

Abstract
We report on a patient with NF1 microdeletion and clinical manifestations that fulfill the diagnostic criteria for neurofibromatosis type 1 but also presenting features reminiscent of Proteus syndrome.
Key Words: Neurofibromatosis type I, NF1 microdeletion, Proteus syndrome
Introduction
Neurofibromatosis type 1 (NF1, MIM 162200) is an autosomal dominant disorder characterized by café-au-lait spots, neurofibromas, Lisch nodules, and ephelids [Lu-Emerson and Plotkin, 2009]. Microdeletions that encompass the entire NF1 gene at 17q11.2 occur in approximately 5% of patients with NF1 [Kluwe et al., 2004], and the phenotype of these patients includes early onset of neurofibromas and tumor malignization and is usually more severe than in patients with intragenic mutations [De Raedt et al., 2003]. Occasionally patients with NF1 might also present asymmetric overgrowth and vascular anomalies as well as skin lesions, a triad of signs present in several syndromes such as hemihyperplasia-multiple lipomatosis syndrome and Proteus syndrome (MIM 176920) [Biesecker, 2006], thus making difficult differential diagnosis. In the patient described here, in spite of the presence of Proteus-like clinical features, the typical manifestations of NF1 prompted the testing of the NF1 gene that was found to be deleted.
Case Report
The boy (fig. 1) was born at term, by C-section, with a weight of 3,900 g. The parents were 2nd degree cousins. At age 5 months, he developed acute pericarditis. At age 7 years, neuropsychomotor development was normal except for deambulation, which was absent, possibly due to lower limb length discrepancy. His weight was 23 kg (50th centile), height 130 cm (90–95th centile) and head circumference (OFC) was 52.5 cm (50–98th centile). He had facial asymmetry, downslanted palpebral fissures, hypertelorism (inner and outer canthal distances >97th centile), frontal hirsutism, short and smooth philtrum, gum hyperplasia; axillary and cervical freckling; pectus excavatum; a large number of café-au-lait spots larger than 0.5 cm all over the body, hyperchromic lesion with hirsutism on the abdominal, lombosacral, perineal and genital regions, and also in the lower limbs. He showed increased growth of the left lower limb and the left foot sole, with the appearance of hypertrophic cerebriform connective tissue nevi; histopathological examination of the tumoral masses in the lower left limb revealed diffuse neurofibromas with extensive adipocitic hyperplasia compromising the connective tissue and peripheral nerve sheaths, thus mischaracterizing the conjunctive nevus in the plantar region. Slit lamp examination disclosed the presence of Lisch nodules. Skeletal X-rays showed lower limb length discrepancy (L > R) and metaphyseal enlargement of the distal femur and proximal tibia. An abdominal CT scan, performed at the age of 5 years, documented the presence of a paravertebral mass in the lumbar and sacral regions; histopathological examination revealed diffuse neurofibroma that was surgically removed.
Fig. 1.
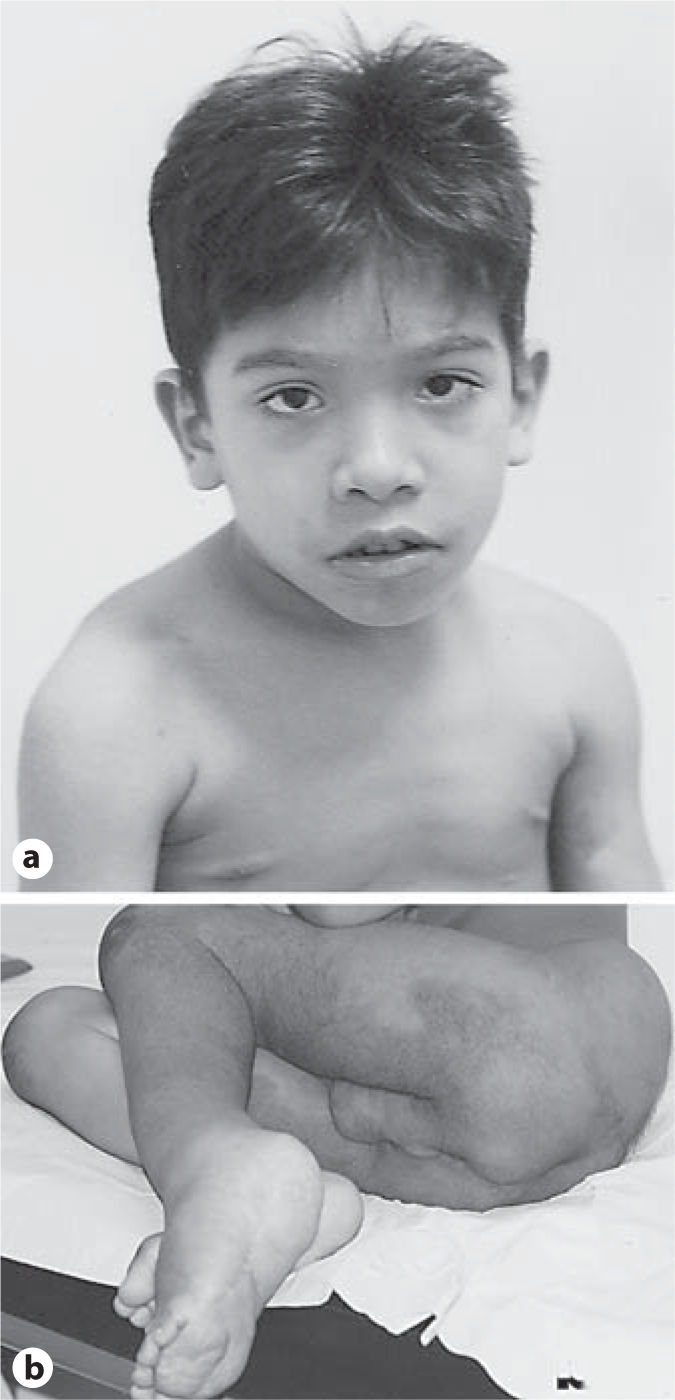
Patient at age 7 years. a Facial dysmorphisms. b Hyperchromic lesion with hirsutism on the abdominal, lombosacral, perineal and genital regions, and in the lower limbs; hypertrophic cerebriform connective tissue nevi-like defect on the left foot sole.
Although the patient's phenotype fulfilled the clinical criteria for NF1 (neurofibromas, axillary freckling, café-au-lait spots and Lisch nodules), he also presented features reminiscent of Proteus syndrome (apparent cerebriform connective tissue nevus, asymmetric disproportionate overgrowth of limbs, linear epidermal nevus, lipomas, capillary and venous malformations, and a facial phenotype including a long face with down slanting of palpebral fissures, ptosis, low nasal bridge, wide and anteverted nares, and an open mouth at rest).
Genetic Analyses and Discussion
Chromosome analysis after G-banding did not show any abnormalities. Fluorescent in situ hybridization was performed on peripheral blood metaphases with probes mapping to the NF1 gene and flanking regions, following standard procedures. The intragenic probes RP11–977D19 and RP11–1093P22 as well as NF1 flanking probes RP11–525H19, RP11–1126E18 and RP11–904A12 were deleted on one chromosome 17. Probes RP11–1108O9 and RP11–96J11 (5′ to NF1REP-P) and RP11–55J8 and RP11–227G15 (3′ to NF1REP-M) hybridized to both chromosomes 17. Probes RP11–946G8 and RP11–1124G9 (NF1REP-P), and RP11–953O6 (NF1REP-M) consistently showed differences in hybridization signal intensities between the 2 chromosomes 17, thus mapping the proximal and distal deletion breakpoints to the NF1 flanking low-copy repeats, NF1REP-P and NF1REP-M, respectively. It corresponds to the most commonly identified deletion in NF1 patients accounting for approximately 80% of all NF1 deletions [Dorschner et al., 2000; López-Correa et al., 2000]. Genotyping of microsatellite marker D17S635 pointed to the maternal origin of the deletion, as observed in 80% of NF1 deletions [Upadhyaya et al., 1998]. Therefore, the severity in the phenotype of our patient cannot be explained by either the deletion size or parental origin. Since PTEN, a tumor suppressor gene, was previously reported as mutated in patients with a Proteus-like syndrome as well as in other hamartomatous disorders [Zhou et al., 2001; Blumenthal and Dennis, 2008], we performed direct sequencing of the promoter and coding region of this gene. No mutations were detected.
Although Proteus and NF1 are likely to be distinct disorders, significant overlap between the two of them may occur. The ‘elephant man’ is a classic case in which the diagnosis of NF1 was reconsidered in favor of Proteus syndrome [Cohen, 1988], and other reports of misdiagnosed cases point to difficulties in clinical differentiation [Cardoso et al., 2003; Biesecker and Cohen, 2005]. In the case reported here, the presence of clinical signs diagnostic of NF1 led to the investigation that identified the causative NF1 mutation.
Acknowledgement
Grant support: FAPESP (CEPID – Center for the Study of the Human Genome 98/14254-2).
Footnotes
L.A.P. and F.M.P. contributed equally to this work.
References
- Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14:1151–1157. doi: 10.1038/sj.ejhg.5201638. [DOI] [PubMed] [Google Scholar]
- Biesecker LG, Cohen MM., Jr Misdiagnosis of neurofibromatosis type 1 as Proteus syndrome. Panminerva Med. 2005;47:197. [PubMed] [Google Scholar]
- Blumenthal GM, Dennis PA. PTEN hamartoma tumor syndromes. Eur J Hum Genet. 2008;16:1289–1300. doi: 10.1038/ejhg.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso MT, de Carvalho TB, Casulari LA, Ferrari I. Proteus syndrome and somatic mosaicism of the chromosome 16. Panminerva Med. 2003;45:267–271. [PubMed] [Google Scholar]
- Cohen MM., Jr Further diagnostic thoughts about the Elephant Man. Am J Med Genet. 1988;29:777–782. doi: 10.1002/ajmg.1320290407. [DOI] [PubMed] [Google Scholar]
- De Raedt T, Brems H, Wolkenstein P, Vidaud D, Pilotti S, et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet. 2003;72:1288–1292. doi: 10.1086/374821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K. NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet. 2000;9:35–46. doi: 10.1093/hmg/9.1.35. [DOI] [PubMed] [Google Scholar]
- Kluwe L, Siebert R, Gesk S, Friedrich RE, Tinschert S, et al. Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum Mutat. 2004;23:111–116. doi: 10.1002/humu.10299. [DOI] [PubMed] [Google Scholar]
- López-Correa C, Brems H, Lázaro C, Marynen P, Legius E. Unequal meiotic crossover: a frequent cause of NF1 microdeletions. Am J Hum Genet. 2000;66:1969–1974. doi: 10.1086/302920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu-Emerson C, Plotkin SR. The Neurofibromatoses. Part 1: NF1. Rev Neurol Dis. 2009;6:E47–E53. [PubMed] [Google Scholar]
- Upadhyaya M, Ruggieri M, Maynard J, Osborn M, Hartog C, et al. Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum Genet. 1998;102:591–597. doi: 10.1007/s004390050746. [DOI] [PubMed] [Google Scholar]
- Zhou X, Hampel H, Thiele H, Gorlin RJ, Hennekam RC, et al. Association of germline mutation in the PTEN tumour suppressor gene and Proteus and Proteus-like syndromes. Lancet. 2001;358:210–211. doi: 10.1016/s0140-6736(01)05412-5. [DOI] [PubMed] [Google Scholar]