

Abstract
Cardiovascular disease (CVD) continues to be a leading cause of death worldwide. Elevated serum cholesterol is one of the classical risk factors for CVD which also include age, hypertension, smoking, diabetes mellitus, obesity and family history. A number of therapeutic drug classes have been developed to treat hypercholesterolemia, yet, an important percentage of patients do not reach their treatment goals. Therefore, new cholesterol-lowering medications, having a site of action different from that of currently available drugs need to be developed. This review summarizes new information about cytochrome P450 enzymes 7A1, 27A1, and 46A1, that play key roles in cholesterol elimination and that have potential to serve as targets for cholesterol-lowering.
Keywords: CYP7A1, CYP27A1, CYP46A1, cholesterol, cholesterol-lowering drugs, cholesterol homeostasis
1. Introduction
Cardiovascular disease (CVD) is the major cause of death all over the world accounting for 36% of death from chronic diseases in the US (data for 2003, http://www.cdc.gov/nccdphp/overview_text.htm). While a number of drugs to treat hyperlipidimea are available, very often, the desirable therapeutic goals are not achieved by a single drug. This warrants the development of additional hypolipidemic agents to be used either as monotherapy or in combination with the currently available medications. By virtue of their function to initiate and control the first steps in all quantitatively important pathways for cholesterol degradation to bile acids, cytochrome P450s enzymes (P450 or CYP) 7A1, 27A1 and 46A1 represent an attractive therapeutic target for cholesterol lowering. Properties of these P450s, their medical significance and transcriptional regulation have been summarized in several recent review papers [1–6]. The purpose of this update is to describe the most recent advances in our knowledge of cholesterol-metabolizing P450s. A brief overview of cholesterol homeostasis and existing lipid-lowering medications is given first followed by the section on the three P450 enzymes and the expert opinion conclusion.
2. Overview of cholesterol homeostasis and key proteins
In humans, cholesterol homeostasis involves cholesterol acquisition from endogenous and exogenous sources, transport in the blood in complex with lipoprotein particles, and elimination through degradation to bile acids and biliary secretion. Pathways of cholesterol input are balanced with the pathways of cholesterol output, therefore, steady state levels of plasma cholesterol do not vary significantly in healthy individuals.
Cholesterol input is provided by de novo synthesis (600–900 mg/day) and diet (300–500 mg/day) [7]. Virtually every tissue synthesizes cholesterol from acetyl coenzyme A (acetyl-CoA). Two acetyl-CoA molecules are condensed to acetoacetyl-CoA by β-ketothiolase followed by a series of enzymatic reactions leading to the formation of cholesterol. 3-Hydroxy-3-methylglutaryl-CoA reductase (HMG-CoA) is the rate-limiting enzyme in the first, squalene, portion of cholesterol biosynthesis, whereas cytochrome P450 51 (CYP51) controls the post-squalene part of the pathway. CYP51 catalyzes the removal of the 14α-methyl group of lanosterol, the first sterol precursor in the pathway. Dietary cholesterol is absorbed from food in the intestine; this process being regulated by Niemann-Pick C1-like protein, which transports cholesterol inside the enterocyte, and the ATP binding cassette (ABC) transporters ABCG5/ABCG8, which efflux cholesterol back into the intestinal lumen. Once in the enterocyte, cholesterol is packaged into chylomicrons, and excreted into the lymph. Upon entering the blood circulation, triglycerides within chylomicrons are hydrolysed by lipases, and cholesterol-enriched chylomicron remnants are taken up by the liver.
In the liver, diet-derived cholesterol or that synthesized de novo are either secreted into the blood in the form of very low-density lipoproteins (VLDL) or in the bile per se. Cholesterol can also be stored as cholesteryl esters (CE) or degraded to bile acids. VLDL secreted in the blood loose their lipid content and are transformed first into intermediate density lipoproteins (IDL) and then into low density lipoproteins (LDL). LDL are the principal carriers of cholesterol to peripheral tissues where they are internalized into cells by the LDL receptors. Uptake of oxidized LDL by macrophages in the arterial wall is an important event in the pathogenesis of atherosclerosis. In the cell, LDL are hydrolyzed by lysosomal enzymes, and this leads to a release of free cholesterol. The appearance of free cholesterol activates acyl-cholesterol acyltransferase (ACAT) which re-esterifies cholesterol for storage. When macrophages become overloaded with CE, they are converted to “foam cells”, the classic component of atherosclerotic plagues. Removal of excess cholesterol from extrahepatic cells is realized either through hydroxylation reaction catalyzed by ubiquitous CYP 27A1 or by CYP46A1, which is expressed in neural tissues, or through the efflux mediated, in part, by the ABCA1 transporter. Effluxed cholesterol is acquired in the blood by nascent HDL and esterified by lecitin-cholesterol acyltransferase (LCAT) present in HDL. The latter is important for maturation of HDL. CE in mature HDL is then transferred to remnant lipoproteins by cholesteryl ester transfer protein (CETP), and remnant particles are cleared from the circulation by the liver. About a half of cholesterol in the liver (~600 mg/day) is then secreted in bile and the other half (~600 mg/day) is degraded to bile acids. CYP7A1 is the key enzyme involved in the latter process.
3. Existing lipid-lowering drugs
The major lipids present in the plasma are fatty acids, triglycerides, cholesterol and phospholipids. Epidemiological surveys indicate that elevated plasma cholesterol and in particular cholesterol carried in complex with LDL (LDL-C) is a major risk factor for coronary heart disease (CHD): every 30-mg/dL increase in LDL-C corresponds to a 30%-increase in the relative risk for CHD [8, 9]. Therefore, in the US, the primary target of cholesterol-lowering therapy is LDL-C as identified by the National Cholesterol Education Program (NCEP) [9]. In contrast to LDL-C, the importance of treating low levels of cholesterol in complex with HDL (HDL-C) is currently less well appreciated, although the potential benefit of HDL-raising therapy [10–12] has evoked considerable interest. It is estimated that risk of CVD increases by 1–3% for every 1% reduction in HDL-C [13]. HDL-C raising remains a secondary goal in the NCEP guidelines because current documentation of risk reduction through controlled clinical trials is not sufficient to warrant setting such a specific goal [9]. Accumulating evidence suggests that elevated triglycerides (TG) levels may pose a significant independent risk for CVD [14]. To reflect a growing awareness of the importance of even moderate TG elevations, the NCEP lowered the ranges for the categorization of TG levels as normal, borderline, high, and very high [8]. Currently, there are five classes of drugs on the market to lower plasma lipid levels: statins, bile acid sequestrants, ezetimibe, nicotinic acid, and fibrates.
Statins are the most effective and most widely prescribed cholesterol-lowering drugs. They inhibit HMG-CoA reductase, which catalyzes the rate-limiting step in cholesterol synthesis in all nucleated cells. Inhibition of cholesterol synthesis leads to reduced cholesterol content and increased expression of LDL-receptor [15]. The upregulation of LDL-receptors lowers concentrations of TG-rich lipoproteins because IDL and VLDL remnants are also removed from the circulation via the LDL-receptor. At maximum approved doses, the LDL-C-lowering effects range from 35% to 55%, and the incidence of CHD can be reduced by 25–60% [16]. All statins lower TG levels up to 20–30%, and, thus, are useful in treatment of moderate hypertriglyceridemia [16]. The overall benefits observed with statins appear to be greater than what might be expected from changes in lipid levels alone, suggesting effects beyond cholesterol lowering. Recent studies indicate that some of the cholesterol-independent effects of statins involve improved endothelial function, enhanced stability of atherosclerotic plaques, decreased oxidative stress and inflammation, and inhibition of the thrombogenic response [17]. As a class, statins seem to be a remarkably safe group of drugs when used at their standard doses. They are well tolerated. The adverse effects include myopathy (muscle pain or weakness, occurs in fewer than one in 10,000 patients), rhabdomyolysis (a rarer and more severe form of myopathy), and increased levels of the liver enzymes transaminases (indicators of liver function, observed in a small percentage of patients) [18].
Bile acid sequestrants or resins bind bile acids in the intestine and, thus, increase hepatic conversion of cholesterol to bile [19]. Hepatic pool of cholesterol is depleted, which in turn stimulates the LDL receptor activity leading to an increased uptake of plasma LDL-C and lowering LDL-C by up to 20% [20]. Currently, this class of drugs is used either as monotherapy or more often as combination therapy to further reduce LDL-C in patients who are already receiving a statin. Side effects include abdominal fullness, gas, and constipation that are observed in 30% of patients. Bile acid-binding resins may also reduce the absorption of lipophilic vitamins and can bind and inactivate polar drugs such as statins, warfarin, digoxin, and folic acid [21]. To avoid such an effect, these substances are given one hour before or four hours after the resin.
Ezetimibe is the first and only member of a novel class of lipid-lowering drugs that block the intestinal absorption of cholesterol, both dietary and biliary. Ezetimibe inhibits Niemann-Pick C1-like protein, located on the brush border membrane of intestinal epithelial cells, although other proteins may be involved [22]. Inhibition of cholesterol absorption leads to a decrease in cholesterol delivery to the liver, resulting in a reduction of hepatic cholesterol and increase of cholesterol clearance from the blood. Ezetimibe does not affect the uptake of TG, fatty and bile acids and fat-soluble vitamins [16], and, thus, exhibits a more favorable adverse event profile compared with bile acid-binding resins. In clinical trials with hypercholesterolemic patients, ezetemide monotherapy resulted in a reduction of LDL-C by ~20% [23]. In combination therapy, ezetimibe provided an additional 18–20%-reduction of LDL-C compared with that attainable with statins alone and also had favorable effects on HDL-C and TG [16]. Ezetemide is generally well tolerated, demonstrating an excellent safety profile [24]. Side effects have been infrequently reported, mainly in combination treatment with statins. Long-term safety data or outcome studies for ezetimibe, however, are not yet available.
Nicotinic acid, also known as niacin or vitamin B3, exerts a variety of effects on lipoprotein metabolism. By binding to the G protein-coupled receptor GPR109A on adipocytes and inhibiting adenylate cyclase, nicotinic acid blocks hormone-sensitive lipase-dependent lipolysis in adipose tissue, thereby lowering the concentration of free fatty acids in the plasma. This leads to a substrate shortage for hepatic TG syntheses and reduces the production of VLDL and LDL [25, 26]. Nicotinic acid also increases HDL-C and is currently the most effective HDL-C-raising agent. The exact mechanism of the HDL-C increase, however, is still not clear. At >1.5 g/day dose, nicotinic acid decreases total cholesterol by 4–16%, VLDL by 25%–40%, LDL-C by 6–28%, and TG by 21–44%. The HDL-C is increased by 18–35% [26]. Combined treatment with simvastatin was reported to reduce LDL-C by 42% and cause a 26% increase in HDL-C and a 60–90% reduction in the incidence of cardiovascular events [27]. The clinical use of nicotinic acid is, however, hindered by harmless but unpleasant effect skin flushing seen in 70% of patients. Other adverse effects and their reported frequency include headaches, gastrointestinal symptoms, hepatoxicity (up to 7%), elevated fasting glucose levels (7%), elevated uric acid levels (9%) that may have clinical relevance in selected patients [26, 28, 29].
Fibrates (derivatives of fibric acid) are agonists of peroxisome proliferator-activated receptor -alpha (PPAR-α), which regulates the expression of many genes involved in lipid metabolism (lipoprotein lipase, apolipoproteins A-I, A-II, and C-III, ABC transporters ABCA1 and ABCG1, and a number of other proteins) [30]. Fibrates are very effective in TG lowering. Activation of PPAR-α results in increased lipolysis and plasma clearance of TG via the activation of lipoprotein lipase [31, 32]. The HDL-C increase is due not only to the reduction of TG, but also secondary to the PPAR-α-mediated stimulation of the apo A-I and apo A-II, the major proteins in HDL [30]. Depending on lipid phenotype and baseline concentrations, fibrates reduce plasma TG by 30–50% and increase HDL-C by 5–15%. The reduction of LDL-C is variable and could be 10–20% in individuals with elevated LDL-C [33]. Fibrates are generally well tolerated; side effects include gastrointestinal and dermatologic, erectile dysfunction, and reactions related to musculoskeletal and neurologic systems [21].
Additional cholesterol-lowering interventions based on new therapeutic targets are under investigation. They include inhibitors of ACAT, CETP and squalene synthase. ACAT is responsible for the conversion of the free intracellular cholesterol into CE; CETP promotes the transfer of cholesteryl esters from antiatherogenic HDL to proatherohgenic VLDL and LDL; and squalene synthase catalyzes the formation of squalene, an intermediate step in the pathway for cholesterol biosynthesis. The results of human trials with these inhibitors, however, have been disappointing [34–36]. The ACAT inhibitor avasimibe failed to show lipid profile changes as well as reductions in surrogate markers for coronary artery disease [34]. The trial with the CETP inhibitor torcetrapib was terminated prematurely because of an unexplained increased risk of death and cardiac events despite increase of HDL-C and decrease of LDL-C [37]. Phase II and phase III trials with the squalene synthase inhibitor lapaquistat raised some safety issues [36]. Two additional phase III clinical trials with lapaquistat are underway (http://www.clinicaltrials.gov).
4. Implications of CYPs 7A1, 27A1, and 46A1 for cholesterol lowering
Bile acid biosynthesis represents the major route for cholesterol disposal from the body [38]. It involves 17 different enzymes and is tightly regulated to ensure that sufficient amounts of cholesterol are catabolized to maintain homeostasis and to provide adequate emulsification in the intestine. When an organism is replete, excess bile acids repress further synthesis, and conversely when bile acids are in short supply, their synthesis is increased [5]. Several metabolic routes led to the formation of bile acids.
4.1. CYP7A1
The metabolic route that dominates under normal physiological conditions and accounts for the elimination of 400–600 mg cholesterol/day takes place in the liver. This route, called the classical bile acid biosynthetic pathway, is initiated by CYP7A1, which converts cholesterol to 7α-hydroxycholesterol, the rate-limiting step in this pathway. Evidence for the critical role of CYP7A1 in whole body cholesterol homeostasis is provided by the clinical phenotype of the three individuals that have been found to have a complete lack of cholesterol 7α-hydroxylase activity due to the inactivating mutation in the CYP7A1 gene (CYP7A1) [39]. These individuals had high total plasma cholesterol (306–419 mg/dL vs <200 mg/dL considered as desirable by the American Heart Association, http://www.americanheart.org/presenter.jhtml?identifier=183). The LDL-C was also high (151–213 mg/dL vs <100 mg/dL considered as optimal) and resistant to the statin treatment. In addition, two male subjects had significantly elevated TG levels (410 mg/dL and 919 mg/dL vs < 150 mg/dL considered as normal) and premature gallstone disease [39]. Thus, CYP7A1 is an important determinant of plasma cholesterol levels and should certainly be considered as target for cholesterol lowering. In fact, there are already drugs that increase cholesterol 7α-hydroxylase activity. The bile acid-binding resin cholestyramine was reported to increase CYP7A1 activity ~5-fold [40]. This increase likely occurs via upregulation of the transcription of CYP7A1 because bile acids are known to suppress the expression of CYP7A1 [5]. Tolerability and compliance issues have limited the use of bile-acid binding resins. Therefore, alternative manipulations of CYP7A1 activity should be considered.
The caveat with CYP7A1 is that its activity varies over a 5–10-fold range in healthy individuals [40–43]. The underlying reason for this interindividual variability is currently unknown. Probably, it is a combination of CYP7A1 genotype, dietary habits, age, and alcohol consumption; each of these factors has been reported to have an effect on cholesterol 7α-hydroxylation [44–50]. CYP7A1 activity is known to be regulated at transcriptional level through the orphan nuclear receptors which are responsive to many stimuli including bile acids, hormones (thyroid, steroid, pituitary, and insulin), glucose and high fat diet [5–7]. The sensitivity of CYP7A1 transcription to different stimuli could explain why in several studies the carriers of the frequent (40%) -204A/C promoter polymorphism in CYP7A1 were found to have elevated LDL-C [51–53], whereas in other studies an association between the polymorphism and plasma LDL-C was either not found or was inconsistent [54, 55]. Also, there are studies in which individuals with the -204A/C polymorphism were shown to have a higher mean increase in total cholesterol after increased intake of dietary cholesterol or intervention with cafestol (the cholesterol-raising factor in boiled coffee) [56, 57]. Finally, the CC homozygotes demonstrated a lower LDL-C reduction upon treatment with atorvastatin than the subjects homozygous for the wild type AA allele [58, 59]. Thus, when evaluating CYP7A1 as a drug target, one needs to keep in mind that effectiveness of CYP7A1 manipulation will probably be at least determined by the CYP7A1 genetic background, hormonal status and nutritional factors such as composition of dietary fat and intake of cholesterol and glucose. Therefore, to identify the most responsive individuals to the CYP7A1-targeted drugs, a knowledge of the CYP7A1 genotype and basal level of the enzyme activity will likely be required. The latter will be especially necessary for subjects who do not have polymorphisms in CYP7A1 to serve as an indicator of dietary preferences and lifestyle. While genotyping becomes a routine procedure in medical practice, direct measurement of cholesterol 7α-hydroxylation is difficult because CYP7A1 is only expressed in the liver. Liver biopsies are necessary to carry out the enzyme assay. Plasma levels of the product, 7α-hydroxycholesterol, were shown to reflect the activity of CYP7A1 [40]. However, 7α-hydroxycholesterol may be formed non-enzymatically and is measured by sophisticated and expensive methods based on isotope dilution-mass spectrometry [40, 60]. To overcome these limitations, another sterol, 7α-hydroxy-4-cholesten-3-one, formed enzymatically from 7α-hydroxycholesterol was tested and shown to be a suitable marker for CYP7A1 activity and bile acid synthesis [61–65]. Thus, to better understand the potential of CYP7A1 as a target for cholesterol lowering, further studies are required in which known modulators of CYP7A1 activity, both positive and negative, are evaluated for their effect on serum lipids based on the knowledge of CYP7A1 genotype and enzyme activity.
4.2. CYP27A1
Under normal conditions, the pathway of bile acid biosynthesis initiated by CYP27A1 accounts for elimination of only 18–20 mg cholesterol/day [66]. This pathways, often called as alternative, begins in extrahepatic tissues and complements the HDL-mediated reverse cholesterol transport [67]. This alternative pathway becomes upregulated when the classical pathway is suppressed. Studies of an individual with complete CYP7A1 deficiency demonstrated that he had a 2-fold-increased CYP27A1 activity compared with control subjects carrying no mutation in CYP7A1 [39]. CYP27A1 converts cholesterol to 27-hydroxycholesterol. This oxygenation reaction is suggested to be important for cholesterol elimination from human lung macrophages and cells in arterial endothelium [68]. CYP27A1 is also involved in the classical pathway of bile acid biosynthesis in the liver, where it hydroxylates bile acid intermediates [3]. The products of CYP27A1 activities 27-hydroxycholesterol and 3β-hydroxy-5-cholestenoic acid are the ligands for the nuclear liver X receptors (LXR) [69, 70]. Yet, several lines of evidence argue against a regulatory role of CYP27A1 [71, 72]. Deficiency of the enzyme activity due to mutations in CYP27A1 leads to a slowly progressive disease cerebrotendinous xanthomatosis (CTX), which is characterized by a variety of manifestations [73]. One of them is premature atherosclerosis. Individuals with CTX usually have normal plasma levels of cholesterol, however, cholesterol and cholestanol (reduced form of cholesterol) are accumulated in almost every tissue [73]. CTX is believed to be a rare disease, but its incidence may be underestimated, as suggested by the recent studies, and equal to 3–5 per 100,000 [74]. CYP27A1 was sequenced only in patients with CTX, therefore, it is currently not known whether there are polymorphisms that result in only moderate decrease of cholesterol 27-hydroxylse activity. This could provide insight into why a number of patients with CHD have, like CTX patients, normal levels of plasma cholesterol [75, 76]. Similar to CYP7A1, CYP27A1 is transcriptionally regulated by bile acids but responses are less prominent than those of CYP7A1 [77, 78]. Consequently, there should be less interindividual variability in the enzyme activity, although such studies have not yet been carried out. While structure/function relationships have been intensively elucidated in CYP27A1 [3, 4], crystal structure of the enzyme is not available. Thus, focusing on crystallographic characterization of CYP27A1 as well as on CYP27A1 pharmacogenomics could be directions that will help to better understand how to manipulate the enzyme activity and whether interindividual variability will contribute to the effectiveness of this manipulation.
4.3. CYP46A1
Circulating in the blood cholesterol can not cross the blood-brain barrier, therefore most of the cholesterol in the central nervous system (CNS) is derived from local synthesis [79]. In the adult brain, cholesterol is primarily synthesized in astrocytes [80] that also synthesize apolipoprotein E (apoE). The two molecules complexed together are secreted into interstitial fluid of the brain and taken up by other glial cells and by neurons [80, 81]. To maintain steady state levels of cholesterol, neurons have an enzyme, CYP46A1, that hydroxylates cholesterol on position 24 [82–84]. Unlike cholesterol, 24(S)-hydroxycholesterol can traverse the blood brain barrier, enter the circulation and be delivered to the liver for further degradation to bile acids [85]. About 6–7 mg of cholesterol is converted to 24(S)-hydroxycholesterol every day [86]. CYP46A1 initiates the major pathway for cholesterol elimination from the brain, and thereby controls cholesterol turnover in the CNS [87]. The latter is important for memory and learning, as indicated by animal studies [87]. Data keep accumulating that polymorphisms in CYP46A1 (only intronic polymorphism has been identified so far) may be a risk factor for Alzheimer’s disease (AD) (www.alzforum.org). The CY46A1-AD link, however, has not yet been unambiguously proven [4]. Significance of CYP46A1 may extend beyond its involvement in bile acid biosyhthesis. Experiments with cell cultures and mice suggest that the product of CYP46A1 activities 24(S)-hydroxycholesterol is the endogenous ligand for LXR, master transcriptional factors that regulate many genes involved in cholesterol homeostasis including ABC transporters, apolipoproteins and HDL modifying enzymes [88, 89]. Evidence is also provided that 24(S)-hydroxycholesterol induces apoE-mediated efflux of cholesterol in astrocytes via an LXR-controlled pathway and, thus, may affect the progression of neurodegeneration [90]. In addition to this regulatory function, cell culture studies imply that 24(S)-hydroxycholesterol may inhibit the formation of amyloid β-peptide [91, 92], a molecule that self-aggregates and forms extracellular amyloid plaques in the brain of AD-affected people. Unlike CYPs 7A1 and 27A1, regulation of CYP46A1 does not seem to be significantly controlled at transcriptional level [93], although recent studies suggest a role of the Sp family of transcriptional factors in CYP46A1 regulation [94]. Protein expression of CYP46A1 and plasma levels of 24(S)-hydroxycholesterol are highly stable in adults [93].
Recently, structures of substrate-free and substrate-bound forms of CYP46A1 have been determined by X-ray crystallography [95]. The active site of CYP46A1 was found to be conformationally flexible with substrate binding eliciting an induced fit (Fig. 1) [95]. This conformational flexibility as well as prior in vitro studies demonstrating that a number of structurally distinct compounds can be metabolized by CYP46A1 [96] prompted an evaluation of whether some marketed drugs can inhibit CYP46A1-mediated cholesterol hydroxylation. These experiments were carried out in vitro using purified recombinant CYP46A1 [95]. Of ~50 drugs tested, seven were found to have a significant inhibitory effect under the experimental conditions used (Table 1). Of these seven, four were CNS active drugs that cross the blood-brain barrier. Based on subsequent estimation of the binding affinities of these drugs and knowledge of pharmacokinetics, CYP46A1 was concluded to have potential to be inhibited by some marketed drugs [95]. Further studies, utilizing CYP46A1-containing human brain microsomes and then, if the inhibition is confirmed, tests on animals, are required to clarify whether CYP46A1 can indeed be an off-site target for some of the pharmaceuticals. Screening of the drugs has also led to an unanticipated finding. Two structurally similar compounds, nonsteroidal anti-inflammatory drugs phenacetin and acetaminophen, were found to modestly, up to 35%, stimulate CYP46A1 activity [95]. The mechanism of this stimulation is currently not clear. One of the possibilities is that stimulation occurs via simultaneous binding of cholesterol and the co-activator molecule in the CYP46A1 active site [95]. This putative mechanism is proposed based on the previous studies in the P450 field investigating the stimulatory effects of the antibacterial compound dapsone on a drug-metabolizing P450 2C9 [97–99]. Although a larger stimulation of CYP46A1 would probably be required to significantly affect cholesterol turnover in the brain, the data obtained warrant further investigation. In-silico and high-throughput screening will be conducted in the author’s laboratory to identify more potent CYP46A1 co-activators. While, it remains to be determined whether such compounds will be identified, the example of CYP2C9 indicates that this goal is reasonable. Hydroxylation of phenytoin by CYP2C9 in vitro has been found to be activated by lansoprazole ~8 fold [100]. Both lansoprazole and phenytoin are marketed drugs. To manipulate CYP46A1 activity, an understanding of how cholesterol and inhibitor/co-activator enter the enzyme active site will also likely be required. All eukaryotic P450s including CYP46A1 are membrane-bound proteins residing either in the endoplasmic reticulum or mitochondrion. The active site in membrane-bound P450s is not located on the surface of the molecule but buried inside the enzyme and connected to the surface by the substrate access channel [101]. Studies in the P450 field suggest that in some P450s the entrance to the substrate access channel is embedded in the lipid bilayer and hydrophobic substrates enter the P450 directly from the membrane [102]. We investigated membrane topology of CYP46A1 and cholesterol access to the enzyme active site and obtained experimental evidence supporting this notion [103]. However, if cholesterol comes from the membrane, How do drugs that are less hydrophobic than cholesterol reach the P450 active site? Crystal structures of CYP46A1 may provide some insight. We analyzed them for the presence of channels connecting the protein surface and enzyme active site. In both substrate-free and substrate bound CYP46A1 structures there is a substrate access channel (shown in blue in Fig. 2), and in both structures it branches near the surface of the molecule. Most of the branching in substrate-free CYP46A1 is likely an artifact because the openings on the surface that initiate this branching are defined in part by the truncated or unmodeld part of the molecule (for clarity, these branches are omitted in Fig. 2). In substrate-bound CYP46A1, however, one of the branches could be real and deserves consideration because it is formed as a result of conformational changes occurring upon substrate binding (shown in light blue in Fig. 2B). In addition to the substrate access channel, there is also a second channel in both CYP46A1 structures. In substrate-free structure, this channel is located on the same side of the molecule where the substrate access channel is. However, unlike the substrate access channel, this second channel does not appear to be embedded in the membrane (shown in green in Fig. 2A) and could be the route whereby different drugs reach the enzyme active site. This channel is closed in substrate-bound CYP46A1 structure, and, instead, a channel beginning on the cytosolic or proximal side of the molecule is opened (shown in orange in Fig. 2B). This proximal channel is filled with a network of hydrogen-bonded water molecules and could play a role in the mechanism of water and proton delivery to the active site of CYP46A1 during the catalysis. Thus, crystallographic studies and studies of CYP46A1 inhibition, membrane topology and substrate access are all in a good agreement and suggest that CYP46A1 activity could indeed be altered (increased or reduced) by exposure to some of the pharmaceuticals.
Fig. 1.

Active site (shown in yellow) of the substrate-free (A) and substrate-bound (B) CYP46A1. The substrate-free form is colored in grey and substrate-bound form is in cyan. Solvent accessible surface of the active site calculated by VOIDOO (http://xray.bmc.uu.se/usf/voidoo.html). The heme group is in pink and substrate (cholesterol sulfate) is in blue except the sulfur and oxygen atoms which are in orange and red, respectively.
Table 1.
Binding and effect of different drugs on cholesterol hydroxylase activity of recombinant CYP46A11
| Compound (indication or use) | Structure | Cholesterol hydroxylation, % | Kd, nM (IC50, nM) |
|---|---|---|---|
| No additive Ranitidine (H2 receptor-antagonist, antiulcerative) |
100±4 9±2 |
NSR2 | |
| Clotrimazole (CYP51 inhibitor, antifungal) | 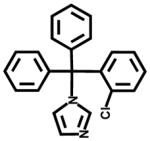 |
16±3 | 11±2 |
| Cimetidine (H2 receptor-antagonist, antiulcerative) | 8±2 | NSR | |
| Clobenpropit (H3 receptor-antagonist, experimental anticonvulsant) | ND3 | 80±9 | |
| Thioperamide (H3-/H4 receptor-antagonist, experimental anticonvulsant) | 34±4 | 50±10 | |
| Tranylcypromine (monoamine oxidase inhibitor, antidepressant) | 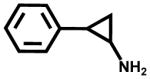 |
ND | 7±2 |
| Selegiline (monoamine oxidase B inhibitor, antidepressant) | 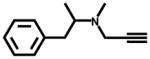 |
18±3 | 63,000 ±3,500 (140±11) |
| Phenacetin (analgesic) | 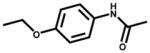 |
135±5 | NSR |
| Acetaminophen (analgesic) | 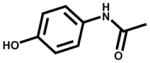 |
132±4 | NSR |
This data are compiled from ref. 99.
NSR, no spectral response. The Kd value was not determined because compound under study did not induce spectral response.
ND, not detectable.
Fig. 2.

Channels in substrate-free (A) and substrate-bound (B) CYP46A1 as identified by Caver (http://loschmidt.chemi.muni.cz/caver/online.php). The substrate-free form is colored in grey and substrate-bound form is in cyan. The heme group is in pink. In both structures, the substrate access channel is shown in blue, and additional channels are colored in green in substrate-free and in orange in substrate-bound CYP46A1. Branching of the substrate access channel in substrate-bound CYP46A1 is shown in light blue. The black line is drawn to illustrate the surface of the membrane.
One may wonder, Why maintenance of cholesterol homeostasis in the brain is so important if the brain does not contribute significantly to the whole-body cholesterol balance? The reasons is that the brain is highly enriched in cholesterol compared with other tissues, and tight regulation of CNS cholesterol is required to maintain membrane integrity of nerve cells and their proper function. Development of neurological disorder such as Alzheimer’s disease appears to be associated with disruption of cholesterol homeostasis in the brain [79, 104–107], and cholesterol reduction may represent an important pharmacological gateway to amyloid β modulation. While statins seem to be the first choice of drugs, there has been persistent controversy regarding possible favorable or adverse effects of statins on cognition, mood and behavior (including aggressive or violent behavior). The possible underlying problem is that statins inhibit the formation of compounds other than sterols, and these compounds may be important for memory and learning [87]. To ascertain the effect on cognitive function as well as to clarify some other non-cardiac issues, the University of California-San Diego and National Institutes of Health initiated a randomized double-blind, placebo-controlled statin study [108]. The results of this study should be reported shortly. Yet, drugs other than statins should be made available on the market to prevent cholesterol accumulation in the brain.
5. Expert opinion
P450s 7A1, 27A1, and 46A1 are important enzymes controlling cholesterol levels in the periphery and brain. They certainly have a potential to serve as targets for cholesterol lowering. However, development of drugs affecting the activity of cholesterol-metabolizing P450s is a challenging goal. First, P450s 7A1, 27A1, and 46A1 are not the only P450 enzymes expressed in humans; there are 54 other human isoforms that have distinct functions and substrate specificities [109]. Despite differences, P450s have a similar overall fold and active site formed by the same set of secondary structural elements [101]. Because of the structural similarities, designing of a very selective P450 drug is difficult and requires knowledge of the enzyme crystal structure. Determination of substrate-bound and substrate-free crystal structures of CYPs 7A1 and 27A1 will undoubtedly improve chances of these P450s to become real drug targets. The second challenge is that drugs targeted at cholesterol-metabolizing P450s should stimulate, not inhibit, the enzyme activity. In principal, stimulation could be accomplished either at transcriptional or post-translational levels. Significant efforts are currently focused on studies of nuclear receptors that control transcription of many proteins involved in lipid metabolism [7, 89, 110, 111]. In contrast, development of approaches to stimulate P450 activity post-translationally is not intensively pursued. The author’s laboratory is investigating this by trying to establish an optimal composition of fat in our diet and specifically a ratio of n-3 to n-6 polyunsaturated fatty acids which maximally stimulates cholesterol degradation and has a strong overall hypocholesterolemic effect [3, 4].
Of the three P450s, 7A1, 27A1, and 46A1, the latter is currently in the best position to become a real drug target. Crystal structures of CYP46A1 are determined, highly encouraging preliminary data is obtained, and experiments elucidating the physiological relevance of the in vitro findings are underway. The example of CYP46A1 proves one more time that the more an enzyme is studied, the higher the chances are that something unexpected and having a pharmacological relevance will be discovered. Investigations of cholesterol-metabolizing P450s may lead to new therapeutic opportunities in cholesterol-lowering and should certainly be continued.
Acknowledgments
Studies in the author’s laboratory described in this paper are supported by National Institutes of Health Grants GM62882, AG024336, EY018383 and by the grant from Merck Research Laboratories
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.BJORKHEM I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260(6):493–508. doi: 10.1111/j.1365-2796.2006.01725.x. [DOI] [PubMed] [Google Scholar]
- 2.NORLIN M, WIKVALL K. Enzymes in the conversion of cholesterol into bile acids. Curr Mol Med. 2007;7(2):199–218. doi: 10.2174/156652407780059168. [DOI] [PubMed] [Google Scholar]
- 3•.PIKULEVA IA. Cholesterol-metabolizing cytochromes P450. Drug Metab Dispos. 2006;34(4):513–520. doi: 10.1124/dmd.105.008789. A summary of medical significance and structure/function characterization of cholesterol-metabolizing cytochormes P450 7A1, 27A1, 11A1 and 46A1. [DOI] [PubMed] [Google Scholar]
- 4•.PIKULEVA IA. Cytochrome P450s and cholesterol homeostasis. Pharmacol Ther. 2006;112:761–773. doi: 10.1016/j.pharmthera.2006.05.014. A summary of physiological roles, medical significance and biochemical properties of nine cytochome P450 enzymes that participate in cholesterol biosynthesis and metabolism. [DOI] [PubMed] [Google Scholar]
- 5•.RUSSELL DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. A comprehensive review of the regulation and genetics of bile acid biosynthesis. [DOI] [PubMed] [Google Scholar]
- 6•.CHIANG JY. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J Hepatol. 2004;40(3):539–551. doi: 10.1016/j.jhep.2003.11.006. A comprehensive review of the regulation of bile acid biosynthesis. [DOI] [PubMed] [Google Scholar]
- 7•.REPA JJ, MANGELSDORF DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. 2000;16:459–481. doi: 10.1146/annurev.cellbio.16.1.459. A comprehensive review of orphan nuclear receptors. [DOI] [PubMed] [Google Scholar]
- 8•.Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143–3421. Updated US guidelines for lipid lowering targets. [PubMed] [Google Scholar]
- 9.GRUNDY SM, CLEEMAN JI, MERZ CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110(2):227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- 10.BODEN WE. High-density lipoprotein cholesterol as an independent risk factor in cardiovascular disease: assessing the data from Framingham to the Veterans Affairs High--Density Lipoprotein Intervention Trial. Am J Cardiol. 2000;86(12A):19L–22L. doi: 10.1016/s0002-9149(00)01464-8. [DOI] [PubMed] [Google Scholar]
- 11.GORDON T, CASTELLI WP, HJORTLAND MC, KANNEL WB, DAWBER TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977;62(5):707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 12.ROBINS SJ, RUBINS HB, FAAS FH, et al. Insulin resistance and cardiovascular events with low HDL cholesterol: the Veterans Affairs HDL Intervention Trial (VA-HIT) Diabetes Care. 2003;26(5):1513–1517. doi: 10.2337/diacare.26.5.1513. [DOI] [PubMed] [Google Scholar]
- 13.FORRESTER JS, SHAH PK. Emerging strategies for increasing high-density lipoprotein. Am J Cardiol. 2006;98(11):1542–1549. doi: 10.1016/j.amjcard.2006.06.059. [DOI] [PubMed] [Google Scholar]
- 14.HOKANSON JE, AUSTIN MA. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk. 1996;3(2):213–219. [PubMed] [Google Scholar]
- 15.BILHEIMER DW, GRUNDY SM, BROWN MS, GOLDSTEIN JL. Mevinolin and colestipol stimulate receptor-mediated clearance of low density lipoprotein from plasma in familial hypercholesterolemia heterozygotes. Proc Natl Acad Sci U S A. 1983;80(13):4124–4128. doi: 10.1073/pnas.80.13.4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DAVIDSON MH, TOTH PP. Comparative effects of lipid-lowering therapies. Prog Cardiovasc Dis. 2004;47(2):73–104. doi: 10.1016/j.pcad.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 17.LIAO JK, LAUFS U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.ARMITAGE J. The safety of statins in clinical practice. Lancet. 2007;370(9601):1781–1790. doi: 10.1016/S0140-6736(07)60716-8. [DOI] [PubMed] [Google Scholar]
- 19.SHEPHERD J, PACKARD CJ, BICKER S, LAWRIE TD, MORGAN HG. Cholestyramine promotes receptor-mediated low-density-lipoprotein catabolism. N Engl J Med. 1980;302(22):1219–1222. doi: 10.1056/NEJM198005293022202. [DOI] [PubMed] [Google Scholar]
- 20.STEINMETZ KL. Colesevelam hydrochloride. Am J Health Syst Pharm. 2002;59 (10):932–939. doi: 10.1093/ajhp/59.10.932. [DOI] [PubMed] [Google Scholar]
- 21.KNOPP RH. Drug treatment of lipid disorders. N Engl J Med. 1999;341(7):498–511. doi: 10.1056/NEJM199908123410707. [DOI] [PubMed] [Google Scholar]
- 22.BAYS HE, NEFF D, TOMASSINI JE, TERSHAKOVEC AM. Ezetimibe: cholesterol lowering and beyond. Expert Rev Cardiovasc Ther. 2008;6(4):447–470. doi: 10.1586/14779072.6.4.447. [DOI] [PubMed] [Google Scholar]
- 23.SUDHOP T, LUTJOHANN D, KODAL A, et al. Inhibition of intestinal cholesterol absorption by ezetimibe in humans. Circulation. 2002;106(15):1943–1948. doi: 10.1161/01.cir.0000034044.95911.dc. [DOI] [PubMed] [Google Scholar]
- 24.FLORENTIN M, LIBEROPOULOS EN, ELISAF MS. Ezetimibe-associated adverse effects: what the clinician needs to know. Int J Clin Pract. 2008;62(1):88–96. doi: 10.1111/j.1742-1241.2007.01592.x. [DOI] [PubMed] [Google Scholar]
- 25.MALIK S, KASHYAP ML. Niacin, lipids, and heart disease. Curr Cardiol Rep. 2003;5(6):470–476. doi: 10.1007/s11886-003-0109-x. [DOI] [PubMed] [Google Scholar]
- 26.GILLE A, BODOR ET, AHMED K, OFFERMANNS S. Nicotinic acid: pharmacological effects and mechanisms of action. Annu Rev Pharmacol Toxicol. 2008;48:79–106. doi: 10.1146/annurev.pharmtox.48.113006.094746. [DOI] [PubMed] [Google Scholar]
- 27.BROWN BG, ZHAO XQ, CHAIT A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345(22):1583–1592. doi: 10.1056/NEJMoa011090. [DOI] [PubMed] [Google Scholar]
- 28.BAYS H. Safety of niacin and simvastatin combination therapy. Am J Cardiol. 2008;101(8A):3B–8B. doi: 10.1016/j.amjcard.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 29.VENKATESH PK, CASKEY D, REDDY PC. Therapies to increase high-density lipoprotein cholesterol and their effect on cardiovascular outcomes and regression of atherosclerosis. Am J Med Sci. 2008;336(1):64–68. doi: 10.1097/MAJ.0b013e31815d4419. [DOI] [PubMed] [Google Scholar]
- 30.GOLDENBERG I, BENDERLY M, GOLDBOURT U. Update on the use of fibrates: focus on bezafibrate. Vasc Health Risk Manag. 2008;4(1):131–141. doi: 10.2147/vhrm.2008.04.01.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.STAELS B, DALLONGEVILLE J, AUWERX J, et al. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98(19):2088–2093. doi: 10.1161/01.cir.98.19.2088. [DOI] [PubMed] [Google Scholar]
- 32.CHAPMAN MJ. Fibrates in 2003: therapeutic action in atherogenic dyslipidaemia and future perspectives. Atherosclerosis. 2003;171(1):1–13. doi: 10.1016/s0021-9150(03)00156-4. [DOI] [PubMed] [Google Scholar]
- 33.BACKES JM, GIBSON CA, HOWARD PA. Optimal lipid modification: the rationale for combination therapy. Vasc Health Risk Manag. 2005;1(4):317–331. doi: 10.2147/vhrm.2005.1.4.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MEUWESE MC, FRANSSEN R, STROES ES, KASTELEIN JJ. And then there were acyl coenzyme A:cholesterol acyl transferase inhibitors. Curr Opin Lipidol. 2006;17(4):426–430. doi: 10.1097/01.mol.0000236369.50378.6e. [DOI] [PubMed] [Google Scholar]
- 35.VAN LEUVEN SI, STROES ES, KASTELEIN JJ. High-density lipoprotein: A fall from grace? Ann Med. 2008:1–10. doi: 10.1080/07853890802082104. [DOI] [PubMed] [Google Scholar]
- 36.ELSAYED RK, EVANS JD. Emerging lipid-lowering drugs: squalene synthase inhibitors. Expert Opin Emerg Drugs. 2008;13(2):309–322. doi: 10.1517/14728214.13.2.309. [DOI] [PubMed] [Google Scholar]
- 37.BARTER PJ, CAULFIELD M, ERIKSSON M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 38.MYANT NB, MITROPOULOS KA. Cholesterol 7a-hydroxylase. J Lipid Res. 1977;18:135–153. [PubMed] [Google Scholar]
- 39.PULLINGER CR, ENG C, SALEN G, et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110(1):109–117. doi: 10.1172/JCI15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.BJORKHEM I, REIHNER E, ANGELIN B, et al. On the possible use of the serum level of 7 alpha-hydroxycholesterol as a marker for increased activity of the cholesterol 7 alpha-hydroxylase in humans. J Lipid Res. 1987;28(8):889–894. [PubMed] [Google Scholar]
- 41.NICOLAU VA, SHEFER S, SALEN G, MOSBACH EH. Determination of hepatic cholesterol 7a-hydroxylase activity in man. J Lipid Res. 1974;15:146–151. [PubMed] [Google Scholar]
- 42.REIHNER E, ANGELIN B, RUDLING M, et al. Regulation of hepatic cholesterol metabolism in humans: stimulatory effects of cholestyramine on HMG-CoA reductase activity and low density lipoprotein receptor expression in gallstone patients. J Lipid Res. 1990;31 (12):2219–2226. [PubMed] [Google Scholar]
- 43.ODA H, YAMASHITA H, KOSAHARA K, KUROKI S, NAKAYAMA F. Esterified and total 7 alpha-hydroxycholesterol in human serum as an indicator for hepatic bile acid synthesis. J Lipid Res. 1990;31(12):2209–2218. [PubMed] [Google Scholar]
- 44.HUBACEK JA, BOBKOVA D. Role of cholesterol 7alpha-hydroxylase (CYP7A1) in nutrigenetics and pharmacogenetics of cholesterol lowering. Mol Diagn Ther. 2006;10(2):93–100. doi: 10.1007/BF03256448. [DOI] [PubMed] [Google Scholar]
- 45.BERTOLOTTI M, ABATE N, BERTOLOTTI S, et al. Effect of aging on cholesterol 7 alpha-hydroxylation in humans. J Lipid Res. 1993;34(6):1001–1007. [PubMed] [Google Scholar]
- 46.CHEEMA SK, CIKALUK D, AGELLON LB. Dietary fats modulate the regulatory potential of dietary cholesterol on cholesterol 7 alpha-hydroxylase gene expression. J Lipid Res. 1997;38 (2):315–323. [PubMed] [Google Scholar]
- 47.KURUSHIMA H, HAYASHI K, SHINGU T, et al. Opposite effects on cholesterol metabolism and their mechanisms induced by dietary oleic acid and palmitic acid in hamsters. Biochim Biophys Acta. 1995;1258(3):251–256. doi: 10.1016/0005-2760(95)00122-s. [DOI] [PubMed] [Google Scholar]
- 48.BRAVO E, FLORA L, CANTAFORA A, et al. The influence of dietary saturated and unsaturated fat on hepatic cholesterol metabolism and the biliary excretion of chylomicron cholesterol in the rat. Biochim Biophys Acta. 1998;1390(2):134–148. doi: 10.1016/s0005-2760(97)00174-4. [DOI] [PubMed] [Google Scholar]
- 49.LAKSHMANAN MR, VEECH RL. Short- and long-term effects of ethanol administration in vivo on rat liver HMG-CoA reductase and cholesterol 7alpha-hydroxylase activities. J Lipid Res. 1977;18(3):325–330. [PubMed] [Google Scholar]
- 50.STAHLBERG D, RUDLING M, ANGELIN B, et al. Hepatic cholesterol metabolism in human obesity. Hepatology. 1997;25(6):1447–1450. doi: 10.1002/hep.510250623. [DOI] [PubMed] [Google Scholar]
- 51.WANG J, FREEMAN DJ, GRUNDY SM, et al. Linkage between cholesterol 7alpha-hydroxylase and high plasma low-density lipoprotein cholesterol concentrations. J Clin Invest. 1998;101(6):1283–1291. doi: 10.1172/JCI1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.COUTURE P, OTVOS JD, CUPPLES LA, et al. Association of the A-204C polymorphism in the cholesterol 7alpha-hydroxylase gene with variations in plasma low density lipoprotein cholesterol levels in the Framingham Offspring Study. J Lipid Res. 1999;40(10):1883–1889. [PubMed] [Google Scholar]
- 53.HOFMAN MK, GROENENDIJK M, VERKUIJLEN PJJH, et al. Modulating effect of the A-278 promoter polymorphism in the cholesterol 7alpha-hydroxylase gene on serum lipid levels in normolipidaemic and hypertriglyceridaemic individuals. Eur J Hum Genet. 2004;12:935–941. doi: 10.1038/sj.ejhg.5201236. [DOI] [PubMed] [Google Scholar]
- 54.ABRAHAMSSON A, KRAPIVNER S, GUSTAFSSON U, et al. Common polymorphisms in the CYP7A1 gene do not contribute to variation in rates of bile acid synthesis and plasma LDL cholesterol concentration. Atherosclerosis. 2005;182:37–45. doi: 10.1016/j.atherosclerosis.2005.01.032. [DOI] [PubMed] [Google Scholar]
- 55.HEGELE RA, WANG J, HARRIS SB, et al. Variable association between genetic variation in the CYP7 gene promoter and plasma lipoproteins in three Canadian populations. Atherosclerosis. 2001;154(3):579–587. doi: 10.1016/s0021-9150(00)00419-6. [DOI] [PubMed] [Google Scholar]
- 56.HOFMAN MK, WEGGEMANS RM, ZOCK PL, et al. CYP7A1 A-278C polymorphism affects the response of plasma lipids after dietary cholesterol or cafestol interventions in humans. J Nutr Biochem. 2004;134:2200–2204. doi: 10.1093/jn/134.9.2200. [DOI] [PubMed] [Google Scholar]
- 57.KOVAR J, SUCHANEK P, HUBACEK JA, POLEDNE R. The A-204C polymorphism in the cholesterol 7alpha-hydroxylase (CYP7A1) gene determines the cholesterolemia responsiveness to a high-fat diet. Physiol Res. 2004;53(5):565–568. [PubMed] [Google Scholar]
- 58.KAJINAMI K, BROUSSEAU ME, ORDOVAS JM, SCHAEFER EJ. Interactions between common genetic polymorphisms in ABCG5/G8 and CYP7A1 on LDL cholesterol-lowering response to atorvastatin. Atherosclerosis. 2004;175(2):287–293. doi: 10.1016/j.atherosclerosis.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 59.KAJINAMI K, BROUSSEAU ME, ORDOVAS JM, SCHAEFER EJ. A promoter polymorphism in cholesterol 7alpha-hydroxylase interacts with apolipoprotein E genotype in the LDL-lowering response to atorvastatin. Atherosclerosis. 2005;180(2):407–415. doi: 10.1016/j.atherosclerosis.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 60.EINARSSON K, ANGELIN B, EWERTH S, NILSELL K, BJORKHEM I. Bile acid synthesis in man: assay of hepatic microsomal cholesterol 7 alpha-hydroxylase activity by isotope dilution-mass spectrometry. J Lipid Res. 1986;27(1):82–88. [PubMed] [Google Scholar]
- 61.AXELSON M, BJORKHEM I, REIHNER E, EINARSSON K. The plasma level of 7 alpha-hydroxy-4-cholesten-3-one reflects the activity of hepatic cholesterol 7 alpha-hydroxylase in man. FEBS Lett. 1991;284(2):216–218. doi: 10.1016/0014-5793(91)80688-y. [DOI] [PubMed] [Google Scholar]
- 62.EUSUFZAI S, AXELSON M, ANGELIN B, EINARSSON K. Serum 7 alpha-hydroxy-4-cholesten-3-one concentrations in the evaluation of bile acid malabsorption in patients with diarrhoea: correlation to SeHCAT test. Gut. 1993;34(5):698–701. doi: 10.1136/gut.34.5.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.SAUTER GH, MUNZING W, VON RITTER C, PAUMGARTNER G. Bile acid malabsorption as a cause of chronic diarrhea: diagnostic value of 7alpha-hydroxy-4-cholesten-3-one in serum. Dig Dis Sci. 1999;44(1):14–19. doi: 10.1023/a:1026681512303. [DOI] [PubMed] [Google Scholar]
- 64.GALMAN C, ARVIDSSON I, ANGELIN B, RUDLING M. Monitoring hepatic cholesterol 7alpha-hydroxylase activity by assay of the stable bile acid intermediate 7alpha-hydroxy-4-cholesten-3-one in peripheral blood. J Lipid Res. 2003;44(4):859–866. doi: 10.1194/jlr.D200043-JLR200. [DOI] [PubMed] [Google Scholar]
- 65.LOVGREN-SANDBLOM A, HEVERIN M, LARSSON H, et al. Novel LC-MS/MS method for assay of 7alpha-hydroxy-4-cholesten-3-one in human plasma. Evidence for a significant extrahepatic metabolism. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;856(1–2):15–19. doi: 10.1016/j.jchromb.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 66.DUANE WC, JAVITT NB. 27-hydroxycholesterol: production rates in normal human subjects. J Lipid Res. 1999;40(7):1194–1199. [PubMed] [Google Scholar]
- 67.LUND E, ANDERSSON O, ZHANG J, et al. Importance of a novel oxidative mechanism for elimination of intracellular cholesterol in humans. Arterioscler Thromb Vasc Biol. 1996;16 (2):208–212. doi: 10.1161/01.atv.16.2.208. [DOI] [PubMed] [Google Scholar]
- 68.BABIKER A, ANDERSSON O, LUND E, et al. Elimination of cholesterol in macrophages and endothelial cells by the sterol 27-hydroxylase mechanism. Comparison with high density lipoprotein-mediated reverse cholesterol transport. J Biol Chem. 1997;272(42):26253–26261. doi: 10.1074/jbc.272.42.26253. [DOI] [PubMed] [Google Scholar]
- 69.SONG C, LIAO S. Cholestenoic acid is a naturally occurring ligand for liver X receptor alpha. Endocrinology. 2000;141(11):4180–4184. doi: 10.1210/endo.141.11.7772. [DOI] [PubMed] [Google Scholar]
- 70.FU X, MENKE JG, CHEN Y, et al. 27-hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells. J Biol Chem. 2001;276(42):38378–38387. doi: 10.1074/jbc.M105805200. [DOI] [PubMed] [Google Scholar]
- 71.BJORKHEM I, DICZFALUSY U. Oxysterols: friends, foes, or just fellow passengers? Arterioscler Thromb Vasc Biol. 2002;22(5):734–742. doi: 10.1161/01.atv.0000013312.32196.49. [DOI] [PubMed] [Google Scholar]
- 72.JAVITT NB. 25R,26-Hydroxycholesterol revisited: synthesis, metabolism, and biologic roles. J Lipid Res. 2002;43(5):665–670. [PubMed] [Google Scholar]
- 73.BJORKHEM I, BOBERG KM, LEITERSDORF E. Inborn errors in bile acid biosynthesis and storage of sterols other than cholesterol. In: Scriver AL, Beaudet AL, Sly WS, Valle D, Childs B, editors. The metabolic and molecular bases of inherited disease. 7. New York: McGraw-Hill; 1995. pp. 2073–2099. [Google Scholar]
- 74.LORINCZ MT, RAINIER S, THOMAS D, FINK JK. Cerebrotendinous xanthomatosis: possible higher prevalence than previously recognized. Arch Neurol. 2005;62(9):1459–1463. doi: 10.1001/archneur.62.9.1459. [DOI] [PubMed] [Google Scholar]
- 75.GENEST J, JR, MCNAMARA JR, ORDOVAS JM, et al. Lipoprotein cholesterol, apolipoprotein A-I and B and lipoprotein (a) abnormalities in men with premature coronary artery disease. J Am Coll Cardiol. 1992;19(4):792–802. doi: 10.1016/0735-1097(92)90520-w. [DOI] [PubMed] [Google Scholar]
- 76.SHUKLA A, SHARMA MK, JAIN A, GOEL PK. Prevention of atherosclerosis progression using atorvastatin in normolipidemic coronary artery disease patients--a controlled randomized trial. Indian Heart J. 2005;57(6):675–680. [PubMed] [Google Scholar]
- 77.CHEN W, CHIANG JY. Regulation of human sterol 27-hydroxylase gene (CYP27A1) by bile acids and hepatocyte nuclear factor 4alpha (HNF4alpha) Gene. 2003;313:71–82. doi: 10.1016/s0378-1119(03)00631-0. [DOI] [PubMed] [Google Scholar]
- 78.ELLIS E, AXELSON M, ABRAHAMSSON A, et al. Feedback regulation of bile acid synthesis in primary human hepatocytes: evidence that CDCA is the strongest inhibitor. Hepatology. 2003;38(4):930–938. doi: 10.1053/jhep.2003.50394. [DOI] [PubMed] [Google Scholar]
- 79.DIETSCHY JM, TURLEY SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12(2):105–112. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 80.PFRIEGER FW. Outsourcing in the brain: do neurons depend on cholesterol delivery by astrocytes? Bioessays. 2003;25(1):72–78. doi: 10.1002/bies.10195. [DOI] [PubMed] [Google Scholar]
- 81.VANCE JE, HAYASHI H, KARTEN B. Cholesterol homeostasis in neurons and glial cells. Semin Cell Dev Biol. 2005;16(2):193–212. doi: 10.1016/j.semcdb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 82.BJORKHEM I, LUTJOHANN D, DICZFALUSY U, et al. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res. 1998;39(8):1594–1600. [PubMed] [Google Scholar]
- 83.LUTJOHANN D, BREUER O, AHLBORG G, et al. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci U S A. 1996;93(18):9799–9804. doi: 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.LUND EG, GUILEYARDO JM, RUSSELL DW. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci U S A. 1999;96(13):7238–7243. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.BJORKHEM I, ANDERSSON U, ELLIS E, et al. From brain to bile. Evidence that conjugation and omega-hydroxylation are important for elimination of 24S-hydroxycholesterol (cerebrosterol) in humans. J Biol Chem. 2001;276(40):37004–37010. doi: 10.1074/jbc.M103828200. [DOI] [PubMed] [Google Scholar]
- 86.HEVERIN M, BOGDANOVIC N, LUTJOHANN D, et al. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J Lipid Res. 2004;45 (1):186–193. doi: 10.1194/jlr.M300320-JLR200. [DOI] [PubMed] [Google Scholar]
- 87••.KOTTI TJ, RAMIREZ DM, PFEIFFER BE, HUBER KM, RUSSELL DW. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc Natl Acad Sci U S A. 2006;103(10):3869–3874. doi: 10.1073/pnas.0600316103. The landmark paper describing that CYP46A1 knockout mice exhibit severe deficiencies in spatial, associative, and motor learning. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.CHEN W, CHEN G, HEAD DL, MANGELSDORF DJ, RUSSELL DW. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab. 2007;5(1):73–79. doi: 10.1016/j.cmet.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.CAO G, BALES KR, DEMATTOS RB, PAUL SM. Liver X receptor-mediated gene regulation and cholesterol homeostasis in brain: relevance to Alzheimer’s disease therapeutics. Curr Alzheimer Res. 2007;4(2):179–184. doi: 10.2174/156720507780362173. [DOI] [PubMed] [Google Scholar]
- 90.ABILDAYEVA K, JANSEN PJ, HIRSCH-REINSHAGEN V, et al. 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J Biol Chem. 2006;281(18):12799–12808. doi: 10.1074/jbc.M601019200. [DOI] [PubMed] [Google Scholar]
- 91.BROWN J, 3RD, THEISLER C, SILBERMAN S, et al. Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J Biol Chem. 2004;279(33):34674–34681. doi: 10.1074/jbc.M402324200. [DOI] [PubMed] [Google Scholar]
- 92.FAMER D, MEANEY S, MOUSAVI M, et al. Regulation of alpha- and beta-secretase activity by oxysterols: cerebrosterol stimulates processing of APP via the alpha-secretase pathway. Biochem Biophys Res Commun. 2007;359(1):46–50. doi: 10.1016/j.bbrc.2007.05.033. [DOI] [PubMed] [Google Scholar]
- 93.OHYAMA Y, MEANEY S, HEVERIN M, et al. Studies on the transcriptional regulation of cholesterol 24-hydroxylase (CYP46A1): marked insensitivity toward different regulatory axes. J Biol Chem. 2006;281(7):3810–3820. doi: 10.1074/jbc.M505179200. [DOI] [PubMed] [Google Scholar]
- 94.MILAGRE I, NUNES MJ, GAMA MJ, et al. Transcriptional regulation of the human CYP46A1 brain-specific expression by Sp transcription factors. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05442.x. [DOI] [PubMed] [Google Scholar]
- 95.MAST N, WHITE MA, BJORKHEM I, et al. Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc Natl Acad Sci U S A. 2008;105(28):9546–9551. doi: 10.1073/pnas.0803717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.MAST N, NORCROSS R, ANDERSSON U, et al. Broad substrate specificity of human cytochrome P450 46A1 which initiates cholesterol degradation in the brain. Biochemistry. 2003;42 (48):14284–14292. doi: 10.1021/bi035512f. [DOI] [PubMed] [Google Scholar]
- 97.HUTZLER JM, HAUER MJ, TRACY TS. Dapsone activation of CYP2C9-mediated metabolism: evidence for activation of multiple substrates and a two-site model. Drug Metab Dispos. 2001;29(7):1029–1034. [PubMed] [Google Scholar]
- 98.WESTER MR, YANO JK, SCHOCH GA, et al. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-A resolution. J Biol Chem. 2004;279(34):35630–35637. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]
- 99.LOCUSON CW, GANNETT PM, AYSCUE R, TRACY TS. Use of simple docking methods to screen a virtual library for heteroactivators of cytochrome P450 2C9. J Med Chem. 2007;50 (6):1158–1165. doi: 10.1021/jm060706p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.LIU KH, KIM MJ, JUNG WM, et al. Lansoprazole enantiomer activates human liver microsomal CYP2C9 catalytic activity in a stereospecific and substrate-specific manner. Drug Metab Dispos. 2005;33(2):209–213. doi: 10.1124/dmd.104.001438. [DOI] [PubMed] [Google Scholar]
- 101••.POULOS TL, JOHNSON EF. Structures of Cytochrome P450 Enzymes. In: Montellano PROd., editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. New York: Kluwer Academic/Plenum Publishers; 2005. pp. 87–114. A summary and analysis of crystallographic studies of different P450s. [Google Scholar]
- 102••.WILLIAMS PA, COSME J, SRIDHAR V, JOHNSON EF, MCREE DE. Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell. 2000;5(1):121–131. doi: 10.1016/s1097-2765(00)80408-6. This paper describes the first crystal structure of the membrane-bound P450 CYP2C5. [DOI] [PubMed] [Google Scholar]
- 103.MAST N, LIAO W-L, PIKULEVA IA, TURKO IV. Mass spectrometry identification of membrane-interacting peptides suggests membrane topology of cytochrome P450 46A1 and NADPH-cytochrome P450 oxidoreductase. Biochemistry. 2008 doi: 10.1016/j.abb.2009.01.002. (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.BJORKHEM I, MEANEY S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24(5):806–815. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- 105.WOLOZIN B. Cholesterol and the biology of Alzheimer’s disease. Neuron. 2004;41 (1):7–10. doi: 10.1016/s0896-6273(03)00840-7. [DOI] [PubMed] [Google Scholar]
- 106.REISS AB. Cholesterol and apolipoprotein E in Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2005;20(2):91–96. doi: 10.1177/153331750502000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.CARTER CJ. Convergence of genes implicated in Alzheimer’s disease on the cerebral cholesterol shuttle: APP, cholesterol, lipoproteins, and atherosclerosis. Neurochem Int. 2007;50 (1):12–38. doi: 10.1016/j.neuint.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 108.GOLOMB BA, CRIQUI MH, WHITE HL, DIMSDALE JE. The UCSD Statin Study: a randomized controlled trial assessing the impact of statins on selected noncardiac outcomes. Control Clin Trials. 2004;25(2):178–202. doi: 10.1016/j.cct.2003.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.GUENGERICH FP. Human cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450. 3. New York, Boston, Dordrecht, London, Moscow: Kluwer Academic/Plenum Publishers; 2005. pp. 377–530. [Google Scholar]
- 110.MAKISHIMA M. Nuclear receptors as targets for drug development: regulation of cholesterol and bile acid metabolism by nuclear receptors. J Pharmacol Sci. 2005;97(2):177–183. doi: 10.1254/jphs.fmj04008x4. [DOI] [PubMed] [Google Scholar]
- 111.MICHAEL LF, SCHKERYANTZ JM, BURRIS TP. The pharmacology of LXR. Mini Rev Med Chem. 2005;5(8):729–740. doi: 10.2174/1389557054553767. [DOI] [PubMed] [Google Scholar]