insight they provide is invaluable to dissecting the molecular mechanisms of disease. They highlight the interplay of the skin, the vasculature, and the immune system.
Psoriasis risk factors shared in other autoimmune or inflammatory diseases
Overlap between some psoriasis loci and those identified in other autoimmune or inflammatory diseases has been reported (Table 2). The same variant of IL23R is associated with Crohn’s disease, psoriatic arthritis, and ankylosing spondylitis [33, 73, 74]. This is consistent with the role of IL23R in Th17 cell activation, and the fact that these cells have a pathogenic role in several other inflammatory diseases including Crohn’s disease and multiple sclerosis [40]. A haplotype harboring IL2 and IL21 is associated with many autoimmune diseases (Table 2). It is also a risk factor for psoriatic arthritis and potentially psoriasis [33]. However, the subtle differences in frequencies of risk alleles in cases versus controls, and the change in allele frequencies in a north to south latitude means that this association is not always easy to detect [33].
Table 2.
Genes in risk loci shared by autoimmune or inflammatory diseases
| Gene | PsA | CD | UC | Asthma | MS | AD | RA | SLE | AS | T1D | CeD | T2D |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IL12B | X | X | X | X | X | |||||||
| 5q31 | X | X | X | X | ||||||||
| TNFAIP3 | X | X | X | X | ||||||||
| IL23R | X | X | X | |||||||||
| IL2/IL21 | X | X | X | X | X | X | X | X | ||||
| COG6 | X | |||||||||||
| PTPN22 | X | X | X | |||||||||
| ADAM33 | X | |||||||||||
| CDKAL1 | X | X |
PsA: Psoriatic arthritis; CD: Crohn’s disease, UC: Ulcerative Colitis; MS: multiple sclerosis; AD: Atopic dermatitis; RA: Rheumatoid arthritis; SLE: Systemic lupus erythematosus; AS: Ankylosing spondylitis; T1D: Type 1 diabetes; CeD: Coeliac disease; T2D: Type 2 diabetes
The NFκB-pathway-related TNFAIP3 association is linked to risk for rheumatoid arthritis, systemic lupus erythematosus and type-1 diabetes although the psoriasis association is distinct from that for rheumatoid arthritis and system lupus erythematosus [34, 75–78]. These and other shared regions of association (Table 2) might reflect general upregulation of immune system pathways, leaving open the question of what alleles specify the organ targeted by the immune system.
Concluding remarks and future perspectives
Identifying the genetic basis of a complex disease like psoriasis is challenging. The task is further complicated by the fact that psoriasis clearly involves an interaction between the immune system and the skin, leading early on to questions about where the primary defect resides. Although many early genetic studies were inconclusive, recent genome-wide association studies have started to identify specific genetic components of both the immune system and the epidermis that affect disease risk. Genetic risk factors such as IL12B and IL23R affect pathways mediated by IL12 and IL-23 which are crucial for the development of the particular immune cell subsets that drive the epidermal component of this skin disease. Other factors such as TNFAIP3 and TNIP1 affect pathways mediated by NFκB such as immunity and inflammation, cell proliferation and apoptosis. These could affect both immune cells and keratinocytes, in part leading to the rate of epidermal proliferation. These risk factors and pathways are distinct from those found in AD where different inflammatory cells infiltrate the skin. By contrast, separate psoriasis genetic risk factors such as deletions of LCE3 genes of the EDC are likely to have a direct effect on skin barrier formation and are also distinct from those found in AD. However, it is still unclear how these pathways intersect and contribute to the vicious cycle of inflammation, proliferation and altered differentiation in psoriasis. For example, how do environmental triggers such as tape stripping, infection, or a deletion of components of the CE lead to an increase in epidermal proliferation and inflammation? One clue is that calcium is critical for controlling the balance of proliferation and differentiation in epidermal keratinocytes [79] [80]. In the case of the Cx26 transgenic mouse which has features of psoriasis including an altered barrier, this increase in epidermal proliferation is thought to be due to increased ATP release which activates purinergic receptors and regulates keratinocyte calcium flux [81]. Findings from mouse models also support a key role of innate barrier dysfunction triggering inflammation, but currently these pathways remain an enigma that needs to be resolved.
As with most other complex diseases, a substantial proportion of the genetic contribution to psoriasis has not yet been identified. Even in the largest GWAS of psoriasis performed to date, association signals can only account for a sibling recurrence risk of 1.35 including 1.25 due to HLA [34]. Consequently, much of the overall sibling recurrence risk for psoriasis, which has been estimated at approximately three- to six-fold [26], remains unexplained. Despite this, genetic findings highlight altered biological pathways leading to the development of pathogenic immune cells in the skin. They have provided a rationale for immunosuppression using monoclonal antibodies directed against TNF-α and p40 (encoded by IL12B and a component of both IL12 and IL23) which have proved to be effective psoriasis therapies for many patients [82].
In the near future it is likely that advances in next-generation sequencing technologies will help in the pinpointing of causative variants from genome wide association studies. It is also possible that the “missing” genetic risk factors may be due to rare variants with higher penetrance. Searches for such genetic factors are underway and might require complete genomic DNA sequencing of patients, or at least the single copy DNA. The identification of new psoriasis susceptibility genes may lead to the discovery of new pathways contributing to psoriasis and ultimately the development of new forms of treatment. However, if these postulated rare variants, that have eluded us so far, occur in intergenic regions of the genome (which have not been comprehensively surveyed) then dissecting their functional consequences will present a whole new series of challenges. Presumably, many of these non-genic sequences will have regulatory functions in either immune or epidermal differentiation providing novel regulatory mechanisms affecting the interplay between the two systems.
Acknowledgments
Dr. Alan Menter kindly provided psoriasis images. We thank Dr. Michael Lovett, Cailin Joyce and Catherine Jordan for critical comments on the manuscript. The studies described here were supported in part by NIH grant R01 AR050266 (to AMB). ER is supported by NIH training grant T32AR007279.
Glossary
- AGR129 mice
A mouse strain engineered to have a deficiency in type I and type II interferon receptors and the recombination activating gene 2. They therefore lack functional B-cells, T-cells and have impaired innate immunity related to interferons
- Cornified envelope
A structure formed in the outermost layers of stratified squamous epithelia that provide a physical barrier against environmental insults. It is composed of several structural proteins, which are irreversibly crosslinked by calcium-activated transglutaminases. Small proline rich proteins (SPRRs) and late cornified envelope proteins (LCEs) are CE precursors
- Corneodesmosomes
Specialized desmosomes that hold the corneocytes together. Corneodesmosomes are the major structure that must be degraded for the skin to undergo desquamation (see Glossary)
- Desmosomes
Adhesion molecules that join adjacent cells together and that provide anchoring points for intermediate filaments to help build a strong structural framework
- Desquamation
The normal process through which terminally differentiated squamous keratinocytes (in the stratum corneum) are sloughed off into the environment and replaced
- Epidermal differentiation complex
a cluster of genes on human chromosome 1 that are critical to the development, maturation and crosslinking of the epidermis. They become activated in terminal stages of keratinocyte differentiation and include involucrin, loricrin, filaggrin, late cornified envelope genes, and small proline-rich protein genes
- Genome-wide association studies
Genetic studies which test for non-random association between genetic markers representing regions of linkage disequilibrium in the genome and disease status
- Hyperkeratosis
Overgrowth and thickening of the outer layer of the skin
- Keratinocytes
Stratified squamous epithelial cells of the skin. Keratinocytes lying in the stratum germinativum (basal layer) proliferate continuously and differentiate to form the skin barrier or cornified layer/stratum corneum
- Lamellar bodies/granules
Secretory organelles filled with a mixture of lipids and immunologically active molecules
- Linkage disequilibrium
The non-random association of alleles from two independent loci due to their close physical proximity
- Parakeratosis
Retention of the nucleus in keratinocytes of the cornified envelope
- Plaque
A raised, flat topped lesion on the skin that is ≥1 cm in diameter that typifies psoriasis vulgaris. Smaller sized lesions (papules) characterize guttate psoriasis. Pustular or erythrodermatous forms of psoriasis might not show well formed plaques, but instead have pustules or extensive inflammation in the skin
- Psoriatic arthritis
A form of inflammatory arthritis that occurs in patients with psoriasis
- SNP
Single nucleotide polymorphism, where more than one base can exist at a particular site in the genome in a particular population. Generally alleles of any polymorphism are present at a frequency of 1% or greater
- Squamous
A cell morphology where the cells are thin and flattened in appearance that fit together into a continuous layer
- T helper cells
Any one of several T lymphocytes that belong to the CD4+ subset that, when stimulated by a specific antigen, release secreted cytokines that promote the activation and function of B cells and killer T cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bhalerao J, Bowcock A. The genetics of psoriasis: a complex disorder of the skin and immune system. Hum Mol Genet. 1998;7:1537–1545. doi: 10.1093/hmg/7.10.1537. [DOI] [PubMed] [Google Scholar]
- 2.Boyman O, et al. Spontaneous Development of Psoriasis in a New Animal Model Shows an Essential Role for Resident T Cells and Tumor Necrosis Factor-α. The Journal of Experimental Medicine. 2004;199:731–736. doi: 10.1084/jem.20031482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnett FC, et al. Psoriasis and psoriatic arthritis associated with human immunodeficiency virus infection. Rheumatic Diseases Clinics of North America. 1991;17:59–78. [PubMed] [Google Scholar]
- 4.Rongioletti F, et al. Psoriasis Induced or Aggravated by Drugs. The Journal of Rheumatology. 2009;83:59–61. doi: 10.3899/jrheum.090227. [DOI] [PubMed] [Google Scholar]
- 5.Nograles KE, et al. New insights into the pathogenesis and genetics of psoriatic arthritis. Nat Clin Pract Rheum. 2009;5:83–91. doi: 10.1038/ncprheum0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gelfand JM, et al. Risk of myocardial infarction in patients with psoriasis. JAMA. 2006;296:1735–1741. doi: 10.1001/jama.296.14.1735. [DOI] [PubMed] [Google Scholar]
- 7.Gelfand JM, et al. The risk of stroke in patients with psoriasis. J Invest Dermatol. 2009;129:2411–2418. doi: 10.1038/jid.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffiths CEM, Barker JNWN. Pathogenesis and clinical features of psoriasis. The Lancet. 2007;370:263–271. doi: 10.1016/S0140-6736(07)61128-3. [DOI] [PubMed] [Google Scholar]
- 9.Eyre RW, Krueger GG. Response to injury of skin involved and uninvolved with psoriasis, and its relation to disease activity: Koebner and ‘reverse’ Koebner reactions. Br J Dermatol. 1982;106:153–159. doi: 10.1111/j.1365-2133.1982.tb00924.x. [DOI] [PubMed] [Google Scholar]
- 10.Zhou X, et al. Novel mechanisms of T-cell and dendritic cell activation revealed by profiling of psoriasis on the 63,100-element oligonucleotide array. Physiol Genomics. 2003;13:69–78. doi: 10.1152/physiolgenomics.00157.2002. [DOI] [PubMed] [Google Scholar]
- 11.Gudjonsson JE, et al. Assessment of the psoriatic transcriptome in a large sample: additional regulated genes and comparisons with in vitro models. J Invest Dermatol. 130:1829–1840. doi: 10.1038/jid.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mee JB, et al. The psoriatic transcriptome closely resembles that induced by interleukin-1 in cultured keratinocytes: dominance of innate immune responses in psoriasis. Am J Pathol. 2007;171:32–42. doi: 10.2353/ajpath.2007.061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nomura I, et al. Distinct patterns of gene expression in the skin lesions of atopic dermatitis and psoriasis: a gene microarray analysis. J Allergy Clin Immunol. 2003;112:1195–1202. doi: 10.1016/j.jaci.2003.08.049. [DOI] [PubMed] [Google Scholar]
- 14.Guttman-Yassky E, et al. Major differences in inflammatory dendritic cells and their products distinguish atopic dermatitis from psoriasis. J Allergy Clin Immunol. 2007;119:1210–1217. doi: 10.1016/j.jaci.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Kamsteeg M, et al. Increased expression of carbonic anhydrase II (CA II) in lesional skin of atopic dermatitis: regulation by Th2 cytokines. J Invest Dermatol. 2007;127:1786–1789. doi: 10.1038/sj.jid.5700752. [DOI] [PubMed] [Google Scholar]
- 16.Sugiyama H, et al. Dysfunctional Blood and Target Tissue CD4+CD25high Regulatory T Cells in Psoriasis: Mechanism Underlying Unrestrained Pathogenic Effector T Cell Proliferation. J Immunol. 2005;174:164–173. doi: 10.4049/jimmunol.174.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halprin KM. Epidermal “turnover time”--a re-examination. Br J Dermatol. 1972;86:14–19. doi: 10.1111/j.1365-2133.1972.tb01886.x. [DOI] [PubMed] [Google Scholar]
- 18.de Jongh GJ, et al. High expression levels of keratinocyte antimicrobial proteins in psoriasis compared with atopic dermatitis. J Invest Dermatol. 2005;125:1163–1173. doi: 10.1111/j.0022-202X.2005.23935.x. [DOI] [PubMed] [Google Scholar]
- 19.Mansbridge JN, et al. Evidence for an alternative pathway of keratinocyte maturation in psoriasis from an antigen found in psoriatic but not normal epidermis. J Invest Dermatol. 1984;83:296–301. doi: 10.1111/1523-1747.ep12340429. [DOI] [PubMed] [Google Scholar]
- 20.Pellegrini, et al. Expression, topography, and function of integrin receptors are severely altered in keratinocytes from involved and uninvolved psoriatic skin. J Clin Invest. 1992;89:1783–1795. doi: 10.1172/JCI115782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guttman-Yassky E, et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J Allergy Clin Immunol. 2009;124:1235–1244. e1258. doi: 10.1016/j.jaci.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 22.Van Ruissen F, et al. Induction of normal and psoriatic phenotypes in submerged keratinocyte cultures. Journal of Cellular Physiology. 1996;168:442–452. doi: 10.1002/(SICI)1097-4652(199608)168:2<442::AID-JCP23>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 23.Lowes MA, et al. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–873. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- 24.Lowes MA, et al. Current concepts in the immunopathogenesis of psoriasis. Dermatol Clin. 2004;22:349–369. vii. doi: 10.1016/j.det.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 25.Tiilikainen A, et al. Psoriasis and HLA-Cw6. Br J Dermatol. 1980;102:179–184. doi: 10.1111/j.1365-2133.1980.tb05690.x. [DOI] [PubMed] [Google Scholar]
- 26.Elder JT, et al. The genetics of psoriasis. Arch Dermatol. 1994;130:216–224. [PubMed] [Google Scholar]
- 27.Bowcock AM, Cookson WO. The genetics of psoriasis, psoriatic arthritis and atopic dermatitis. Hum Mol Genet. 2004;13(Spec No 1):R43–55. doi: 10.1093/hmg/ddh094. [DOI] [PubMed] [Google Scholar]
- 28.Tomfohrde J, et al. Gene for Familial Psoriasis Susceptibility Mapped to the Distal End of Human Chromosome 17q. Science. 1994;264:1141–1145. doi: 10.1126/science.8178173. [DOI] [PubMed] [Google Scholar]
- 29.Hwu WL, et al. Mapping of psoriasis to 17q terminus. J Med Genet. 2005;42:152–158. doi: 10.1136/jmg.2004.018564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hewett D, et al. Identification of a Psoriasis Susceptibility Candidate Gene by Linkage Disequilibrium Mapping with a Localized Single Nucleotide Polymorphism Map. Genomics. 2002;79:305–314. doi: 10.1006/geno.2002.6720. [DOI] [PubMed] [Google Scholar]
- 31.Cargill M, et al. A Large-Scale Genetic Association Study Confirms IL12B and Leads to the Identification of IL23R as Psoriasis-Risk Genes. The American Journal of Human Genetics. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capon F, et al. Identification of ZNF313/RNF114 as a novel psoriasis susceptibility gene. Hum Mol Genet. 2008;17:1938–1945. doi: 10.1093/hmg/ddn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, et al. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008;4:e1000041. doi: 10.1371/journal.pgen.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nair RP, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang XJ, et al. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat Genet. 2009;41:205–210. doi: 10.1038/ng.310. [DOI] [PubMed] [Google Scholar]
- 36.Nair RP, et al. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet. 2006;78:827–851. doi: 10.1086/503821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng BJ, et al. Multiple Loci within the major histocompatibility complex confer risk of psoriasis. PLoS Genet. 2009;5:e1000606. doi: 10.1371/journal.pgen.1000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang XJ, et al. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat Genet. 2009;41:205–210. doi: 10.1038/ng.310. [DOI] [PubMed] [Google Scholar]
- 39.Tsunemi Y, et al. Interleukin-12 p40 gene (IL12B) 3′-untranslated region polymorphism is associated with susceptibility to atopic dermatitis and psoriasis vulgaris. Journal of Dermatological Science. 2002;30:161–166. doi: 10.1016/s0923-1811(02)00072-5. [DOI] [PubMed] [Google Scholar]
- 40.van Beelen AJ, et al. Stimulation of the Intracellular Bacterial Sensor NOD2 Programs Dendritic Cells to Promote Interleukin-17 Production in Human Memory T Cells. Immunity. 2007;27:660–669. doi: 10.1016/j.immuni.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 41.Lizzul PF, et al. Differential expression of phosphorylated NF-kappaB/RelA in normal and psoriatic epidermis and downregulation of NF-kappaB in response to treatment with etanercept. J Invest Dermatol. 2005;124:1275–1283. doi: 10.1111/j.0022-202X.2005.23735.x. [DOI] [PubMed] [Google Scholar]
- 42.Capon F, et al. Identification of ZNF313/RNF114 as a novel psoriasis susceptibility gene. Hum Mol Genet. 2008;17:1938–1945. doi: 10.1093/hmg/ddn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hollox EJ, et al. Psoriasis is associated with increased β-defensin genomic copy number. Nat Genet. 2008;40:23–25. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kryczek I, et al. Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. J Immunol. 2008;181:4733–4741. doi: 10.4049/jimmunol.181.7.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huh WK, et al. Dynamic alteration of human beta-defensin 2 localization from cytoplasm to intercellular space in psoriatic skin. J Mol Med. 2002;80:678–684. doi: 10.1007/s00109-002-0373-z. [DOI] [PubMed] [Google Scholar]
- 46.Nograles KE, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008;159:1092–1102. doi: 10.1111/j.1365-2133.2008.08769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mischke D, et al. Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex (“epidermal differentiation complex”) on human chromosome 1q21. J Invest Dermatol. 1996;106:989–992. doi: 10.1111/1523-1747.ep12338501. [DOI] [PubMed] [Google Scholar]
- 48.Bowcock AM, et al. Insights into psoriasis and other inflammatory diseases from large-scale gene expression studies. Hum Mol Genet. 2001;10:1793–1805. doi: 10.1093/hmg/10.17.1793. [DOI] [PubMed] [Google Scholar]
- 49.Chen H, et al. Association of skin barrier genes within the PSORS4 locus is enriched in Singaporean Chinese with early-onset psoriasis. J Invest Dermatol. 2009;129:606–614. doi: 10.1038/jid.2008.273. [DOI] [PubMed] [Google Scholar]
- 50.Jackson B, et al. Late cornified envelope family in differentiating epithelia--response to calcium and ultraviolet irradiation. J Invest Dermatol. 2005;124:1062–1070. doi: 10.1111/j.0022-202X.2005.23699.x. [DOI] [PubMed] [Google Scholar]
- 51.de Cid R, et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat Genet. 2009;41:211–215. doi: 10.1038/ng.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Guzman Strong C, et al. A milieu of regulatory elements in the epidermal differentiation complex syntenic block: implications for atopic dermatitis and psoriasis. Hum Mol Genet. 19:1453–1460. doi: 10.1093/hmg/ddq019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palmer CN, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 54.Zhao Y, et al. Filaggrin null alleles are not associated with psoriasis. J Invest Dermatol. 2007;127:1878–1882. doi: 10.1038/sj.jid.5700817. [DOI] [PubMed] [Google Scholar]
- 55.Bergboer JG, et al. Deletion of Late Cornified Envelope 3B and 3C Genes Is Not Associated with Atopic Dermatitis. J Invest Dermatol. doi: 10.1038/jid.2010.88. [DOI] [PubMed] [Google Scholar]
- 56.Gudjonsson JE, et al. Mouse models of psoriasis. J Invest Dermatol. 2007;127:1292–1308. doi: 10.1038/sj.jid.5700807. [DOI] [PubMed] [Google Scholar]
- 57.Cook PW, et al. Transgenic expression of the human amphiregulin gene induces a psoriasis-like phenotype. J Clin Invest. 1997;100:2286–2294. doi: 10.1172/JCI119766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xia YP, et al. Transgenic delivery of VEGF to mouse skin leads to an inflammatory condition resembling human psoriasis. Blood. 2003;102:161–168. doi: 10.1182/blood-2002-12-3793. [DOI] [PubMed] [Google Scholar]
- 59.Kopp T, et al. IL-23 production by cosecretion of endogenous p19 and transgenic p40 in keratin 14/p40 transgenic mice: evidence for enhanced cutaneous immunity. J Immunol. 2003;170:5438–5444. doi: 10.4049/jimmunol.170.11.5438. [DOI] [PubMed] [Google Scholar]
- 60.Sano S, et al. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med. 2005;11:43–49. doi: 10.1038/nm1162. [DOI] [PubMed] [Google Scholar]
- 61.Hida S, et al. CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity. 2000;13:643–655. doi: 10.1016/s1074-7613(00)00064-9. [DOI] [PubMed] [Google Scholar]
- 62.de Guzman Strong C, et al. Lipid defect underlies selective skin barrier impairment of an epidermal-specific deletion of Gata-3. J Cell Biol. 2006;175:661–670. doi: 10.1083/jcb.200605057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Segre JA, et al. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999;22:356–360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 64.Zenz R, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–375. doi: 10.1038/nature03963. [DOI] [PubMed] [Google Scholar]
- 65.Carroll JM, et al. Suprabasal integrin expression in the epidermis of transgenic mice results in developmental defects and a phenotype resembling psoriasis. Cell. 1995;83:957–968. doi: 10.1016/0092-8674(95)90211-2. [DOI] [PubMed] [Google Scholar]
- 66.Schon MP, et al. Murine psoriasis-like disorder induced by naive CD4+ T cells. Nat Med. 1997;3:183–188. doi: 10.1038/nm0297-183. [DOI] [PubMed] [Google Scholar]
- 67.Taurog JD, et al. Inflammatory disease in HLA-B27 transgenic rats. Immunol Rev. 1999;169:209–223. doi: 10.1111/j.1600-065x.1999.tb01317.x. [DOI] [PubMed] [Google Scholar]
- 68.Blumberg H, et al. Interleukin 20: discovery, receptor identification, and role in epidermal function. Cell. 2001;104:9–19. doi: 10.1016/s0092-8674(01)00187-8. [DOI] [PubMed] [Google Scholar]
- 69.Guo L, et al. Targeting expression of keratinocyte growth factor to keratinocytes elicits striking changes in epithelial differentiation in transgenic mice. EMBO J. 1993;12:973–986. doi: 10.1002/j.1460-2075.1993.tb05738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boyman O, et al. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med. 2004;199:731–736. doi: 10.1084/jem.20031482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lew W, et al. Psoriasis vulgaris: cutaneous lymphoid tissue supports T-cell activation and “Type 1” inflammatory gene expression. Trends Immunol. 2004;25:295–305. doi: 10.1016/j.it.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 72.Leclerc EA, et al. Corneodesmosin gene ablation induces lethal skin-barrier disruption and hair-follicle degeneration related to desmosome dysfunction. J Cell Sci. 2009;122:2699–2709. doi: 10.1242/jcs.050302. [DOI] [PubMed] [Google Scholar]
- 73.Begovich AB, et al. The autoimmune disease-associated IL12B and IL23R polymorphisms in multiple sclerosis. Hum Immunol. 2007;68:934–937. doi: 10.1016/j.humimm.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 74.Duerr RH, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fung EY, et al. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun. 2009;10:188–191. doi: 10.1038/gene.2008.99. [DOI] [PubMed] [Google Scholar]
- 76.Coenen MJ, et al. Common and different genetic background for rheumatoid arthritis and coeliac disease. Hum Mol Genet. 2009;18:4195–4203. doi: 10.1093/hmg/ddp365. [DOI] [PubMed] [Google Scholar]
- 77.Graham RR, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059–1061. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Plenge RM, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tu CL, et al. The role of the calcium sensing receptor in regulating intracellular calcium handling in human epidermal keratinocytes. J Invest Dermatol. 2007;127:1074–1083. doi: 10.1038/sj.jid.5700633. [DOI] [PubMed] [Google Scholar]
- 80.Xie Z, et al. Calcium-induced human keratinocyte differentiation requires src- and fyn-mediated phosphatidylinositol 3-kinase-dependent activation of phospholipase C-gamma1. Mol Biol Cell. 2005;16:3236–3246. doi: 10.1091/mbc.E05-02-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Djalilian AR. Connexin 26 regulates epidermal barrier and wound remodeling and promotes psoriasiform response. The Journal of Clinical Investigation. 2006;116:1243–1253. doi: 10.1172/JCI27186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Elder JT. Genome-wide association scan yields new insights into the immunopathogenesis of psoriasis. Genes Immun. 2009;10:201–209. doi: 10.1038/gene.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Prinz JC, et al. Selection of conserved TCR VDJ rearrangements in chronic psoriatic plaques indicates a common antigen in psoriasis vulgaris. Eur J Immunol. 1999;29:3360–3368. doi: 10.1002/(SICI)1521-4141(199910)29:10<3360::AID-IMMU3360>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 84.Johnston A, et al. Peripheral blood T cell responses to keratin peptides that share sequences with streptococcal M proteins are largely restricted to skin-homing CD8(+) T cells. Clin Exp Immunol. 2004;138:83–93. doi: 10.1111/j.1365-2249.2004.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huffmeier U, et al. Systematic linkage disequilibrium analysis of SLC12A8 at PSORS5 confirms a role in susceptibility to psoriasis vulgaris. J Invest Dermatol. 2005;125:906–912. doi: 10.1111/j.0022-202X.2005.23847.x. [DOI] [PubMed] [Google Scholar]
- 86.Segre J. Complex redundancy to build a simple epidermal permeability barrier. Curr Opin Cell Biol. 2003;15:776–782. doi: 10.1016/j.ceb.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 87.Broome AM, et al. S100 protein subcellular localization during epidermal differentiation and psoriasis. J Histochem Cytochem. 2003;51:675–685. doi: 10.1177/002215540305100513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee KC, Eckert RL. S100A7 (Psoriasin)--mechanism of antibacterial action in wounds. J Invest Dermatol. 2007;127:945–957. doi: 10.1038/sj.jid.5700663. [DOI] [PubMed] [Google Scholar]
- 89.Elias PM. Journal of Investigative Dermatology. Nature Publishing Group; 2005. Stratum Corneum Defensive Functions: An Integrated View; pp. 183–200. [DOI] [PubMed] [Google Scholar]
- 90.Odland GF, Holbrook K. The lamellar granules of epidermis. Curr Probl Dermatol. 1981;9:29–49. doi: 10.1159/000403343. [DOI] [PubMed] [Google Scholar]