

Abstract
The enantioselective intramolecular aminative functionalization of unactivated alkenes and related π-systems is a straight-forward and atom economical strategy for the synthesis of chiral nitrogen heterocycles. These reactions can be categorized as oxidatively neutral, such as alkene hydroamination, or as oxidative reactions, such as alkene difunctionalization, e.g. aminooxygenation and carboamination. This perspective reviews the current work in the field and explores mechanistic trends that are common among the different catalysts and reaction types.
1 Introduction
The asymmetric synthesis of chiral nitrogen heterocycles by aminative functionalization of unactivated π-bonds has been traditionally challenging as the process involves stereocontrolled addition of the amine onto the alkene or equivalent unsaturated bond. This review will discuss recent progress in the use of chiral metal catalysts to enable these cyclization reactions and control the three-dimensional orientation of the C–N bond-forming step. We will focus on the intramolecular addition of amines to unactivated, non-polarized π-systems, which offer different synthetic challenges than polarized π bonds, those conjugated to electron-withdrawing groups, as the latter can undergo intrinsically favorable 1,4-addition processes as well as benefit from Lewis acid activation of the electron withdrawing group (EWG).
We will present a variety of different metal-catalyzed reactions for the cyclization of amines onto unactivated π-systems that none-the-less share similarities in transition state conformational preferences related to the cyclization process. Lewis acidic π-catalysts are necessary to promote the otherwise intrinsically unfavorable nucleophilic addition of amines onto these electron-rich π systems.
2 Oxidatively neutral cyclizations
Oxidatively neutral aminative π-bond functionalizations are reactions where the substrate and product are in the same net oxidation state. These include the hydroamination of alkenes, dienes, allenes and alkynes and the SN2′ type displacement of allylic leaving groups.
2.1 Enantioselective intramolecular hydroamination of alkenes and dienes
The inter- and intramolecular enantioselective catalytic hydroamination of alkenes, dienes, allenes and alkynes has been reviewed previously.1-5 This review focuses only on instructive and recent developments. A number of metals can catalyze the enantioselective hydroamination/cyclization of alkenes. It is interesting to note that for these reactions, all of the proposed enantioinduction models have involved “inner sphere” or syn aminometallation processes (vide infra). Many of these reactions also occur more readily on terminal rather than internal alkenes, indicating that steric hindrance affects the metal–carbon bond-forming process.
2.1.1 Hydroaminations catalyzed by rare earth metals
The pioneering enantioselective intramolecular hydroaminations of alkenes catalyzed by chiral rare-earth metal catalysts has been reviewed previously.1-5 Although early studies were conducted with configurationally unstable cyclopentadienyl-based ligands4, in 2001, Livinghouse and co-workers demonstrated that cyclopentadienyl ligands are not necessary for catalytic hydroamination6,7 and new, more stable chiral catalysts were subsequently developed.
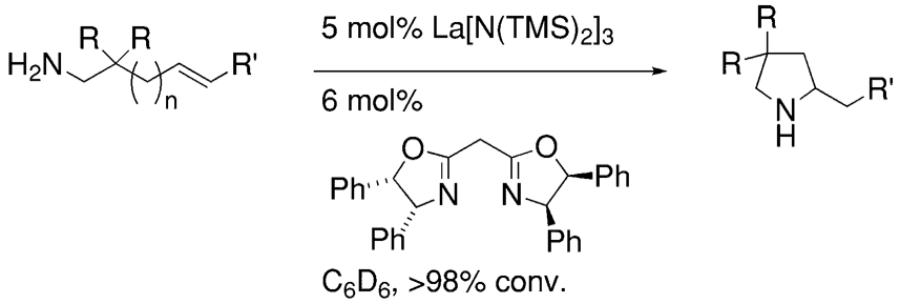
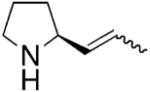
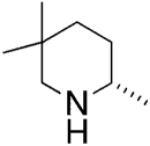
In 2002, Marks and co-workers reported that upon screening a number of chiral bis(oxazoline) ligands and several lanthanide metals, the [(4R,5S)-Ph2Box]La[N(TMS)2]2 catalyst provided efficient and moderately enantioselective (up to 67% ee at 23 °C) hydroamination of terminal alkenes and dienes (Table 1).8
Table 1.
Lanthanide-catalyzed enantioselective hydroamination of alkenes and dienes8
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Nt(h−1) | Temp (°C) | ee (%)a |
| 1 |
|

|
25 | 23 | 67 |
| 2 |
|

|
0.09 | 23 | 40 |
| 3 |
|
|
3.0 | 23 | 17 |
| 4 |
|
|
4.0 | 60 | 56 |
| 5 |
|

|
0.6 | 60 | 54 |
Determined by chiral HPLC analysis.
The reaction mechanism was examined in depth.8 Kinetic studies indicated the hydroamination rate is zero-order in [amine substrate] and first-order in [catalyst]. This is consistent with rate-determining Ln–N alkene insertion. A model that rationalizes the observed pyrrolidine stereochemistry is shown in Scheme 1. In the favored transition state, the metal is trigonal bipyramidyl and the ligand and the approaching olefin adopt equatorial positions while the two amine substituents adopt apical positions. Interestingly, piperidine rings are formed with the opposite enantioinduction (Table 1).
Scheme 1.

Proposed mechanism of Marks’ enantioselective lanthanide catalyzed alkene hydroamination with a C2-symmetric bis(oxazoline) ligand.
A number of other research groups, including those of Scott9, Livinghouse10, Hultzsch11 and Schulz12 have also identified chiral and configurationally stable, non-cyclopentadienyl ligands that coordinate with lanthanides and whose complexes thereof catalyze the enantioselective hydroamination of alkenes. The catalyst structures and the enantioselectivity are given for the quantitative cyclizations of 2,2-dimethyl-4-pentenylamine to 2,4,4-trimethylpyrrolidine (Fig. 1).
Fig. 1.

Other lanthanide–ligand complexes used for asymmetric alkene hydroamination.
2.1.2 Hydroaminations catalyzed by group 4 metals
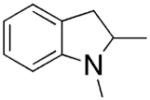
Group 4 metal catalysts are attractive because of their low cost and toxicity.13 The first enantioselective hydroamination/cyclization of alkenes catalyzed by a chiral zirconium catalyst was reported by Scott and co-workers in 2004.14 The cationic zirconium catalyst [ZrL2CH2Ph][B(C6F5)4], 5–10 mol%, enabled the quantitative cyclization of several secondary amines in 20–82% ee (Table 2). Primary amines were unreactive with this catalyst, presumably because the in situ-formed zirconium imido species is catalytically inactive.
Table 2.
Hydroamination/cyclization catalyzed by Scott’s zirconium catalyst14




Conditions: C6D5Br at 100 °C, ca. 10 mol% catalyst, 3–4 h.
NMR analysis of (R)-(+)-Mosher’s acid salt.

A few years after Scott’s report, Schafer and Bergman independently reported the successful enantioselective cyclization/hydroamination of primary amines.13,15 Schafer and co-workers synthesized an axially chiral amidate ligand and treated the ligand with Zr(NMe2)4 to obtain the zirconium amidate precatalyst (Table 3). Hydroamination/cyclization of a number of primary 4-pentenyl amines was achieved with 10 mol% of Schafer’s catalyst in refluxing toluene for 2–12 h, providing isolated pyrrolidines in 82–96% yield and 62–93% ee (Table 3). All of the substrates contain β-quaternary carbons, which is likely important to both the rate and selectivity of the reaction. The authors note that piperidines could also be formed, but with much lower enantioselectivity.
Table 3.
Hydroamination/cyclization catalyzed by Shafer’s zirconium catalyst13


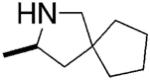
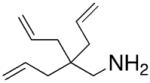

Conditions: toluene at 110 °C, 10 mol% catalyst, 2–12 h.
Isolated yield.
Determined by NMR analysis following derivatization with (S)-(+)-Mosher’s acid chloride.

The absolute stereochemistry of the major products was rationalized as having been formed via a proposed transition state which minimizes steric interactions between the amine backbone and the catalyst during the stereochemistry-determining [2 + 2] syn aminozirconation step (Fig. 2).
Fig. 2.
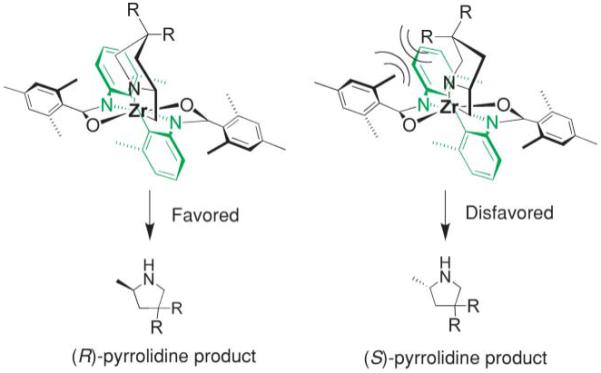
Origin of the enantioselectivity in the hydroamination reactions with Schafer’s catalyst.
Bergman and co-workers synthesized a chiral neutral zirconium catalyst by reaction of chiral diphosphinic amide ligands with Zr(NMe2)4 (Table 4).15 They demonstrated that indolines, pyrrolidines and piperidines can be cyclized using 10–20 mol% of their catalyst in 33–99% isolated yield and 33–80% ee (Table 4).
Table 4.
Hydroamination/cyclization catalyzed by Bergman’s zirconium catalyst15
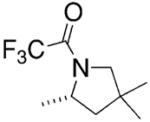





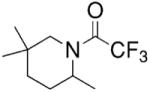
Conditions: toluene at 85–135 °C, 10–20 mol% catalyst, 24–72 h, then trifluoroacetic anhydride.
ee determined by chiral GC or HPLC.
Both Schafer and Bergman believe their reactions involve a [2 + 2] addition of a zirconium imido intermediate to the alkene. In support of this, Schafer demonstrated that secondary amines are unreactive with her catalyst.13
2.1.3 Main group metals: organolithium catalysts
The first lithium-based chiral hydroamination reaction was reported by Hultzsch and co-workers in 2006 using diamidobinaphthyl ligands as the source of chirality.16 Shortly thereafter, Tomioka and co-workers reported an organolithium-catalyzed reaction with chiral bis(oxazoline) ligands.17 The stereoselective C–N bond formation is thought to occur via a syn aminometallation mechanism.17
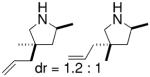
Hultzsch demonstrated that treatment of the L-proline derived axially chiral (S,S,S)-diaminobinaphthyl ligand (Table 5) with two equivalents of n-BuLi provided the corresponding dilithium salt as a yellow-orange powder. The crystal structure indicated a dimeric structure involving four highly coordinated lithium atoms. Treatment of the primary amine substrates with 5 mol% of Hultzsch’s lithium catalyst in toluene at room temperature provided efficient cyclization in 79–86% isolated yield (96–98% NMR yield) of various pyrrolidines with % ee’s ranging from 17–74% (Table 5).16 Although the styrenyl substrate (entry 4) was considerably more reactive than the substrates with terminal olefins, the enantioselectivity was significantly diminished. In these reactions it was noted that addition of THF reduced catalytic performance and eroded enantioselectivity.
Table 5.
Hydroamination/cyclization catalyzed by Hultzsch’s lithium catalyst16
| Entrya | Substrate | Product(s) | Time (h) | Yield (NMR, isolated)b | ee (%), config. |
|---|---|---|---|---|---|
| 1 |
|

|
42 | 96, 86 | 68, S |
| 2 |

|

|
0.8 | 97 | 31, S |
| 3d |

|

|
2 | 98, 82 | 74, S |
| 4 |
|
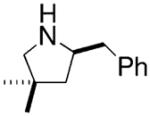
|
0.08 | 98 | 17 |
| 5 |
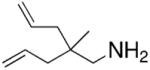
|
|
2 | 98, 79 | 64, 72 |
Conditions: 5 mol% catalyst, C6D6, 22 °C, Ar atm.
NMR yield relative to ferrocene internal standard.
Enantiomeric excess was determined by 19F NMR of the Mosher amides.
Reaction in toluene.

Hultzsch et al. demonstrated that the presence of multiple lithium and amine atoms in the catalyst is necessary for high catalyst reactivity. While LiN(SiMe3)2 was a poor catalyst, a catalyst system comprised of LiN(SiMe3)2 and (−)-sparteine gave improved rates, though poor selectivity.
Tomioka and co-workers demonstrated the enantioselective hydroamination/cyclization of the secondary amine styrenyl substrates 1 and 3 (Scheme 2).17 Under optimal conditions, using chiral bis(oxazoline) ligands (S,S)-i-PrCH2-Box and (4R,2′S)-i-Bu-Box, they obtained high yields of isoindoline 2 and tetrahydroisoquinoline 4 with 91% and 84% ee, respectively. The (S)-i-PrCH2-Box ligand favored formation of the S-product while the pseudo-enantiomeric (4R,2′S)-i-Bu-Box ligand favored formation of the R-adduct. The reactions proceeded at low temperature (−60 °C) and with high yield (>90%) with a catalyst mixture comprised of a 1:1:2 ratio of n-BuLi to i-Pr2NH to bis(oxazoline). Chiral ligand loading was typically 40 mol% but could be as low as 10 mol% with substrate 2. A transition state that could rationalize the formation of the S-stereoisomer using the (S)-i-PrCH2-Box ligand with substrate 2 is shown in Scheme 2, where a dominant stereocontrol element could be minimization of steric interactions between the alkene undergoing addition and the nearest oxazoline ring (lower right quadrant).
Scheme 2.

Chiral bis(oxazoline) ligands in enantioselective lithium-catalyzed hydroamination reactions.
2.2 Enantioselective intramolecular hydroamination of allenes
Allenes are often more reactive than alkenes in addition chemistry due to the 10 kcal of strain associated with the cumulated double bond.18,19 A number of transition metals have demonstrated utility in the intramolecular aminofunctionalization of allenes.20 Highly enantioselecctive hydroamination of allenes have only been reported using gold catalysts (vide infra). Although chiral palladium21 and titanium-catalyzed22 intramolecular enantioselective hydroaminations have been tried, the levels of asymmetric induction obtained were quite low.
The use of chiral gold(I) complexes has enabled the enantioselective cyclization of heteroatoms onto allenes.23-25 Toste and co-workers reported the first intramolecular enantioselective hydroamination of γ- and δ-N-allenyl sulfonamides catalyzed by gold(I) complexes in 2007.26,27 Widenhoefer and coworkers reported their results with γ-N-allenyl carbamates in the same year.28,29 An outer-sphere, trans aminometallation has been proposed for the stereochemistry-determining C–N bond forming step (Scheme 3).24,25,29
Scheme 3.

Proposed catalytic cycle for the gold-catalyzed hydroamination of allenes.
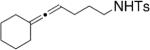
Toste and co-workers explored the use of two kinds of chiral catalysts in the intramolecular hydroamination of allenes. In their first report, chiral dinuclear gold(I)-phosphine complexes such as 5–7, where chirality is derived from the phosphine ligand, catalyzed the enantioselective hydroamination/cyclization of a number of γ- and δ-N-allenyl sulfonamides (Table 6).26 Isolated yields ranged from 41–99% and the ee’s ranged from 70–99%. They found that the nature of the achiral counterion influenced the efficiency and selectivity of the reactions; the optimized catalysts had the electron-withdrawing p-nitrobenzoate counterions. To obtain high enantioselectivities, highly substituted allene substrates were necessary (Table 6).
Table 6.
Au(I)-catalyzed hydroamination/cyclization of N-sulfonylallenes26
| Entrya | Substrate | Conditions | Product | Yield (%) | ee (%) |
|---|---|---|---|---|---|
| 1 |
|
A |
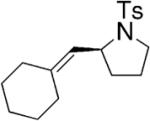
|
88 | 98 |
| 2 |
|
A |

|
75 | 83 |
| 3 |

|
B |

|
94 | 93 |
| 4 |
|
C |
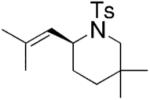
|
70 | 98 |
Conditions: A = 3 mol% of 5, 0.3 M in DCE, 23 °C; B = 5 mol% of 6, 0.3 M in MeNO2, 50 °C; C = 5 mol% of 7, 0.3 M in MeNO2, 50 °C.

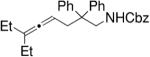
In a similar manner, Widenhoefer and co-workers also employed chiral dinuclear gold(I)-phosphine catalysts in their hydroamination/cyclization reactions of N-carbamoyl allenes (Table 7).28,29 Interestingly, allenylcarbamates failed to undergo hydroamination in Toste’s reaction,26 while allenylsulfonamides failed to give appreciable enantioselectivity in Widenhoefer’s reaction.28 In Widenhoefer’s reaction, the allene substrates are not required to be highly substituted to obtain respectable enantioselectivities.
Table 7.
Au(I)-catalyzed hydroamination/cyclization of N-carbamoyl allenes28
| Entrya | Substrate | Conditions | Product | Yield (%) |
ee (%) |
|---|---|---|---|---|---|
| 1 |
|
A |

|
97 | 81 |
| 2 |
|
B |

|
99 | 34 |
| 3 |
|
C |

|
83 | 91 |
| 4 |

|
A |

|
98 | 50 |
Conditions: A = 2.5 mol% [(S)-DTBM-MeOBIPHEP]Au2Cl2, 5 mol% AgClO4, 0.30 M in toluene, −40 °C, 24 h; B = same as A except −20 °C; C = same as A except 0 °C, 24 h then rt, 24 h.

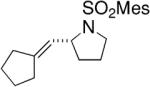
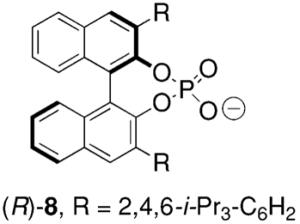
Toste and co-workers subsequently reported that a mononuclear gold(I) complex with an achiral phosphine ligand [Ph(CH3)2P] and a chiral binaphthol-derived phosphate counterion proved highly effective in catalyzing the enantioselective cyclization of γ-N-allenyl sulfonamides in benzene at 23 °C (Table 8).27
Table 8.
Chiral anion strategy for Au(I)-catalyzed hydroamination/cyclization of allenes27
| Entrya | Substrate | Product | Yield (%) |
ee(%) |
|---|---|---|---|---|
| 1 |
|

|
97 | 96 |
| 2 |
|
|
88 | 98 |
| 3 |
|

|
84 | 99 |
Conditions: 5 mol% Ph(CH3)2PAuCl, 5 mol% Ag-(R)-8, benzene, 23 °C, 48 h.
2.3 Enantioselective intramolecular hydroamination of alkynes
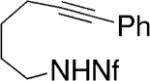
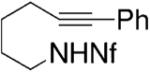
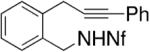
Yamamoto and co-workers have reported the enantioselective synthesis of pyrrolidines and piperidines via palladium-catalyzed intramolecular alkyne hydroamination.21,30-32 The reactions occur in good yield (87–93%) and enantioselectivity (79–95% ee) in benzene at 100 °C for 48 h. These conditions require 10 mol% palladium catalyst loading and 20 mol% of the methyl Norphos bisphosphine ligand (Table 9). Generally, the (R,R)-ligand provides the S-product. The nonafluorobutanesulfonyl (Nf) nitrogen substituent provided the highest yields and selectivities. Interestingly, neither the corresponding allene or diene substrates provided analogous enantioselectivities.
Table 9.
Palladium-catalyzed hydroamination/cyclization of alkynes32
| Entrya | Substrate | Product | Yield (%) | ee (%) |
|---|---|---|---|---|
| 1 |
|

|
93 | 92 |
| 2 |

|

|
90 | 88 |
| 3 |
|

|
92 | 79 |
| 4 |
|

|
90 | 95 |
Conditions: 5 mol % Pd2(dba)3·CHCl3, 10 mol% PhCO2H, 20 mol% ligand in PhH at 100 °C, 48 h.

The reaction is thought to involve addition of a palladium hydride onto the alkyne followed by beta-hydride elimination to form the allene. Subsequent hydropalladation would provide the π-allyl species, which may undergo nucleophilic trans addition by the amine to provide the nitrogen heterocycle and re-generate the catalyst (Scheme 4).
Scheme 4.

Proposed catalytic cycle for the intramolecular hydroamination of alkynes.
2.4 Enantioselective intramolecular aminative SN2′ displacement of allylic acetates
In 2002, Overman and co-workers reported a novel nitrogen heterocycle forming reaction that involves a formal enantioselective intramolecular SN2′ displacement of an allylic acetate, catalyzed by chiral palladium(II) catalyst 9 (Scheme 5).33,34 In this reaction, allylic tosylamides afford chiral vinyl-substituted nitrogen heterocycles. The reaction is proposed to involve a syn aminopalladation onto the alkene followed by reductive elimination of a palladium(II) acetate complex.33
Scheme 5.

Palladium-catalyzed enantioselective SN2′-type cyclization.
3 Oxidative cyclizations
When a substrate is oxidatively cyclized, it experiences a net oxidation upon conversion to the product. Reactions that fit into this category include carboamination, aminooxygenation, oxidative amination, diamination and aminohalogenation of alkenes. Enantioselective versions of only the first two cyclization reactions have been developed. Racemic metal-catalyzed/promoted intramolecular alkene oxidative aminations,35 diaminations36-40 and aminohalogenations41-43 have been reported, but no enantioselective versions of these reactions have yet appeared.
3.1 Enantioselective intramolecular carboamination of alkenes
The intramolecular carboamination of alkenes is an efficient method for the synthesis of nitrogen heterocycles from alkenyl amines. In these reactions, a new carbon–nitrogen and a new carbon–carbon bond are formed in the alkene addition reaction. Thus far, two examples of enantioselective intramolecular alkene carboaminations have been published, one using a chiral palladium catalyst44 and the other using a chiral copper catalyst45 (vide infra).
3.1.1 Palladium-catalyzed enantioselective carboamination
The first intramolecular catalytic enantioselective alkene carboamination was reported by Dan Yang and co-workers in 2006.44 This palladium-catalyzed reaction provides chiral indolines in moderate to good yields and 75–91% enantiomeric excess using 5–20 mol% Pd(TFA)2 and 20–80 mol% (−)-sparteine as the chiral catalyst (Table 10). The absolute configuration of one of the products was determined to be S. This reaction involves an intramolecular aminopalladation followed by intramolecular alkene insertion and terminates with a beta-hydride elimination. O2 (1 atm) was used as the stoichiometric oxidant. No model for the enantioselective C–N bond formation has been put forth but the basic reaction conditions46 andgood %eeatelevated (80 °C) temperature may indicate a tight, syn-aminopalladation enantiodetermining transition state.
Table 10.
Palladium-catalyzed enantioselective alkene carboamination44






Conditions: 10 mol% Pd(TFA)2, 40 mol% (−)-sparteine, 26–48 h.
Yield of isolated product.
Enantiomeric excess was determined by HPLC analysis using a Chiralcel OD column.
20 mol% Pd(TFA)2 and 80 mol% (−)-sparteine was used.
Absolute configuration determined by X-ray crystallography.

3.1.2 Copper-catalyzed enantioselective carboamination of alkenes
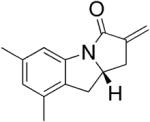
We reported a copper-catalyzed enantioselective alkene carboamination in 2007.45 This reaction provides chiral polycyclic sultams in moderate to good yields and 46–94% ee (Table 11). The pre-complexed catalyst is composed of 20 mol% Cu(OTf)2 and 20 mol% (R,R)-Ph-Box, a bis(oxazoline) ligand, and the stoichiometric oxidant is MnO2. 4-Pentenylarylsulfonamides provided the highest enantioselectivity.
Table 11.
Copper-catalyzed enantioselective intramolecular carboamination45
 | ||||
|---|---|---|---|---|
| Entrya | Substrate | Product | Yield (%)b | ee (%)c |

|

|
|||
| 1 | R1 = Me, R2 = Me | 85 | 92 | |
| 2 | R1 = Me, R2 = H | 73 | 92 | |
| 3 | R1 = Me, R2 = Cl | 45 | 92 | |
| 4 | R1 = Me, R2 = OMe | 75 | 94 | |
| 5 | R1 = Ph, R2 = Me | 78 | 94 | |
| 6 | R1 = H, R2 = H | 77 | 82 | |
| 7 |

|
|
75 | 46 |
| 8d |

|
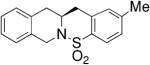
|
63 | 72 |
Conditions: Cu(OTf)2 (20 mol%) and (R,R)-Ph-box (20 mol%) were complexed in PhCF3 at 50 °C for 1 h (sealed tube). Substrate, MnO2 (300 mol%) and K2CO3 (100 mol%) were added, the reaction tube was sealed and heated at 120 °C for 24 h.
Yields refer to isolated product.
Enantiomeric excess was measured by chiral HPLC.
Reaction was run for 96 h.
The absolute configurations of two products (not shown) were determined by X-ray crystallography to be S; the rest are assigned by analogy. The enantioinduction is rationalized as shown in Scheme 6, which involves a presumed rate-determining synaminocupration transition state. In this transition state, steric interactions between the substrate’s N-substituent and the oxazoline substituent on the ligand that it is nearest to are minimized. The carbon–carbon bond is subsequently formed via homolysis of the unstable carbon–copper(II) bond to form a primary radical that adds to the neighboring aryl ring.47 Rearomatization, probably facilitated by oxidation of the aryl radical to the aryl cation, then provides the sultam product.
Scheme 6.

Proposed catalytic cycle for the enantioselective copper-catalyzed alkene carboamination.
Since both enantiomers of the Ph-box ligand are commercially available, this protocol provides access to either enantiomer of the product. The SO2 group in the sultam products can be reductively removed (Li, NH3) to provide the chiral pyrrolidine.45
This enantioselective carboamination reaction was used in our concise total synthesis of (S)-(+)-tylophorine (Scheme 7).48 Copper-catalyzed enantioselective carboamination of sulfonamide 10 provided sultam 11 in 64% yield. Reductive removal of SO2 followed by Pictet–Spengler reaction of the resulting pyrrolidine with formaldehyde provided (S)-(+)-tylophorine in 81% ee as determined by chiral HPLC. The yield in the SO2 removal is low due to concurrent loss of methoxy groups under the reaction conditions.
Scheme 7.

Application of the copper-catalyzed carboamination to the total synthesis of (+)-tylophorine.
3.2 Catalytic enantioselective intramolecular aminooxygenation of alkenes
We reported the first enantioselective intramolecular alkene aminooxygenation in 2008.49 This reaction is also catalyzed by chiral copper-bis(oxazoline) complexes and provides access to methyleneoxy-functionalized indolines and pyrrolidines in good to excellent yields and with good enantioselectivity (Table 12). In these reactions, tetramethylaminopyridyl radical (TEMPO) serves as both the oxygen source and as the oxidant. However, an O2 atmosphere is required for complete conversion of the 4-pentenylsulfonamides to their corresponding pyrrolidines. For this reaction, the (4R,5S)-diphenylbis(oxazoline) ligand 12 provided the highest enantioselectivity. Arylsulfonyl N-substituents gave the highest reactivity and selectivity (compare entries 5 and 10). The TEMPO adducts can be subsequently reduced to provide the corresponding aminoalcohols or oxidized to provide the aldehydes without reduction of enantiomeric excess.49
Table 12.
Copper-catalyzed enantioselective intramolecular aminooxy-genation49
 | ||||
|---|---|---|---|---|
| Entrya | Substrate | Product | Yield (%)b |
ee (%)c |

|

|
|||
| 1d | R1 = Me, R2 = Ts | 97 | 88 | |
| 2d | R1 = Ph, R2 = Ts | 97 | 92 | |
| 3d,e | R1 = H, R2 = Ts | 74 | 75 | |
| 4d,e | R1 = Me, R2 = Ns | 86 | 89 | |
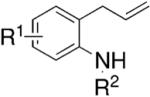
|

|
|||
| 5 | R1 = H, R2 = Ts | 97 | 90 | |
| 6 | R1 = p-F, R2 = Ts | 83 | 89 | |
| 7 | R1 = p-Cl, R2 = Ts | 73 | 91 | |
| 8 | R1 = p-OMe, R2 = Ts | 82 | 86 | |
| 9 | R1 = H, R2 = Ns | 75 | 61 | |
| 10 | R1 = H, R2 = Ms | 57 | 50 | |
| 11d,e |

|

|
61 | 82 |
Conditions: Cu(OTf)2 (20 mol%) and (4R,5S)-12(25 mol%) were complexed in PhCF3 at 50 °C for 1 h (sealed tube). Substrate, TEMPO (300 mol%) and K2CO3 (100 mol%) were added, the reaction tube was sealed and heated at 110 °C for 24 h.
Yields refer to isolated product.
Enantiomeric excess was measured by chiral HPLC analysis.
Reaction was run at 120 °C under O2 (1 atm).
40 mol% Cu(OTf)2 and 50 mol% ligand was used.
A rate-determining syn-aminocupration is invoked to rationalize the observed product stereoselectivity (Scheme 8). Homolysis of the resulting carbon–copper bond then provides a primary radical which combines with TEMPO radical to give the observed aminooxygenation product. TEMPO and/or O2 serve to reoxidize Cu(I) to Cu(II).
Scheme 8.

Proposed cycle for the copper-catalyzed enantioselective alkene aminooxygenation.
4 Summary and future outlook
As can be seen from the new methodologies summarized in this review, the catalytic asymmetric amination of alkenes, dienes, allenes and alkynes has experienced a flurry of progress in recent years. It is interesting that most, but not all of these transformations are thought to occur via syn aminometallation processes, which can offer tight transition states for translation of the ligand chirality to the forming nitrogen-bearing stereocenter. Notable exceptions are the gold-catalyzed allene hydroaminations and the palladium-catalyzed alkyne hydroamination. It is expected that application of these methods in the context of fine chemical synthesis and medicinal chemistry, and in the total synthesis of natural products will continue and likely will drive further advances in the field. In this respect, a number of challenges still remain. The intramolecular catalytic enantioselective oxidative amination, diamination and aminohalogenation of alkenes have yet to be reported. Additionally, many of the existing reactions still require significant thermal activation to occur, indicating the need for more active catalysts to be developed. More active catalysts could also further expand the substrate scope. Furthermore, a more rigorous understanding of the factors involved in determining enantioselectivity could lead to more rationally designed ligands and better pairing of substrates and catalysts. Advances in these areas and others will further establish the enantioselective amination of unactivated π bonds as a method of choice for the synthesis of chiral nitrogen heterocycles.
Acknowledgements
Enantioselective nitrogen heterocycle synthesis in our lab has been supported by the National Institutes of Health (NIGMS RO1 078383).
Biography

Sherry R. Chemler received a B.A. from Boston University in 1994 and a Ph.D. in 1999 under the direction of Professor William R. Roush at Indiana University. She continued her studies as a postdoctoral fellow with Professor Samuel J. Danishefsky at the Memorial Sloan-Kettering Cancer Institute in New York. In 2002 she began her academic career as an Assistant Professor at the University at Buffalo, The State University of New York. She was promoted to Associate Professor in 2008. Her research interests include the discovery and development of new reactions, asymmetric catalysis, and the synthesis of biologically active small molecules.
References
- 1.Roesky PW, Muller TE. Angew. Chem., Int. Ed. 2003;42:2708–2710. doi: 10.1002/anie.200301637. [DOI] [PubMed] [Google Scholar]
- 2.Hultzsch KC. Adv. Synth. Catal. 2005;347:367–391. [Google Scholar]
- 3.Hultzsch KC. Org. Biomol. Chem. 2005;3:1819–1824. doi: 10.1039/b418521h. [DOI] [PubMed] [Google Scholar]
- 4.Hong S, Marks TJ. Acc. Chem. Res. 2004;37:673–686. doi: 10.1021/ar040051r. [DOI] [PubMed] [Google Scholar]
- 5.Muller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem. Rev. 2008;108:3795–3892. doi: 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- 6.Kim YK, Livinghouse T, Bercaw JE. Tetrahedron Lett. 2001;42:2933–2935. [Google Scholar]
- 7.Kim YK, Livinghouse T. Angew. Chem., Int. Ed. 2002;41:3645–3647. doi: 10.1002/1521-3773(20021004)41:19<3645::AID-ANIE3645>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 8.Hong S, Tian S, Metz MV, Marks TJ. J. Am. Chem. Soc. 2003;125:14768–14783. doi: 10.1021/ja0364672. [DOI] [PubMed] [Google Scholar]
- 9.O’Shaughnessy PN, Knight PD, Morton C, Gillespie KM, Scott P. Chem. Commun. 2003:1770–1771. [Google Scholar]
- 10.Kim JY, Livinghouse T. Org. Lett. 2005;7:1737–1739. doi: 10.1021/ol050294z. [DOI] [PubMed] [Google Scholar]
- 11.Gribkov DV, Hultzsch KC, Hampel F. J. Am. Chem. Soc. 2006;128:3748–3759. doi: 10.1021/ja058287t. [DOI] [PubMed] [Google Scholar]
- 12.Riegert D, Collin J, Meddour A, Schulz E, Trifonov A. J. Org. Chem. 2006;71:2514–2517. doi: 10.1021/jo052322x. [DOI] [PubMed] [Google Scholar]
- 13.Wood MC, Leitch DC, Yeung CS, Kozak JA, Schafer L. Angew. Chem., Int. Ed. 2007;46:354–358. doi: 10.1002/anie.200603017. [DOI] [PubMed] [Google Scholar]
- 14.Knight PD, Munslow I, O’Shaughnessy PN, Scott P. Chem. Commun. 2004:894–895. doi: 10.1039/b401493f. [DOI] [PubMed] [Google Scholar]
- 15.Watson DA, Chiu M, Bergman RG. Organometallics. 2006;25:4731–4733. doi: 10.1021/om0606791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez PH, Hultzsch KC, Hampel F. Chem. Commun. 2006:2221–2223. doi: 10.1039/b518360j. [DOI] [PubMed] [Google Scholar]
- 17.Ogata T, Ujihara A, Tsuchida S, Shimizu T, Kaneshige A, Tomioka K. Tetrahedron Lett. 2007;48:6648–6650. [Google Scholar]
- 18.Schuster HF, Coppola GM. Allenes in Organic Synthesis. Wiley-Interscience; New York: 1984. [Google Scholar]
- 19.Arredondo VM, McDonald FE, Marks TJ. Organometallics. 1999;18:1949–1960. [Google Scholar]
- 20.Bates RW, Satcharoen V. Chem. Soc. Rev. 2002;31:12–21. doi: 10.1039/b103904k. [DOI] [PubMed] [Google Scholar]
- 21.Lutete LM, Kadota I, Yamamoto Y. J. Am. Chem. Soc. 2004;126:1622–1623. doi: 10.1021/ja039774g. [DOI] [PubMed] [Google Scholar]
- 22.Hoover JM, Petersen JR, Pikul JH, Johnson AR. Organometallics. 2004;23:4614–4620. [Google Scholar]
- 23.Shen HC. Tetrahedron. 2008;64:3885–3903. [Google Scholar]
- 24.Widenhoefer RA, Han X. Eur. J. Org. Chem. 2006:4555–4563. [Google Scholar]
- 25.Widenhoefer RA. Chem. Eur. J. 2008;14:5382–5391. doi: 10.1002/chem.200800219. [DOI] [PubMed] [Google Scholar]
- 26.LaLonde RL, Sherry BD, Kang EJ, Toste FD. J. Am. Chem. Soc. 2007;129:2452–2453. doi: 10.1021/ja068819l. [DOI] [PubMed] [Google Scholar]
- 27.Hamilton GL, Kang EJ, Mba M, Toste FD. Science. 2007;317:496–499. doi: 10.1126/science.1145229. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z, Bender CF, Widenhoefer RA. Org. Lett. 2007;9:2887–2889. doi: 10.1021/ol071108n. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Z, Bender CF, Widenhoefer RA. J. Am. Chem. Soc. 2007;129:14148–14149. doi: 10.1021/ja0760731. [DOI] [PubMed] [Google Scholar]
- 30.Patil NT, Lutete LM, Wu H, Pahadi NK, Gridnev ID, Yamamoto Y. J. Org. Chem. 2006;71:4270–4279. doi: 10.1021/jo0603835. [DOI] [PubMed] [Google Scholar]
- 31.Patil NT, Wu H, Yamamoto Y. J. Org. Chem. 2007;72:6577–6579. doi: 10.1021/jo0708137. [DOI] [PubMed] [Google Scholar]
- 32.Narsireddy M, Yamamoto Y. J. Org. Chem. 2008;73:9698–9709. doi: 10.1021/jo801785r. [DOI] [PubMed] [Google Scholar]
- 33.Overman LE, Remarchuk TP. J. Am. Chem. Soc. 2002;124:12–13. doi: 10.1021/ja017198n. [DOI] [PubMed] [Google Scholar]
- 34.Kirsch SF, Overman LE. J. Org. Chem. 2005;70:2859–2861. doi: 10.1021/jo047763f. [DOI] [PubMed] [Google Scholar]
- 35.Kotov V, Scarborough CC, Stahl SS. Inorg. Chem. 2007;46:1910–1923. doi: 10.1021/ic061997v. [DOI] [PubMed] [Google Scholar]
- 36.Zabawa TP, Kasi D, Chemler SR. J. Am. Chem. Soc. 2005;127:11250–11251. doi: 10.1021/ja053335v. [DOI] [PubMed] [Google Scholar]
- 37.Zabawa TP, Chemler SR. Org. Lett. 2007;9:2035–2038. doi: 10.1021/ol0706713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Streuff J, Hovelmann CH, Nieger M, Muniz K. J. Am. Chem. Soc. 2005;127:14586–14587. doi: 10.1021/ja055190y. [DOI] [PubMed] [Google Scholar]
- 39.Muniz K, Streuff J, Hovelmann CH, Nunez A. Angew. Chem., Int. Ed. 2007;46:7125–7127. doi: 10.1002/anie.200702160. [DOI] [PubMed] [Google Scholar]
- 40.Sibbald PA, Michael FE. Org. Lett. 2009;11:1147–1149. doi: 10.1021/ol9000087. [DOI] [PubMed] [Google Scholar]
- 41.Lei AW, Lu XY, Liu GS. Tetrahedron Lett. 2004;45:1785–1788. [Google Scholar]
- 42.Manzoni MR, Zabawa TP, Kasi D, Chemler SR. Organometallics. 2004;23:5618–5621. [Google Scholar]
- 43.Michael FE, Sibbald PA, Cochran BM. Org. Lett. 2008;10:793–796. doi: 10.1021/ol702922c. [DOI] [PubMed] [Google Scholar]
- 44.Yip K-T, Yang M, Law K-L, Zhu N-Y, Yang D. J. Am. Chem. Soc. 2006;128:3130–3131. doi: 10.1021/ja060291x. [DOI] [PubMed] [Google Scholar]
- 45.Zeng W, Chemler SR. J. Am. Chem. Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu G, Stahl SS. J. Am. Chem. Soc. 2007;129:6328–6335. doi: 10.1021/ja070424u. [DOI] [PubMed] [Google Scholar]
- 47.Sherman ES, Fuller PH, Kasi D, Chemler SR. J. Org. Chem. 2007;72:3896–3905. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeng W, Chemler SR. J. Org. Chem. 2008;73:6045–6047. doi: 10.1021/jo801024h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuller PH, Kim J-W, Chemler SR. J. Am. Chem. Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m. [DOI] [PMC free article] [PubMed] [Google Scholar]